动植物全基因组重测序简介
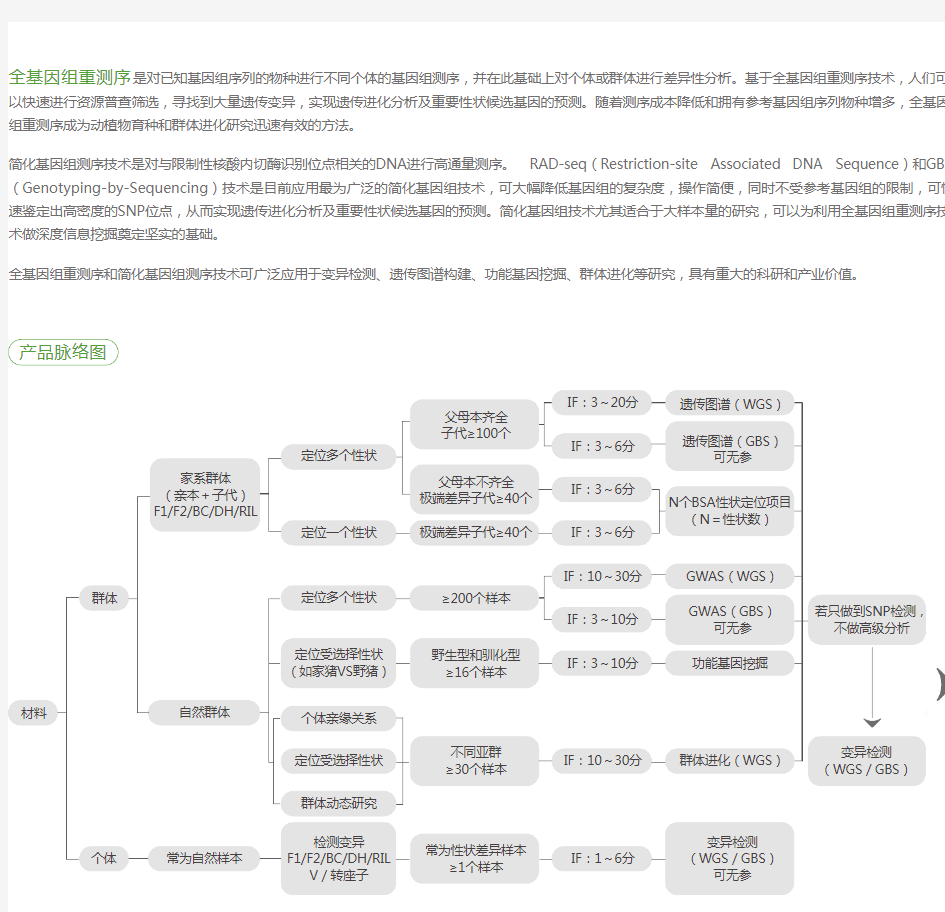
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。 RAD-seq(Restriction-site Associated DNA Sequence)和GBS (Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
产品脉络图
基因组重测序
基因组重测序 背景介绍 全基因组重测序,是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。与已知序列比对,寻找单核苷酸多态性位点(SNP )、插入缺失位点(InDel ,Insertion/Deletion )、结构变异位点(SV ,Structure Variation )位点及拷贝数变化(CNV) 。 可以寻找到大量基因差异,实现遗传进化分析及重要性状候选基因的预测。涉 及临床医药研究、群体遗传学研究、关联分析、进化分析等众多应用领域。 随着测序成本的大幅度降低以及测序效率的数量级提升, 全基因组重测序已经成为研究人类疾病及动植物分子育种最为快速有效的方法之一。利用illumina Hiseq 2000 平台,将不同插入片段文库和双末端测序相结合,可以高效地挖掘基因序列差异和结构变异等信息, 为客户进行疾病研究、分子育种等提供准确依据。 重测序的两个条件:(1)该物种基因组序列已知;(2)所测序群体之间遗传性差异不大( >99% 相似度 ) 在已经完成的全基因组测序及其基因功能注释的基础上,采用全基因组鸟枪法(WGS )对DNA 插入片段进行双末端测序。 技术路线 生物信息学分析
送样要求 1.样品总量:每次样品制备需要大于5ug 的样品。为保证实验质量及延续性,请一次性提供至少20ug的样品。如需多次制备样品,按照制备次数计算样品总量。 2.样品纯度:OD值260/280应在1.8~2.0 之间;无蛋白质、RNA或肉眼可见杂质污染。 3.样品浓度:不低于50 ng/μL。 4.样品质量:基因组完整、无降解,电泳结果基因组DNA主带应在λ‐Hind III digest 最大条带23 Kb以上且主带清晰,无弥散。 5.样品保存:限选择干粉、酒精、TE buffer或超纯水一种,请在样品信息单中注明。 6.样品运输:样品请置于1.5 ml管中,做好标记,使用封口膜封好;基因组DNA如果用乙醇沉淀,可以常温运输;否则建议使用干冰或冰袋运输,并选择较快的运输方式。 提供结果 根据客户需求,提供不同深度的信息分析结果。
人类基因组重测序分析
6 首页 科技服务 医学检测 科学与技术 市场与支持 加入我们 关于我们提供领先的基因组学解决方案 Providing Advanced Genomic Solutions 诺禾致源 人类疾病基因组重测序分析图3 Circos 图 人类基因组重测序分析6项升级 Novo-Zhonghua Genomes 数据库注释 一些位点的突变可能在千人基因组中或在欧美人群中属于低频突变,但是对于中国人群来说却是常见突变。诺禾致源自建中国人数据库 Novo-Zhonghua Genomes,数据库中的所有样本均来自正常中国人群。已有研究表明,与国际通用的多人种数据库相比,使用单一人种数据库进行疾病研究,可以有效减少假阳性现象。 图2 真核生物基因的结构[6] 复杂疾病变异分类标准 DamLevel Variant Calling Variant Annotation Benign Likely Benign VUS Likely Pathogenic Custom knowledge Clinical Data Pathogenic Family Testing Published + in house data Population frequency Predictions: PolyPhen, SIFT, etc Amino acid conservation Published Disease Information Variant classification Candidate Variants Novo-Zhonghua Genomes 数据库注释 复杂疾病突变位点有害性分类 非编码区(Non-coding region)分析 疾病基因组 CNV/SV 分析 基于基因(Gene-based)的 Burden Analysis (复杂疾病散发样本) 可视化的数据结果展示 基于健康中国人群的千人测序数据,测序深度 > 30× 参考 ACMG 等,推出针对复杂疾病变异位点有害性的分类标准 应用 ENCODE 数据库最新内容,并结合国际通用数据库、自建数 复杂疾病突变位点有害性分类 基于美国医学遗传学会 ACMG[2]与 Duzkale H[3]提出的变异分类标准,诺禾致源疾病基因组信息分析团队推出了一套针对复杂疾病变异位点有害性的分类标准 DamLevel(如下图所示)。DamLevel 将变异位点的有害性分为5个层级:Pathogenic、Likely Pathogenic、VUS(Variant of uncertain significance)、Likely Begnin、Begnin,更好地鉴定个体遗传变异与疾病的相关性。 