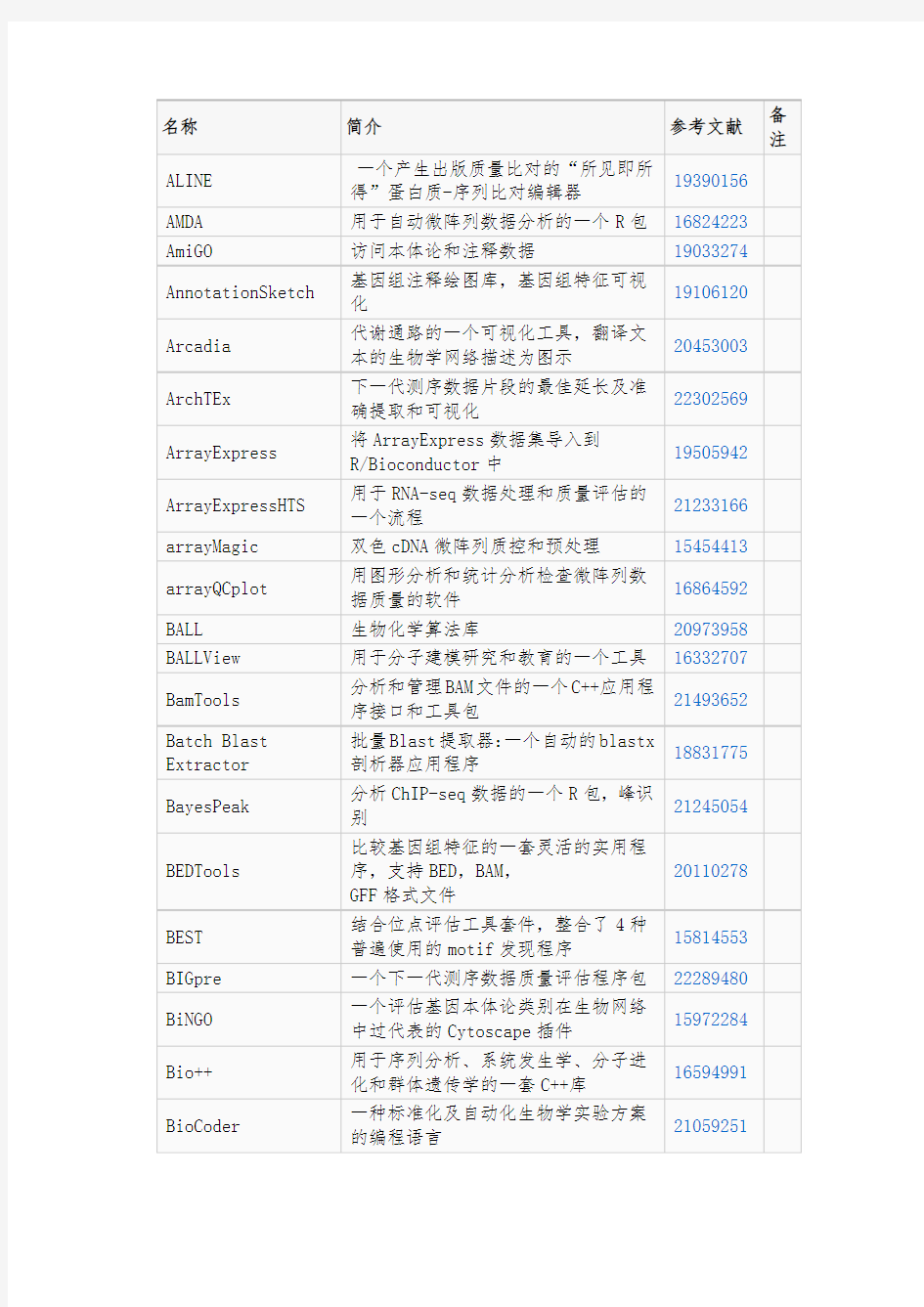
常用的生物信息学软件的介绍和文献依据

生物信息学的主要研究内容
常用数据库 在DNA序列方面有GenBank、EMBL和等 在蛋白质一级结构方面有SWISS-PROT、PIR和MIPS等 在蛋白质和其它生物大分子的结构方面有PDB等 在蛋白质结构分类方面有SCOP和CATH等 生物信息学的主要研究内容 1、序列比对(Alignment) 基本问题是比较两个或两个以上符号序列的相似性或不相似性。序列比对是生物信息学的基础,非常重要。两个序列的比对有较成熟的动态规划算法,以及在此基础上编写的比对软件包BLAST和FASTA,可以免费下载使用。这些软件在数据库查询和搜索中有重要的应用。 2、结构比对 基本问题是比较两个或两个以上蛋白质分子空间结构的相似性或不相似性。已有一些算法。 3、蛋白质结构预测,包括2级和3级结构预测,是最重要的课题之一 从方法上来看有演绎法和归纳法两种途径。前者主要是从一些基本原理或假设出发来预测和研究蛋白质的结构和折叠过程。分子力学和分子动力学属这一范畴。后者主要是从观察和总结已知结构的蛋白质结构规律出发来预测未知蛋白质的结构。同源模建(Homology)和指认(Threading)方法属于这一范畴。虽然经过30余年的努力,蛋白结构预测研究现状远远不能满足实际需要。 4、计算机辅助基因识别(仅指蛋白质编码基因)。最重要的课题之一 基本问题是给定基因组序列后,正确识别基因的范围和在基因组序列中的精确位置.这是最重要的课题之一,而且越来越重要。经过20余年的努力,提出了数十种算法,有十种左右重要的算法和相应软件上网提供免费服务。原核生物计算机辅助基因识别相对容易些,结果好一些。从具有较多内含子的真核生物基因组序列中正确识别出起始密码子、剪切位点和终止密码子,是个相当困难的问题,研究现状不能令人满意,仍有大量的工作要做。 5、非编码区分析和DNA语言研究,是最重要的课题之一 在人类基因组中,编码部分进展总序列的3~5%,其它通常称为“垃圾”DNA,其实一点也不是垃圾,只是我们暂时还不知道其重要的功能。分析非编码区DNA 序列需要大胆的想象和崭新的研究思路和方法。DNA序列作为一种遗传语言,不仅体现在编码序列之中,而且隐含在非编码序列之中。 6、分子进化和比较基因组学,是最重要的课题之一 早期的工作主要是利用不同物种中同一种基因序列的异同来研究生物的进化,构建进化树。既可以用DNA序列也可以用其编码的氨基酸序列来做,甚至于可通过相关蛋白质的结构比对来研究分子进化。以上研究已经积累了大量的工作。近年来由于较多模式生物基因组测序任务的完成,为从整个基因组的角度来研究分子进化提供了条件。 7、序列重叠群(Contigs)装配 一般来说,根据现行的测序技术,每次反应只能测出500或更多一些碱基对的序列,这就有一个把大量的较短的序列全体构成了重叠群(Contigs)。逐步把它们拼接起来形成序列更长的重叠群,直至得到完整序列的过程称为重叠群装配。拼接EST数据以发现全长新基因也有类似的问题。已经证明,这是一个NP-完备
生物信息学软件及使用概述
生物信息学软件及使 刘吉平 liujiping@https://www.360docs.net/doc/163576306.html, 用概述 生 物秀-专心做生物! w w w .b b i o o .c o m
生物信息学是一门新兴的交叉学生物信息学的概念: 科,它将数学和计算机知识应用于生物学,以获取、加工、存储、分类、检索与分析生物大分子的信息,从而理解这些信息的生物学意义。 生 物秀-专心做生物! w w w .b b i o o .c o m
分析和处理实验数据和公共数据,生物信息学软件主要功能 1.2.提示、指导、替代实验操作,利用对实验数据的分析所得的结论设计下一阶段的实验 3.实验数据的自动化管理 4.寻找、预测新基因及其结构、功能 5.蛋白质高级结构及功能预测(三维建模,目前研究的焦点和难点) 生 物秀-专心做生物! w w w .b b i o o .c o m
功能1. 分析和处理实验数据和公共数据,加快研究进度,缩短科研时间 ?核酸:序列同源性比较,分子进化树构建,结构信息分析,包括基元(Motif)、酶切点、重复片断、碱基组成和分布、开放阅读框(ORF ),蛋白编码区(CDS )及外显子预测、RNA 二级结构预测、DNA 片段的拼接; ?蛋白:序列同源性比较,结构信息分析(包括Motif ,限制酶切点,内部重复序列的查找,氨基酸残基组成及其亲水性及疏水性分析),等电点及二级结构预测等等; ?本地序列与公共序列的联接,成果扩大。 生 物秀-专心做生物! w w w .b b i o o .c o m
Antheprot 5.0 Dot Plot 点阵图 Dot plot 点阵图能够揭示多个局部相似性的复杂关系 生 物秀-专心做生物! w w w .b b i o o .c o m
常用生物学软件简介