非编码区(Non-coding region)分析 基因组非编码区变异可以引发多种疾病,包括心脏类疾病、糖尿病、癌症、肥胖症等[4,5],但目前对非编码区突变的筛选和功能描述仍具挑战性。诺禾致源非编码区分析,应用 ENCODE 数据库最新内容对非编码区突变进行注释,通过国际通用数据库和自建的 Novo-Zhonghua Genomes 数据库进行频率筛选以及保守性过滤,精确定位非编码区中低频且保守的突变,筛选到与疾病相关的非编码区突变。 疾病基因组 CNV/SV 分析 CNV/SV 与基因表达、表型、人类疾病发生发展都有着非常密切的关系[7,8],诺禾致源疾病基因组信息分析团队研发了一整套 CNV/SV 筛选方法,包括有害性 CNV/SV 筛选和 de novo CNV/SV 分析(基于成三或成四家系)等。利用 DGV、DECIPHER、CNVD 等数据库对变异检出结果进行标记,从结果中进一步过滤掉良性 CNV/SV,经过一系列筛选后,准确鉴定个体 CNV/SV 遗传变异与疾病的相关性。 图4 CNV 分布图 表1 本次产品升级亮点 图5 Burden 分析结果的热图展示 1 2 3 4 5 Novo-Zhonghua Genomes 数据库注释 Novo-Zhonghua Genomes 数据库是诺禾致源自建针对 中国正常人群的数据库,助 力中国人群基因组信息解析。 复杂疾病突变位点 有害性分类 诺禾致源推出的复杂疾病变 异位点有害性的分类标准 (DamLevel),准确标识复杂 疾病的致病性突变位点。 非编码区 (Non-coding region)分析 应用 ENCODE 数据库最新内 容对非编码区进行注释、筛 选,精确定位非编码区中低 频且保守的突变。 疾病基因组 CNV/SV 分析 完整的有害性 CNV/SV 筛选 和 de novo CNV/SV 分析, 准确鉴定个体 CNV/SV 遗传 变异与疾病的相关性。 基于基因 (Gene-based)的 Burden Analysis 针对复杂疾病的研究,通过 检测疾病状态与基因变异的 相关性,寻找特定疾病(或 性状)的易感基因。 可视化的 数据结果展示 灵活易用的测序数据结果展 示,使大量复杂数据的分析 变得轻松而高效,提高数据 可读性。 ? log 10 ( P ? value ) Mutations of Genes Prioritized by Burden Analysis CIR1 PIGP CTSE PRB2 CYP HDAC1 GRK6 PIGK MYL6B EHD2 0810 246 Mutations 4 3 2 1 基于基因(Gene-based)的 Burden Analysis 关联分析是研究复杂疾病的1个重要方法,其通过检测疾病状态与基因变异的相关性,寻找特定疾病(或性状)的易感基因。通常是在具有不同表型的2组个体(一般为患病者和正常对照者)中,基于遗传位点(或基因、单体型)的频率分布差异,间接反映该遗传位点(或基因)可能与疾病(或性状)存在关联性。 Burden Analysis(Gene-based)基于复杂疾病的 case 和 control 散发样本,通过 Fisher's exact test 以及 SKAT 统计方法分析得到候选基因,针对候选基因可以进行富集分析(KEGG 富集分析和 GO 富集分析)与蛋白网络互作分析。 可视化的结果展示 诺禾致源疾病基因组信息分析团队,会为客户提供不断更新的变异注释、项目特异性分析和灵活易用的“变异-基因-疾病”可视化结果,让科学研究更轻松。 图6 疾病与基因关联性展示图 产品名称升级亮点 引领行 业新 标杆 参考文献 [1] Nagasaki M, Yasuda J, Katsuoka F, et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals.[J]. Nature Communications, 2015, 6. 阅读原文 >> [2] Richards S, Aziz N, Bale S, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genetics in Medicine, 2015. 阅读原文 >> [3] Duzkale H, Shen J, McLaughlin H, et al. A systematic approach to assessing the clinical significance of genetic variants[J]. Clinical genetics, 2013, 84(5): 453-463. 阅读原文 >> [4] Yoshinari M, Akihiko M, Dongquan S, et al. A functional polymorphism in the 5' UTR of GDF5 is associated with susceptibility to osteoarthritis.[J]. Nature Genetics, 2007, 39(4):529-33. 阅读原文 >> [5] Kjong-Van L, Ting C. Exploring functional variant discovery in non-coding regions with SInBaD.[J]. Nucleic Acids Research, 2012, 41 (1):e7-e7. 阅读原文 >> [6] https://https://www.360docs.net/doc/331786788.html,/wiki/Regulatory_sequence 阅读原文 >> [7] Sudmant P H, Rausch T, Gardner E J, et al. An integrated map of structural variation in 2,504 human genomes.[J]. Nature, 2015, 526 (7571):75-81. 阅读原文 >> [8] Birney E, Soranzo N. Human genomics: The end of the start for population sequencing.[J]. Nature, 2015, 526(7571):52-3. 阅读原文 >> 免费升级7-9月 新签合同 免费升级数据分析
高通量测序常用名词解释
什么是高通量测序? 高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。 什么是Sanger法测序(一代测序) Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。直到掺入一种链终止核苷酸为止。每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。由于ddNTP 缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。终止点由反应中相应的双脱氧而定。每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。 什么是基因组重测序(Genome Re-sequencing) 全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。 什么是de novo测序 de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。获得一个物种的全基因组序列是加快对此物种了解的重要捷径。随着新一代测序技术的飞速发展,基因组测序所需的成本和时间较传统技术都大大降低,大规模基因组测序渐入佳境,基因组学研究也迎来新的发展契机和革命性突破。利用新一代高通量、高效率测序技术以及强大的生物信息分析能力,可以高效、低成本地测定并分析所有生物的基因组序列。 什么是外显子测序(whole exon sequencing) 外显子组测序是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel等具有较大的优势,但无法研究基因组结构变异如染色体断裂重组等。 什么是mRNA测序(RNA-seq) 转录组学(transcriptomics)是在基因组学后新兴的一门学科,即研究特定细胞在某一功能状态下所能转录出来的所有RNA(包括mRNA和非编码RNA)的类型与拷贝数。Illumina 提供的mRNA测序技术可在整个mRNA领域进行各种相关研究和新的发现。mRNA测序不对引物或探针进行设计,可自由提供关于转录的客观和权威信息。研究人员仅需要一次试验即可快速生成完整的poly-A尾的RNA完整序列信息,并分析基因表达、cSNP、全新的转录、全新异构体、剪接位点、等位基因特异性表达和罕见转录等最全面的转录组信息。简单的样
全基因组重测序数据分析
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排 突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使 得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组 学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基 因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需 要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。
高通量基因组测序中 测序深度,覆盖度
高通量基因组测序中,什么是测序深度和覆盖度? 1G=1024M 测序深度是指测序得到的总碱基数与待测基因组大小的比值。假设一个基因大小为2M,测序深度为10X,那么获得的总数据量为20M。(测序深度=总数据量20M/基因组大小2M=10X) 覆盖度是指测序获得的序列占整个基因组的比例。由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap。例如一个细菌基因组测序,覆盖度是98%,那么还有2%的序列区域是没有通过测序获得的。 1、全基因组重测序是对已知基因组序列的物种进行不同个体的基因 序的个体,通过序列比对,可以找到大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel,Insertion/Deletion)、结构变异位点(SV, 技术路线 提取基因组DNA,利用Covaris进行随机打断,电泳回收所需长度的DNA片段(0.2~5Kb),加上接头, 进行cluster制备(Solexa)或E-PCR (SOLiD),最后利用Paired-End(Solexa)或者Mate-Pair(SOLiD)的方法对插入片段进行重测序。图1-1,以SOLiD为例,说明整个实验方案。