网址: https://www.360docs.net/doc/163576306.html,/ 1. Oligo 6是目前使用最为广泛的一款引物设计软件,除了可以简单快捷地完成各种引物和探针的设计与分析外,还具有很多其他同类软件所不具有的高级功能:a) 已知一个PC R引物的序列,搜寻和设计另一个引物的序列。b) 按照不同的物种对MM子的偏好性设计简并引物。c) 对环型DNA片段,设计反向PCR引物。d) 设计多重PCR引物。e) 为LCR反应设计探针,以检测某个突变是否出现。f) 分析和评价用其他途径设计的引物是否合理。g) 同源序列查找,并根据同源区设计引物。 h) 增强了的引物/探针搜寻手段。设计引物过程中,可以“Lock”每个参数,如Tm值范围和引物3’端的稳定性等。i) 以多种形式存储结果;支持多用户,每个用户可保存自己的特殊设置。 网址: Oligo 6.71 Demo(引物设计软件):https://www.360docs.net/doc/163576306.html,/Soft/2006/112.htm Oligo—引物设计软件电子教程(引物设计和评估) Oligo 6 Tour 主要功能介绍 https://www.360docs.net/doc/163576306.html,/ 2.Vector NTI Suite是一套功能最全,而且界面最美观,最友好的分子生物学应用软件包。主要包括四个大型软件,它们分别可以对DNA、RNA、蛋白质分子进行各种分析和操作。Vector⑴NTI:作为Vecto r NTI Suite的核心组成部分,它可以在生物研究的全过程中提供数据组织和序列编辑的软件支持。Vector NTI 是以一种窗口形式,且支持项目组织的数据库来完成这一功能的;通过这个数据库,可以保存和组织大部分的实验数据,比如:基因结构、载体、序列片断、引物、蛋白质、多肽、电泳Markers和限制性内切酶等。实际上,该数据库还支持对Vector NTI Suite中各种小型的绘图和结果展示工具的管理。Vector NTI 可以按照用户要求设计克隆策略。用户只需提供克隆载体,外源片断序列,明确载体克隆的大致位置或酶切位点,其它工作由软件完成。设计结果以图文形式输出到屏幕;最后根据客户定制的条件进行模拟电泳。Vector NTI 还具有强大的设计和评估PCR引物、测序引物和杂交探针功能。BioPlot⑵:BioPlot 是一个对蛋白质和核酸序列进行各种理化特性分析的综合性工具,它是一种方便的桌面程序。和其他程序不同的是,BioPlot可以绘制50种以上预定制的蛋白质特征图谱,如疏水性和抗原性;并将序列与特征图谱和活性序列区域一一对应。BioPlot还可以对核酸序列进行8种不同类型的分析,如:退火温度、自由能和GC含量等。AlignX⑶:AlignX可以对多个蛋白质或核酸序列进行同源比较,以寻找不同序列之间的同源区域或相似性很高序列中的不同碱基,并绘制进化树;为下一步设计PCR引物、探针及研究系统发育提供基础。AlignX可以识别所有标准TXT格式,如FASTA、GeneBank、EMBL、SWISS-PROT、GenPept 和ASCII Text。ContigExpress⑷:Contig Express是用来对多个小核酸片段进行拼接而形成连续的长序列。这些小片段可以是Text序列,也可以是直 接从自动测序仪得到的测序图。它用同一个浏览窗口来组织序列片段和拼接结果,同时还有一个由多个子窗口组成的窗口将序列和它们的特性测序图及拼接示意图分别对应。拼接的结果可以直接保存成GeneBan k、EMBL或FASTA文件。Vector NTI Suite⑸其他功能:支持多用户。提供PubMed/Entrez-Search、B last Search、Blast Viewer和3D-Mol(用来看PDB文件)等在线工具。 网址: Vector NTI7.0 中文使用手册 Vector NTI Suite 9.1 Demo(综合性蛋白核酸分析工具包)https://www.360docs.net/doc/163576306.html,/Soft/2007 /460.htm https://www.360docs.net/doc/163576306.html,/solutions/vectornti/index.html 3.DNAStar 是一款基于Windows和Macintosh平台的序列分析软件,特点是操作简单,功能强大。主
生物信息学实验指导讲解
生物信息学实验指导 适用专业:生物技术与制药大类 生物技术 编写:解增言 生物信息学院 2014年9月
目录 实验1 在线BLAST同源序列查询 (3) 实验2 本地BLAST同源序列查询 (8) 实验3 利用ClustalX与MEGA进行多序列比对与分子系统发生树构建 (10) 实验4 利用RNAfold预测RNA二级结构 (14) 实验5 Pfam蛋白质结构域分析 (17) 实验6 利用PSSpred预测蛋白质二级结构 (19) 实验7 利用Cn3D和RasMol分析蛋白质三级结构 (21) 实验8 利用GO及EST数据分析基因功能 (24)