也称目标外显子组捕获,是指利用序列捕获技术将全基因组外显子区域DNA 捕捉并富集后进行高通量测序的基因组分析方法。是一种选择基因组的编码序列的高效策略,外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel 等具有较大的优势。 外显子(expressed region)是真核生物基因的一部分,它在剪接(Splicing)后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质。外显子是最后出现在成熟RNA中的基因序列,又称表达序列。既存在于最初的转录产物中,也存在于成熟的RNA分子中的核苷酸序列。在人类基因中大约有180,000外显子,占人类基因组的1%,约30MB。
群体进化-基于全基因组重测序
DNA样品总量: ≥3 μg 适用范围 样品要求 文库类型测序策略与深度 分析内容项目周期 群体进化(基于全基因组重测序) 标准分析时间为120天,个性化分析需根据项目实际情况进行评估 HiSeq PE150推荐测序深度≥5X/个体350 bp小片段DNA文库 1. 已有参考基因组序列的物种中不同亚群(自然群体) 2. 各亚群间划分明显,同一亚群内的个体有一定代表性 3. 每个亚群选取10个样本左右(推荐动物≥10个,植物≥15个) 4. 总体不少于30个样本与参考基因组比对群体SNP检测、注释及统计系统进化树构建群体遗传结构分析 群体主成分分析连锁不平衡分析选择消除分析候选基因GO和KEGG富集构建单体型图谱种群历史和有效群体大小 技术参数 针对已有参考基因组的物种,对其各亚种进行全基因组重测序获得基因组信息,通过与参考基因组比对,得到大量高准确性的SNP、InDel、SV等变异信息,讨论群体的遗传结构、遗传平衡和影响遗传平衡的因素,从而从分子层面揭示该物种的进化机制、环境适应性等系列问题。该技术能精准地得到全基因组内所有遗传信息,最大程度地挖掘出群体内遗传变异。诺禾具有丰富的群体遗传学项目经验,研究成果发表于Nature Genetics(Li, M, et al. 2013& Zhou, XM, et al. 2014)等。参考文献 [1] Li M, Tian S, Jin L, et al . Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars [J]. Nature genetics, 2013, 45(12): 1431-1438. [2] Zhan S, Zhang W, Niitepo ~ld K, et al . The genetics of monarch butterfly migration and warning colouration [J]. Nature, 2014.案例解析 [案例一] 家猪和藏猪的群体进化分析[1] 2013年,诺禾致源科技服务团队与四川农业大学研究者合作发表 该成果。本研究对6个代表性藏猪群体、5个四川盆地特有猪种, 共48个样本进行全基因组重测序,并结合55个欧亚野猪及家猪的 基因组数据进行群体遗传学分析。在藏猪中鉴定出低氧适应、能 量代谢等共268个适应高原环境的快速进化基因,揭示了藏猪高 原适应性的遗传机制。与自然选择相比,人工选择可更有效地塑 造驯养动物基因组;欧亚猪种存在明显的遗传背景差异,欧亚地 理隔离造成的遗传结构差异甚至超过了野生和驯化的差异。[案例二] 帝王蝶长距离迁飞遗传机制被解密[2] 北美地区的帝王蝶具有迁飞习性,而分布于热带地区的帝王蝶及 其近缘种不具有迁飞特性。该研究从涵盖当今世界上主要的帝王 蝶分布区域中,选取了包括迁飞型和非迁飞型的22个地理种群、 5个近缘种的101只班蝶属蝴蝶进行了全基因组重测序和群体遗传 学分析。结果表明,现存的帝王蝶起源于北美地区,且祖先属于 迁飞型,打破了先前认为包括鸟类等在内的迁飞物种均是热带起 源的普遍认知。其次,利用群体遗传学分析对全基因组进行精细 扫描发现,与飞行相关的肌肉发育进化是帝王蝶实现长距离迁飞 的主要适应性选择。 图1 藏猪及其它猪种的群体遗传结构 图2 帝王蝶样本分布及系统进化树
QTL-seq:用重测序方法进行数量性状基因座(QTL)定位的方法