实验1 在线BLAST同源序列查询 一、实验目的 1.了解同源序列查询的原理和用途; 2.掌握利用NCBI在线BLAST工具查找同源序列的方法。 二、实验原理 在生物学种系发生理论中,若两个或多个结构具有相同的祖先,则称它们同源(homologous)。分子生物学中的同源指两条序列来自于一条共同的祖先序列。一般来说,相似超过一定程度的序列具有同源性。在生物信息学研究中,常用序列比对(alignment)来研究序列的同源性以及推测物种之间的关系。 最常见的比对是蛋白质序列之间或核酸序列之间的两两比对,通过比较两个序列之间的相似区域和保守性位点,寻找二者可能的分子进化关系。进一步的比对是将多个蛋白质或核酸同时进行比较,寻找这些有进化关系的序列之间共同的保守区域或位点,从而探索导致它们产生共同功能的序列模式。此外,还可以把蛋白质序列与核酸序列相比来探索核酸序列可能的表达框架;把蛋白质序列与具有三维结构信息的蛋白质相比,从而获得蛋白质折叠类型的信息。 比对还是数据库搜索算法的基础,将查询序列与整个数据库]的所有序列进行比对,从数据库中获得与其最相似序列的已有的数据,能最快速的获得有关查询序列的大量有价值的参考信息,对于进一步分析其结构和功能都会有很大的帮助。近年来随着生物信息学数据大量积累和生物学知识的整理,通过比对方法可以有效地分析和预测一些新发现基因的功能。 序列两两比对 序列比对的理论基础是进化学说,如果两个序列之间具有足够的相似性,就推测二者可能有共同的进化祖先,经过序列内残基的替换、残基或序列片段的缺失、以及序列重组等遗传变异过程分别演化而来。序列相似和序列同源是不同的概念,序列之间的相似程度是可以量化的参数,而序列是否同源需要有进化事实的验证。在残基-残基比对中,可以明显看到序列中某些氨基酸残基比其它位置上的残基更保守,这些信息揭示了这些保守位点上的残基对蛋白质的结构和功能是至关重要的,例如它们可能是酶的活性位点残基,形成二硫键的半胱氨酸残基,与配体结合部位的残基,与金属离子结合的残基,形成特定结构motif的残基等等。但并不是所有保守的残基都一定是结构功能重要的,可能它们只是由于历史的原因被保留下来,而不是由于进化压力而保留下来。因此,如果两个序列有显著的保守性,要确定二者具有共同的进化历史,进而认为二者有近似的结构和功能还需要更多实验和信息的支持。通过大量实验和序列比对的分析,一般认为蛋白质的结构和功能比序列具有更大的保守性,因此粗略的说,如果序列之间的相似性超过30%,它们就很可能是同源的。 早期的序列比对是全局的序列比较,但由于蛋白质具有的模块性质,可能由于外显子的交换而产生新蛋白质,因此局部比对会更加合理。通常用打分矩阵描述序列两两比对,两条序列分别作为矩阵的两维,矩阵点是两维上对应两个残基的相似性分数,分数越高则说明两个残基越相似。因此,序列比对问题变成在矩阵里寻找最佳比对路径,目前最有效的方法是Needleman-Wunsch动态规划算法,在此基础上又改良产生了 Smith-Waterman算法和SIM算法。在 FASTA程序包中可以找到用动态规划算法进行序列比对的工具LALIGN,它能给出多个不相互交叉的最佳比对结果。
生物信息学复习题及答案(陶士珩)
生物信息学复习题 一、名词解释 生物信息学, 二级数据库, FASTA序列格式, genbank序列格式, Entrez,BLAST,查询序列(query),打分矩阵(scoring matrix),空位(gap),空位罚分,E 值, 低复杂度区域,点矩阵(dot matrix),多序列比对,分子钟,系统发育(phylogeny),进化树的二歧分叉结构,直系同源,旁系同源,外类群,有根树,除权配对算法(UPGMA),邻接法构树,最大简约法构树,最大似然法构树,一致 树(consensus tree),bootstrap,开放阅读框(ORF),密码子偏性(codon bias),基因预测的从头分析法,结构域(domain),超家族,模体(motif),序列表谱(profile),PAM矩阵,BLOSUM,PSI-BLAST,RefSeq,PDB数据库,GenPept, 折叠子,TrEMBL,MMDB,SCOP,PROSITE,Gene Ontology Consortium,表谱(profile)。 二、问答题 1)生物信息学与计算生物学有什么区别与联系? 2)试述生物信息学研究的基本方法。 3)试述生物学与生物信息学的相互关系。 4)美国国家生物技术信息中心(NCBI)的主要工作是什么?请列举3个以上NCBI 维护的数据库。 5)序列的相似性与同源性有什么区别与联系? 6)BLAST套件的blastn、blastp、blastx、tblastn和tblastx子工具的用途 什么? 7)简述BLAST搜索的算法。 8)什么是物种的标记序列? 9)什么是多序列比对过程的三个步骤? 10)简述构建进化树的步骤。 11)简述除权配对法(UPGMA)的算法思想。 12)简述邻接法(NJ)的算法思想。 13)简述最大简约法(MP)的算法思想。 14)简述最大似然法(ML)的算法思想。 15)UPGMA构树法不精确的原因是什么? 16)在MEGA2软件中,提供了多种碱基替换距离模型,试列举其中2种,解释其 含义。 17)试述DNA序列分析的流程及代表性分析工具。 18)如何用BLAST发现新基因? 19)试述SCOP蛋白质分类方案。 20)试述SWISS-PROT中的数据来源。 21)TrEMBL哪两个部分? 22)试述PSI-BLAST 搜索的5个步骤。 三、操作与计算题 1)如何获取访问号为U49845的genbank文件?解释如下genbank文件的LOCUS行提供的信息: LOCUS SCU49845 5028 bp DNA linear PLN 21-JUN-1999 2)利用Entrez检索系统,对核酸数据搜索,输入如下信息,将获得什
生物信息学中的序列比对算法
生物信息学中的序列比对算法 张永1,王瑞2 (1.南昌航空大学计算机学院,江西南昌330063;2.江西大宇职业技术学院,江西南昌330038) 摘要:生物信息学是以计算机为工具对生物信息进行储存、检索和分析的科学。序列比对是生物信息学中的一个基本问题,设计快速而有效的序列比对算法是生物信息学研究的一个重要内容,通过序列比较可以发现生物序列中的功能、结构和进化的信息,序列比较的基本操作是比对。本文介绍了序列比对算法的发展现状,描述了常用的各类序列比对算法,并分析了它们的优劣。 关键词:生物信息学;双序列比对;多序列比对 中图分类号:TP301文献标识码:A文章编号:1009-3044(2008)03-10181-04 SequenceAlignmentAlgorithmsinBioinformatics ZHANGYong1,WANGRui2 (1.SchoolofComputing,NanchangHangkongUniversity,Nanchang330063,China;2.JiangxiDayuVocationalInstitute,Nanchang330038,China) Abstract:Bioinformaticsisthesubjectofusingcomputertostore,retrieveandanalyzebiologicalinformation.Sequencealignmentisaba-sicprobleminBioinformatics,anditsmainresearchworkistodeveloprapidandeffectivesequencealignmentalgorithms.Wemaydiscov-erfunctional,structuralandevolutionaryinformationinbiologicalsequencesbysequencecomparing.Thispaperintroducesthedevelop-mentactualityofsequencealignmentalgorithms,describesvarietyofsequencealignmentalgorithmandanalysestheadvantagesanddisad-vantagesofthem. Keywords:Bioinformatics;PairwiseSequenceAlignment;MultipleSequenceAlignment 1引言 生物信息学是80年代末随着人类基因组计划的启动而兴起的一门新的交叉学科,最初常被称为基因组信息学。生物信息学是在生命科学的研究中,以计算机为工具对生物信息进行储存、检索和分析的科学。它是当今生命科学和自然科学的重大前沿领域之一,同时也将是21世纪自然科学的核心领域之一。其研究重点主要体现在基因组学和蛋白组学两方面,具体说,是从核酸和蛋白质序列出发,分析序列中表达结构与功能的生物信息。 生物信息学的研究重点主要体现在基因组学和蛋白质学两方面,具体地说就是从核酸和蛋白质序列出发,分析序列中表达结构和功能的生物信息。生物信息学的基本任务是对各种生物分析序列进行分析,也就是研究新的计算机方法,从大量的序列信息中获取基因结构、功能和进化等知识。在从事分子生物学研究的几乎所有实验室中,对所获得的生物序列进行生物信息学分析已经成为下一步实验之前的一个标准操作。而在序列分析中,将未知序列同已知序列进行相似性比较是一种强有力的研究手段,从序列的片段测定,拼接,基因的表达分析,到RNA和蛋白质的结构功能预测,物种亲缘树的构建都需要进行生物分子序列的相似性比较。例如,有关病毒癌基因与细胞癌基因关系的研究,免疫分子相互识别与作用机制的研究,就大量采用了这类比较分析方法。这种相似性比较分析方法就称为系列比对(SequenceAlignment)。目前,国际互联网上提供了众多的序列比对分析软件。然而,不同的分析软件会得到不同的结果,同时所使用的参数在很大程度上影响到分析的结果。有时常常会由于采用了不合适的参数而丢失了弱的但却具有统计学显著性意义的主要信息,导致随后的实验研究走弯路。因此,生物信息学中的序列比对算法的研究具有非常重要的理论与实践意义。 序列比对问题根据同时进行比对的序列数目分为双序列比对和多序列比对。双序列比对有比较成熟的动态规划算法,而多序列比对目前还没有快速而又十分有效的方法。一般来说,评价生物序列比对算法的标准有两个:一为算法的运算速度,二为获得最佳比对结果的敏感性或准确性。人们虽已提出众多的多序列比对算法,但由于问题自身的计算复杂性,它还尚未得到彻底解决,是 收稿日期:2007-11-25 基金资助:南昌航空大学校自选(EC200706086) 作者简介:张永(1977-),男,硕士,辽宁铁岭人,南昌航空大学计算机学院讲师,研究方向:生物信息学、信息处理;王瑞(1977-),男,江西大宇职业技术学院外语系助教。
生物信息学数据库或软件