TECHNICAL ADVANCE/RESOURCE QTL-seq:rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations Hiroki Takagi 1,2,Akira Abe 2,3,Kentaro Yoshida 1,Shunichi Kosugi 1,Satoshi Natsume 1,Chikako Mitsuoka 1,Aiko Uemura 1,Hiroe Utsushi 1,Muluneh Tamiru 1,Shohei Takuno 4,Hideki Innan 5,Liliana M.Cano 6,Sophien Kamoun 6and Ryohei Terauchi 1,*1 Iwate Biotechnology Research Center,Kitakami,Iwate,024-0003,Japan,2 United Graduate School of Iwate University,Morioka,Iwate,020-8550,Japan,3 Iwate Agricultural Research Center,Kitakami,Iwate,024-0003,Japan,4 Department of Plant Sciences,University of California,Davis,CA 95616,USA,5 Graduate University for Advanced Studies,Hayama,Japan,and 6 The Sainsbury Laboratory,Norwich Research Park,Norwich,UK Received 7September 2012;revised 13December 2012;accepted 20December 2012;published online 05January 2013.*For correspondence (e-mail terauchi@ibrc.or.jp). SUMMARY The majority of agronomically important crop traits are quantitative,meaning that they are controlled by multiple genes each with a small effect (quantitative trait loci,QTLs).Mapping and isolation of QTLs is important for ef?cient crop breeding by marker-assisted selection (MAS)and for a better understanding of the molecular mechanisms underlying the traits.However,since it requires the development and selection of DNA markers for linkage analysis,QTL analysis has been time-consuming and labor-intensive.Here we report the rapid identi?cation of plant QTLs by whole-genome resequencing of DNAs from two populations each composed of 20–50individuals showing extreme opposite trait values for a given phenotype in a segregating progeny.We propose to name this approach QTL-seq as applied to plant species.We applied QTL-seq to rice recombinant inbred lines and F 2populations and successfully identi?ed QTLs for important agronomic traits,such as partial resistance to the fungal rice blast disease and seedling vigor.Simulation study showed that QTL-seq is able to detect QTLs over wide ranges of experimental variables,and the method can be generally applied in population genomics studies to rapidly identify genomic regions that underwent arti?cial or natural selective sweeps. Keywords:quantitative trait loci,breeding,whole genome sequencing,next generation sequencer,selective sweep,technical advance. INTRODUCTION The world’s population has already exceeded 7billion and is still growing,while the amount of land suitable for agri-culture is decreasing due to a variety of factors such as