一、搜索生物信息学数据库或者软件 数据库是生物信息学的主要内容,各种数据库几乎覆盖了生命科学的各个领域。 核酸序列数据库有GenBank,EMBL,DDB等,核酸序列是了解生物体结构、功能、发育和进化的出发点。国际上权威的核酸序列数据库有三个,分别是美国生物技术信息中心(NCBI)的GenBank ,欧洲分子生物学实验室的EMBL-Bank(简称EMBL),日本遗传研究所的DDBJ 蛋白质序列数据库有SWISS-PROT,PIR,OWL,NRL3D,TrEMBL等, 蛋白质片段数据库有PROSITE,BLOCKS,PRINTS等, 三维结构数据库有PDB,NDB,BioMagResBank,CCSD等, 与蛋白质结构有关的数据库还有SCOP,CATH,FSSP,3D-ALI,DSSP等, 与基因组有关的数据库还有ESTdb,OMIM,GDB,GSDB等, 文献数据库有Medline,Uncover等。 另外一些公司还开发了商业数据库,如MDL等。
生物信息学数据库覆盖面广,分布分散且格式不统一, 因此一些生物计算中心将多个数据库整合在一起提供综合服务,如EBI的SRS(Sequence Retrieval System)包含了核酸序列库、蛋白质序列库,三维结构库等30多个数据库及CLUSTALW、PROSITESEARCH等强有力的搜索工具,用户可以进行多个数据库的多种查询。 二、搜索生物信息学软件 生物信息学软件的主要功能有: 分析和处理实验数据和公共数据,加快研究进度,缩短科研时间; 提示、指导、替代实验操作,利用对实验数据的分析所得的结论设计下一阶段的实验;寻找、预测新基因及预测其结构、功能; 蛋白高级结构预测。 如:核酸序列分析软件BioEdit、DNAClub等;序列相似性搜索BLAST;多重系列比对软件Clustalx;系统进化树的构建软件Phylip、MEGA等;PCR 引物设计软件Primer premier6.0、oligo6.0等;蛋白质二级、三级结构预测及三维分子浏览工具等等。 NCBI的网址是:https://www.360docs.net/doc/163576306.html,。 Entrez的网址是:https://www.360docs.net/doc/163576306.html,/entrez/。 BankIt的网址是:https://www.360docs.net/doc/163576306.html,/BankIt。 Sequin的相关网址是:https://www.360docs.net/doc/163576306.html,/Sequin/。 数据库网址是:https://www.360docs.net/doc/163576306.html,/embl/。
生物信息学考试复习
——古 A.名词解释 1. 生物信息学:广义是指从事对基因组研究相关的生物信息的获取,加工,储存,分配,分析和解释。狭义是指综合应用信息科学,数学理论,方法和技术,管理、分析和利用生物分子数据的科学。 2. 基因芯片:将大量已知或未知序列的DNA片段点在固相载体上,通过物理吸附达到固定化(cDNA芯片),也可以在固相表面直接化学合成,得到寡聚核苷酸芯片。再将待研究的样品与芯片杂交,经过计算机扫描和数据处理,进行定性定量的分析。可以反映大量基因在不同组织或同一组织不同发育时期或不同生理条件下的表达调控情况。 3. NCBI:National Center for Biotechnology Information.是隶属于美国国立医学图书馆(NLM)的综合性数据库,提供生物信息学方面的研究和服务。 4. EMBL:European Molecular Biology Laboratory.EBI为其一部分,是综合性数据库,提供生物信息学方面的研究和服务。 5. 简并引物:PCR引物的某一碱基位置有多种可能的多种引物的混合体。 6. 序列比对:为确定两个或多个序列之间的相似性以至于同源性,而将它们按照一定的规律排列。
7. BLAST:Basic Local Alignment Search Tool.是通过比对(alignment)在数据库中寻找和查询序列(query)相似度很高的序列的工具。 8. ORF:Open Reading Frame.由起始密码子开始,到终止密码子结束可以翻译成蛋白质的核酸序列,一个未知的基因,理论上具有6个ORF。 9. 启动子:是RNA聚合酶识别、结合并开始转录所必须的一段DNA序列。原核生物启动子由上游调控元件和核心启动子组成,核心启动子包括-35区(Sextama box)TTGACA,-10区(Pribnow Box)TATAAT,以及+1区。真核生物启动子包括远上游序列和启动子基本元件构成,启动子基本元件包括启动子上游元件(GC岛,CAAT盒),核心启动子(TATA Box,+1区帽子位点)组成。 10. motif:模体,基序,是序列中局部的保守区域,或者是一组序列中共有的一小段序列模式。 11. 分子进化树:通过比较生物大分子序列的差异的数值重建的进化树。 12. 相似性:序列比对过程中用来描述检测序列和目标序列之间相似DNA碱基或氨基酸残基序列所占的比例。 同源性:两个基因或蛋白质序列具有共同祖先的结论。13.
生物信息学名词解释
名词解释: Consensus sequence:共有序列,指多种原核基因启动序列特定区域内,通常在转录起始点上游-10及-35区域存在一些相似序列。 1、FASTA序列格式:是将DNA或者蛋白质序列表示为一个带有一些标记的核苷酸或者氨基酸字符串,大于号(>)表示一个新文件的开始,其他无特殊要求。 2、Similarity相似性:是直接的连续的数量关系,是指序列比对过程中用来描述检测序列和目标序列之间相同DNA碱基或氨基酸残基顺序所占比列的高低。 3、genbank序列格式:是GenBank 数据库的基本信息单位,是最为广泛的生物信息学序列格式之一。该文件格式按域划分为4个部分:第一部分包含整个记录的信息(描述符);第二部分包含注释;第三部分是引文区,提供了这个记录的科学依据;第四部分是核苷酸序列本身,以“//”结尾。 4、模体(motif):短的保守的多肽段,含有相同模体的蛋白质不一定是同源的,一般10-20个残基。 5、查询序列(query sequence):也称被检索序列,用来在数据库中检索并进行相似性比较的序列。 6、打分矩阵(scoring matrix):在相似性检索中对序列两两比对的质量评估方法。包括基于理论(如考虑核酸和氨基酸之间的类似性)和实际进化距离(如PAM)两类方法。 7、空位(gap):在序列比对时,由于序列长度不同,需要插入一个或几个位点以取得最佳比对结果,这样在其中一序列上产生中断现象,这些中断的位点称为空位。 8、PDB:PDB中收录了大量通过实验(X射线晶体衍射,核磁共振NMR)测定的生物大分子的三维结构,记录有原子坐标、配基的化学结构和晶体结构的描述等。PDB数据库的访问号由一个数字和三个字母组成(如,4HHB),同时支持关键词搜索,还可以FASTA程序进行搜索。 9、Prosite:是蛋白质家族和结构域数据库,包含具有生物学意义的位点、模式、可帮助识别蛋白质家族的统计特征。 PROSITE中涉及的序列模式包括酶的催化位点、配体结合位点、与金属离子结合的残基、二硫键的半胱氨酸、与小分子或其它蛋白质结合的区域等;PROSITE 还包括根据多序列比对而构建的序列统计特征,能更敏感地发现一个序列是否具有相应的特征。 10、PIR:是一个集成了关于蛋白质功能预测数据的公共资源的数据库,其目的是支持基因组蛋白质研究。 11、SWLSS—MODE:是目前最著名的蛋白质三级结构预测服务器,建立在已知生物大分子结构基础上,利用同源建模的方法对未知序列的蛋白质三级结构进行预测。 12、空位罚分:空位罚分是为了补偿插入和缺失对序列相似性的影响,序列中的空位的引入不代表真正的进化事件,所以要对其进行罚分,空位罚分的多少直接影响对比的结果。13、E值:衡量序列之间相似性是否显著的期望值。E值大小说明了可以找到与查询序列(query)相匹配的随机或无关序列的概率,E值越接近零,越不可能找到其他匹配序列,E 值越小意味着序列的相似性偶然发生的机会越小,也即相似性越能反映真实的生物学意义。 14、点矩阵(dot matrix):构建一个二维矩阵,其X轴是一条序列,Y轴是另一个序列,然后在2个序列相同碱基的对应位置(x,y)加点,如果两条序列完全相同则会形成一条主对角线,如果两条序列相似则会出现一条或者几条直线;如果完全没有相似性则不能连成直线。 15、多序列比对:通过序列的相似性检索得到许多相似性序列,将这些序列做一个总体的比对,以观察它们在结构上的异同,来回答大量的生物学问题。 16、MEGA:是一款免费的构树软件,它提供了序列比对、格式转换、数据修订、距离计
启动子生物信息学分析软件
https://www.360docs.net/doc/163576306.html,/seq_tools/promoter.html 2. PlantCARE(plant cis-acting regulatory elements), a database of plant cis-acting regulatory elements http://bioinformatics.psb.ugent.be/webtoo ls/plantcare/html/ 3. promoter 2.0 prediction server http://www.cbs.dtu.dk/services/Promoter/ 4. 启动子分析网址: 1 https://www.360docs.net/doc/163576306.html,/seq_tools/promoter.html 2 http://alggen.lsi.upc.es/recerca/menu_recerca.html 3 http://www.cbs.dtu.dk/services/Promoter/ 4 https://www.360docs.net/doc/163576306.html,/~molb470/ ... s/solorz/index.html 5 https://www.360docs.net/doc/163576306.html,/molbio/proscan/ http://bip.weizmann.ac.il/toolbo ... ters.html#databases https://www.360docs.net/doc/163576306.html,/seq_tools/promoter.html https://www.360docs.net/doc/163576306.html,.sg/promoter/CGrich1_0/CGRICH.htm https://www.360docs.net/doc/163576306.html,/pub/programs.html#pmatch https://www.360docs.net/doc/163576306.html,.hk/~b400559/arraysoft_pathway.html#Promoter http://www.dna.affrc.go.jp/PLACE/signalup.html http://intra.psb.ugent.be:8080/PlantCARE/ http://www.cbs.dtu.dk/services/Promoter/ https://www.360docs.net/doc/163576306.html,/molbio/proscan/ https://www.360docs.net/doc/163576306.html,/molbio/signal/ https://www.360docs.net/doc/163576306.html,/thread-41571-1-1.htm 常用启动子分析网址: http://bip.weizmann.ac.il/toolbox/seq_analysis/promoters.html#databas es
生物信息学软件使用
生物信息学软件的使用(以MC4R基因为例) 第一章从NCBI上查找DNA、mRNA、蛋白质序列 一、以猪的黑素皮质素受体4(MC4R, melanocortin-4 re-ceptor)基因为例,介绍如何从NCBI 上查找DNA、mRNA、氨基酸序列。 1.首先查找MC4R的DNA序列。 在百度里输入NCBI,打开后得到的结果如下网页: 在Search 栏输入“MC4R pig”,在下拉菜单里选择Gene,然后点击Search,得到如下结果:
点击第一个ID为397359的链接,得到如下的结果:
可以看到该基因位于猪的1号染色体上,在右下方有个“Go to nucleotide”即进入核酸序列,有三种格式(用红圈标记的),经常用的是“FASTA”和“GenBank”,“FASTA”格式的比较简洁,不包含任何的数字,就全部是碱基,序列的对比和分析是就要用到这种格式;而“GenBank”格式就比较详细,可以查看到很多信息,比如碱基数、mRNA序列、内含子、外显子、CDS,以及氨基酸序列等等之类的。点击GenBank后得到如下结果: Sus scrofa breed mixed chromosome 1, Sscrofa10.2 DNA LOCUS NC_010443 2265 bp DNA linear CON 29-SEP-2013 DEFINITION Sus scrofa breed mixed chromosome 1, Sscrofa10.2. ACCESSION NC_010443 REGION: complement(178553488..178555752) GPC_000000583 VERSION NC_010443.4 GI:347618793 DBLINK BioProject: PRJNA28993 Assembly: GCF_000003025.5 KEYWORDS RefSeq. SOURCE Sus scrofa (pig) ORGANISM Sus scrofa Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Laurasiatheria; Cetartiodactyla; Suina; Suidae; Sus. COMMENT REFSEQ INFORMATION: The reference sequence is identical to CM000812.4. On Oct 11, 2011 this sequence version replaced gi:333795951. Assembly Name: Sscrofa10.2 The genomic sequence for this RefSeq record is from the genome assembly released by the Swine Genome Sequencing Consortium as Sscrofa10.2 in August 2011 (see https://www.360docs.net/doc/163576306.html,/Projects/S_scrofa). Sscrofa10.2 is a mixed assembly of clones and contigs from the whole-genome shotgun
生物信息学常用工具
常用DNA和蛋白质序列数据分析工具: ●序列比对工具: a)BLAST: ●网络比对,包括基础的Blast比对、参数、特殊Blast如PSI-Blast、Blast2 等; ●本地比对,包括程序下载、安装、数据库的下载及格式化、Blast程序的 运行等。 b)多序列比对ClustalX(Windows系统) 包括程序下载、安装、及程序的运行、结果的输入输出等。 ●真核生物基因结构的预测: a)基因可读框的识别: Genescan; CpG岛、转录终止信号和启动子区域预测; CpGPlot; POLYAH; PromoterScan; b)基因密码子偏好性: CodonW; c)采用mRNA序列预测基因: Spidey; d)ASTD数据库 ●分子进化遗传分析工具 ●MEGA;
●Phylip; ●蛋白质结构和功能预测 a)一级结构 ProtParam蛋白质序列理化参数检索; ProtScale蛋白质疏水性分析; COILS卷曲螺旋预测; b)二级结构 PredictProtein蛋白质结构预测; PSIPRED不同蛋白质结构预测方法; c)InterProScan: 模式和序列谱研究 Prosite:蛋白质结构域、家族和功能为点数据库; Pfam:蛋白质家族比对和HMM数据库; BLOCK:模块搜索数据库; SMART:简单模块架构搜索工具; TMHMM:跨膜结构预测工具; d)三级结构 Swiss-Model Workspace: 同源建模的网络综合服务器; Phyre:线串法预测蛋白质折叠; HMMSTR/Rosetta:从头预测蛋白质结构; Swiss-PdbViewer:分子建模和可视化工具; 序列模体的识别和解析; MEME程序包; ●蛋白质谱数据分析
常用生物信息学软件
常用生物信息学软件 一、基因芯片 1、基因芯片综合分析软件。 ArrayVision 7.0 一种功能强大的商业版基因芯片分析软件,不仅可以进行图像分析,还可以进行数据处理,方便protocol的管理功能强大,商业版正式版:6900美元。 Arraypro 4.0 Media Cybernetics公司的产品,该公司的gelpro, imagepro一直以精确成为同类产品中的佼佼者,相信arraypro也不会差。 phoretix? Array Nonlinear Dynamics公司的基因片综合分析软件。 J-express 挪威Bergen大学编写,是一个用JA V A语言写的应用程序,界面清晰漂亮,用来分析微矩阵(microarray)实验获得的基因表达数据,需要下载安装JA V A运行环境JRE1.2后(5.1M)后,才能运行。 2、基因芯片阅读图像分析软件 ScanAlyze 2.44 ,斯坦福的基因芯片基因芯片阅读软件,进行微矩阵荧光图像分析,包括半自动定义格栅与像素点分析。输出为分隔的文本格式,可很容易地转化为任何数据库。 3、基因芯片数据分析软件 Cluster 斯坦福的对大量微矩阵数据组进行各种簇(Cluster)分析与其它各种处理的软件。 SAM Significance Analysis of Microarrays 的缩写,微矩阵显著性分析软件,EXCEL软件的插件,由Stanford大学编制。 4.基因芯片聚类图形显示 TreeView 1.5 斯坦福开发的用来显示Cluster软件分析的图形化结果。现已和Cluster成为了基因芯片处理的标准软件。 FreeView 是基于JA V A语言的系统树生成软件,接收Cluster生成的数据,比Treeview 增强了某些功能。 5.基因芯片引物设计 Array Designer 2.00 DNA微矩阵(microarray)软件,批量设计DNA和寡核苷酸引物工具 三、序列综合分析 V ector NTI Suite 8.0 不喜欢装备各种专业性强的软件,而希望用一个综合性的软件代替的同志可以选择本软件。本阶段的大部分功能它都有。该软件具体特有良好的数据库管理(增加、修改、查找),对要操作的数据放在一个界面相同的数据库中统一管理。软件中的大部分分析可以通过在数据库中进行选定(数据)->分析->结果(显示、保存和入库)三步完成。在分析主界面,软件可以对核酸蛋白分子进行限制酶分析、结构域查找等多种分析和操作,生成重组分子策略和实验方法,进行限制酶片段的虚拟电泳,新建输入各种格式的分子数据、
生物信息学分析方法