rapid climate change.Therefore there is a great demand for ef?cient crop improvement to increase yield without further expanding farmland and damaging the environ-ment (Godfray,2010;David et al.,2011). In crop plants,multiple genes each with a relatively minor effect control the majority of agronomically impor-tant traits.These genes are called quantitative trait loci (QTLs)(Falconer and Mackay,1996).Identi?cation of QTLs is an important task in plant breeding.Once a QTL control-ling a favorable trait is mapped with closely linked DNA markers,it is introduced into an elite cultivar by crossing of the recurrent elite parent to the donor plant.Following QTL a process marker-assisted selection and Matsuoka,2006).Marker-assisted selection reduces the effort and time needed for phenotype evaluation of the progeny during successive improves introgression ?2013The Authors The Plant Journal ?2013Blackwell Publishing Ltd 174 The Plant Journal (2013)74,174–183doi:10.1111/tpj.12105
高通量测序:第二代测序技术详细介绍
在过去几年里,新一代DNA 测序技术平台在那些大型测序实验室中迅猛发展,各种新技术犹如雨后春笋般涌现。之所以将它们称之为新一代测序技术(next-generation sequencing),是相对于传统Sanger 测序而言的。Sanger 测序法一直以来因可靠、准确,可以产生长的读长而被广泛应用,但是它的致命缺陷是相当慢。十三年,一个人类基因组,这显然不是理想的速度,我们需要更高通量的测序平台。此时,新一代测序技术应运而生,它们利用大量并行处理的能力读取多个短DNA 片段,然后拼接成一幅完整的图画。 Sanger 测序大家都比较了解,是先将基因组DNA 片断化,然后克隆到质粒载体上,再转化大肠杆菌。对于每个测序反应,挑出单克隆,并纯化质粒DNA。每个循环测序反应产生以ddNTP 终止的,荧光标记的产物梯度,在测序仪的96或384 毛细管中进行高分辨率的电泳分离。当不同分子量的荧光标记片断通过检测器时,四通道发射光谱就构成了测序轨迹。 在新一代测序技术中,片断化的基因组DNA 两侧连上接头,随后运用不同的步骤来产生几百万个空间固定的PCR 克隆阵列(polony)。每个克隆由单个文库片段的多个拷贝组成。之后进行引物杂交和酶延伸反应。由于所有的克隆都是系在同一平面上,这些反应就能够大规模平行进行。同样地,每个延伸所掺入的荧光标记的成像检测也能同时进行,来获取测序数据。酶拷问和成像的持续反复构成了相邻的测序阅读片段。
Solexa高通量测序原理 --采用大规模并行合成测序法(SBS,Sequencing-By-Synthesis)和可逆性末端终结技术(ReversibleTerminatorChemistry) --可减少因二级结构造成的一段区域的缺失。 --具有高精确度、高通量、高灵敏度和低成本等突出优势 --可以同时完成传统基因组学研究(测序和注释)以及功能基因组学(基因表达及调控,基因功能,蛋白/核酸相互作用)研究 ----将接头连接到片段上,经PCR扩增后制成Library。 ----随后在含有接头(单链引物)的芯片(flowcell)上将已加入接头的DNA片段变成单链后通过与单链引物互补配对绑定在芯片上,另一端和附近的另外一个引物互补也被固定,形成“桥” ----经30伦扩增反应,形成单克隆DNA簇 ----边合成边测序(Sequencing By Synthesis)的原理,加入改造过的DNA 聚合酶和带有4 种荧光标记的dNTP。这些dNTP是“可逆终止子”,其3’羟基末端带有可化学切割的基团,使得每个循环只能掺入单个碱基。此时,用激光扫描反应板表面,读取每条模板序列第一轮反应所聚合上去的核苷酸种类。之后,将这些基团化学切割,恢复3'端粘性,继续聚合第二个核苷酸。如此继续下去,直到每条模板序列都完全被聚合为双链。这样,统计每轮收集到的荧光信号结果,就可以得知每个模板DNA 片段的序列。目前的配对末端读长可达到2×50 bp,更长的读长也能实现,但错误率会增高。读长会受到多个引起信号衰减的因素所影响,如荧光标记的不完全切割。 Roche 454 测序技术 “一个片段= 一个磁珠= 一条读长(One fragment =One bead = One read)”