核酸和蛋白质序列分析 蛋白质, 核酸, 序列 关键词:核酸序列蛋白质序列分析软件 在获得一个基因序列后,需要对其进行生物信息学分析,从中尽量发掘信息,从而指导进一步的实验研究。通过染色体定位分析、内含子/外显子分析、ORF分析、表达谱分析等,能够阐明基因的基本信息。通过启动子预测、CpG岛分析和转录因子分析等,识别调控区的顺式作用元件,可以为基因的调控研究提供基础。通过蛋白质基本性质分析,疏水性分析,跨膜区预测,信号肽预测,亚细胞定位预测,抗原性位点预测,可以对基因编码蛋白的性质作出初步判断和预测。尤其通过疏水性分析和跨膜区预测可以预测基因是否为膜蛋白,这对确定实验研究方向有重要的参考意义。此外,通过相似性搜索、功能位点分析、结构分析、查询基因表达谱聚簇数据库、基因敲除数据库、基因组上下游邻居等,尽量挖掘网络数据库中的信息,可以对基因功能作出推论。上述技术路线可为其它类似分子的生物信息学分析提供借鉴。本路线图及推荐网址已建立超级链接,放在北京大学人类疾病基因研究中心网站(https://www.360docs.net/doc/163576306.html,/science/bioinfomatics.htm),可以直接点击进入检索网站。 下面介绍其中一些基本分析。值得注意的是,在对序列进行分析时,首先应当明确序列的性质,是mRNA序列还是基因组序列?是计算机拼接得到还是经过PCR扩增测序得到?是原核生物还是真核生物?这些决定了分析方法的选择和分析结果的解释。 (一)核酸序列分析 1、双序列比对(pairwise alignment) 双序列比对是指比较两条序列的相似性和寻找相似碱基及氨基酸的对应位置,它是用计算机进行序列分析的强大工具,分为全局比对和局部比对两类,各以Needleman-Wunsch算法和Smith-Waterman算法为代表。由于这些算法都是启发式(heuristic)的算法,因此并没有最优值。根据比对的需要,选用适当的比对工具,在比对时适当调整空格罚分(gap penalty)和空格延伸罚分(gap extension penalty),以获得更优的比对。 除了利用BLAST、FASTA等局部比对工具进行序列对数据库的搜索外,我们还推荐使用EMBOSS软件包中的Needle软件(http://bioinfo.pbi.nrc.ca:8090/EMBOSS/),和Pairwise BLAST (https://www.360docs.net/doc/163576306.html,/BLAST/)。以上介绍的这些双序列比对工具的使用都比较简单,一般输入所比较的序列即可。 (1)BLAST和FASTA FASTA(https://www.360docs.net/doc/163576306.html,/fasta33/)和BLAST (https://www.360docs.net/doc/163576306.html,/BLAST/)是目前运用较为广泛的相似性搜索工具。这两个工具都采用局部比对的方法,选择计分矩阵对序列计分,通过分值的大小和统计学显著
生物信息学_复习题及答案(打印)
一、名词解释: 1.生物信息学:研究大量生物数据复杂关系的学科,其特征是多学科交叉,以互联网为媒介,数据库为载体。利用数学知识建立各种数学模型; 利用计算机为工具对实验所得大量生物学数据进行储存、检索、处理及分析,并以生物学知识对结果进行解释。 2.二级数据库:在一级数据库、实验数据和理论分析的基础上针对特定目标衍生而来,是对生物学知识和信息的进一步的整理。 3.FASTA序列格式:是将DNA或者蛋白质序列表示为一个带有一些标记的核苷酸或者氨基酸字符串,大于号(>)表示一个新文件的开始,其他无特殊要求。 4.genbank序列格式:是GenBank 数据库的基本信息单位,是最为广泛的生物信息学序列格式之一。该文件格式按域划分为4个部分:第一部分包含整个记录的信息(描述符);第二部分包含注释;第三部分是引文区,提供了这个记录的科学依据;第四部分是核苷酸序列本身,以“//”结尾。 5.Entrez检索系统:是NCBI开发的核心检索系统,集成了NCBI的各种数据库,具有链接的数据库多,使用方便,能够进行交叉索引等特点。 6.BLAST:基本局部比对搜索工具,用于相似性搜索的工具,对需要进行检索的序列与数据库中的每个序列做相似性比较。P94 7.查询序列(query sequence):也称被检索序列,用来在数据库中检索并进行相似性比较的序列。P98 8.打分矩阵(scoring matrix):在相似性检索中对序列两两比对的质量评估方法。包括基于理论(如考虑核酸和氨基酸之间的类似性)和实际进化距离(如PAM)两类方法。P29 9.空位(gap):在序列比对时,由于序列长度不同,需要插入一个或几个位点以取得最佳比对结果,这样在其中一序列上产生中断现象,这些中断的位点称为空位。P29 10.空位罚分:空位罚分是为了补偿插入和缺失对序列相似性的影响,序列中的空位的引入不代表真正的进化事件,所以要对其进行罚分,空位罚分的多少直接影响对比的结果。P37 11.E值:衡量序列之间相似性是否显著的期望值。E值大小说明了可以找到与查询序列(query)相匹配的随机或无关序列的概率,E值越接近零,越不可能找到其他匹配序列,E值越小意味着序列的相似性偶然发生的机会越小,也即相似性越能反映真实的生物学意义。P95 12.低复杂度区域:BLAST搜索的过滤选项。指序列中包含的重复度高的区域,如poly(A)。 13.点矩阵(dot matrix):构建一个二维矩阵,其X轴是一条序列,Y轴是另一个序列,然后在2个序列相同碱基的对应位置(x,y)加点,如果两条序列完全相同则会形成一条主对角线,如果两条序列相似则会出现一条或者几条直线;如果完全没有相似性则不能连成直线。 14.多序列比对:通过序列的相似性检索得到许多相似性序列,将这些序列做一个总体的比对,以观察它们在结构上的异同,来回答大量的生物学问题。 15.分子钟:认为分子进化速率是恒定的或者几乎恒定的假说,从而可以通过分子进化推断出物种起源的时间。 16.系统发育分析:通过一组相关的基因或者蛋白质的多序列比对或其他性状,可以研究推断不同物种或基因之间的进化关系。 17.进化树的二歧分叉结构:指在进化树上任何一个分支节点,一个父分支都只能被分成两个子分支。 系统发育图:用枝长表示进化时间的系统树称为系统发育图,是引入时间概念的支序图。 18.直系同源:指由于物种形成事件来自一个共同祖先的不同物种中的同源序列,具有相似或不同的功能。(书:在缺乏任何基因复制证据的情况下,具有共同祖先和相同功能的同源基因。) 19.旁系(并系)同源:指同一个物种中具有共同祖先,通过基因重复产生的一组基因,这些基因在功能上可能发生了改变。(书:由于基因重复事件产生的相似序列。) 20.外类群:是进化树中处于一组被分析物种之外的,具有相近亲缘关系的物种。 21.有根树:能够确定所有分析物种的共同祖先的进化树。 22.除权配对算法(UPGMA):最初,每个序列归为一类,然后找到距离最近的两类将其归为一类,定义为一个节点,重复这个过程,直到所有的聚类被加入,最终产生树根。 23.邻接法(neighbor-joining method):是一种不仅仅计算两两比对距离,还对整个树的长度进行最小化,从而对树的拓扑结构进行限制,能够克服UPGMA算法要求进化速率保持恒定的缺陷。 24.最大简约法(MP):在一系列能够解释序列差异的的进化树中找到具有最少核酸或氨基酸替换的进化树。 25.最大似然法(ML):它对每个可能的进化位点分配一个概率,然后综合所有位点,找到概率最大的进化树。最大似然法允许采用不同的进化模型对变异进行分析评估,并在此基础上构建系统发育树。