基因组重测序分析流程-代码文件
差异位点分析流程步骤分解 数据准备: mkdir 1.QC cd 1.QC ln -s /root/mdna-data/reseq/1.QC/*.fastq . Ls cd .. mkdir 2.mapping cd 2.mapping ln -s /root/mdna-data/reseq/2.mapping/ref.fasta . 步骤1:参考基因建索引 cd 2.mapping ##bwa建索引: bwa index ref.fasta Expected Result:得到一系列BWA 进行alignment 需要的文件。 ##samtools建索引: samtools faidx ref.fasta Expected Result:生成refgene.fasta.fai。每行都是fasta 文件中每条contig 的record,每条record 由contig name, size, location, basesPerLine 和bytesPerLine 组成。 ##生成字典: java -jar /root/mdna_software/picard-tools-1.102/CreateSequenceDictionary.jar R=ref.fasta O=ref.dict Expected Result:生成refgene.dict。描述fasta 文件内容,类似SAM header 格式。 步骤2:bwa比对 ##用bwa作比对: nohup bwa aln -e 3 -i 10 -t 1 -R 100 -q 20 ref.fasta ../1.QC/test_trim1.fastq -f 1.sai & nohup bwa aln -e 3 -i 10 -t 1 -R 100 -q 20 ref.fasta ../1.QC/test_trim2.fastq -f 2.sai & nohup bwa aln -e 3 -i 10 -t 1 -R 100 -q 20 ref.fasta ../1.QC/test_trim_unpaired.fastq -f s.sai & jobs
测序技术的发展历程
测序技术的发展历程 随着1953年沃森和克里克发现了DNA的双螺旋结构,到2001年,首个人类基因组图谱的绘制完成,人们越来越多的认识到测序在生物医学中的重要作用。 测序技术的发展历史 Sanger测序技术 1975年由桑格和考尔森开创的链终止法测序技术标志着人类第一代DNA测序技术的诞生。1977年,人类历史上第一个基因组序列噬菌体X174由桑格团队测序完成。自此,人类获得了窥探生命遗传差异本质的能力,并以此为开端步入基因组学时代。 SangerJ.D. Waston、F.Crick
虽然第一代测序技术的测序读长可达1000bp,准确性高达99.999%,但其测序成本高,通量低等方面的缺点,严重影响了其真正大规模的应用。因而第一代测序技术并不是最理想的测序方法。从那时起人们开始了二代测序技术的探索。 第二代测序技术 第二代测序技术的核心思想是边合成边测序(Sequencing by Synthesis),在Sanger等测序方法的基础上,通过技术创新,用不同颜色的荧光标记四种不同的dNTP,当DNA聚合酶合成互补链时,每添加一种dNTP就会释放出不同的荧光,根据捕捉的荧光信号并经过特定的计算机软件处理,从而获得待测DNA的序列信息。 现有的技术平台主要包括Roche/454 FLX(已宣布停产)、Illumina Hiseq Miseq等系列和Applied Biosystems SOLID system。 Roche/454 FLX Illumina Hiseq 2500 AB SOLID 第三代测序技术 第二代测序技术虽然较Sanger测序有了巨大的突破,但是其测序的理论基础仍然建立在PCR扩增的基础之上。为了有效的避免测序过程中由于PCR扩增带来的偏差,科学家们积极投身到第三代单分子测序仪研究当中。目前最具代表性的包括Heliscope单分子实时合成测序法,纳米孔测序技术等。
测序常用名词解释整理
高通量测序领域常用名词解释大全 什么是高通量测序? 高通量测序技术(,)是对传统测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术( , )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序( )。 什么是法测序(一代测序) 法测序利用一种聚合酶来延伸结合在待定序列模板上的引物。直到掺入一种链终止核苷酸为止。每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(),并混入限量的一种不同的双脱氧核苷三磷酸()。由于缺乏延伸所需要的3基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。每一种和的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用光胶片放射自显影或非同位素标记进行检测。 什么是基因组重测序() 全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。 什么是测序
测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。获得一个物种的全基因组序列是加快对此物种了解的重要捷径。随着新一代测序技术的飞速发展,基因组测序所需的成本和时间较传统技术都大大降低,大规模基因组测序渐入佳境,基因组学研究也迎来新的发展契机和革命性突破。利用新一代高通量、高效率测序技术以及强大的生物信息分析能力,可以高效、低成本地测定并分析所有生物的基因组序列。 测序名词关系图 什么是
基于全基因组重测序获得的具LRR结构域基因的抗黄瓜白粉病功能鉴定
基于全基因组重测序获得的具LRR结构域基因的抗黄瓜白粉病 功能鉴定 黄瓜白粉病是黄瓜(CucumissativusL.)生产上的三大主要病害之一,发病时不但降低植株的光合效能,同时影响植株产量和果实品质,发病严重时常常引起30%左右的减产。黄瓜抗白粉病新品种选育及应用是克服白粉病危害的根本技术途径。 基于基因组测序技术和生物信息学的方法探究抗病基因已成为可能。本研究利用高通量Illumina测序技术,对实验室多年筛选获得的一个具有高抗白粉病且能稳定遗传的片段代换系SSL508-28和高感白粉病受体亲本D8进行了全基因组重测序,对比黄瓜9930参考基因组信息,在SSL508-28中发现了 468,616 个单核苷酸多态性位点(single nucleotide polymorphisms,SNPs)和 67,259小片段插入缺失位点(insertion/deletion,InDel),在D8 中获得了 537,352 个 SNPs 和 91,698个InDels。 通过对比SSL508-28与D8基因组,共得到了 15,682个SNPs和6,262个InDels,这些SNPs和InDels趋向于集中分布在五号染色体上。基于以上结果,我们对获得的SNPs和InDels进行了功能注释,发现有120个SNPs为非同义(non-synonymous)突变,30个InDels为移码突变(frameshift mutation),这些非同义突变SNPs和移码突变InDels分布在94个基因当中。 为了进一步验证94个突变基因对SSL508-28抗白粉病表型的贡献,我们对这94个基因进行了功能分类,其中有5个基因属于抗病(resistance,R)基因家族中NBS-LRR(Nucleotide binding site-leucine-rich repeats)类,利用 qRT-PCR 对这 5 个NBS-LRR基因在D8和SSL508-28中接种白粉菌前后的表达量进行检测,
重测序分析简介
重测序参考手册
目录 目录 (1) 1. 重测序简介 (3) 2. 重测序实验方法 (3) 基因组DNA抽提 (3) 基因组DNA样品建库 (3) 上机前定量 (4) 3. 重测序分析内容 (4) 重测序分析流程 (5) 重测序分析内容 (5) 4. 重测序重要技术参数 (6) 5. 重测序分析内容解释 (6) 6. 重测序分析内容示例 (6) SNP、INDEL的样本差异分析 (12) 7. 成功分析案例/或已发表论文 (14) 8. 概念及常用工具链接 (14)
1. 重测序简介 全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。全基因组重测序的个体,通过序列比对,可以找到大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel,Insertion/Deletion)、结构变异位点(SV,Structure Variation)位点。众信可以协助客户,通过生物信息手段,分析不同个体基因组间的结构差异,同时完成注释。 2. 重测序实验方法 提取基因组DNA,利用Covaris进行随机打断,电泳回收所需长度的DNA片段(0.2~5Kb),加上接头, 进行cluster制备(Solexa)或E-PCR (SOLiD),最后利用Paired-End或者Mate-Pair的方法对插入片段进行重测序。 实验步骤主要包括以下几点: 基因组DNA抽提 不同生物(植物、动物、微生物)的基因组DNA的提取方法有所不同; 不同种类或同一种类的不同组织因其细胞结构及所含的成分不同,分离方法也有差异。在提取某种特殊组织的DNA时必须参照文献和经验建立相应的提取方法, 以获得可用的DNA大分子。尤其是组织中的多糖和酶类物质对随后的酶切、PCR反应等有较强的抑制作用,因此用富含这类物质的材料提取基因组DNA时, 应考虑除去多糖和酚类物质。 基因组DNA样品建库 这是样品准备过程中最主要的环节,也就是真正意义上的建库(通常我们所说的建库包括整个样品准备的过程)。 样品片段化(Covaris) Covaris利用超声波剪切DNA,并将传统超声波法可控制化、精确化。DNA可以在小体积中被剪切,减少了因为蒸发带来的样品损耗,并且被剪切的DNA片段大小之间的偏差较小。Covaris剪切的片段大小较小,并且片段大小范围较传统超声波法窄。选择合适的打断参数条件,使最后打断的DNA片段大小集中在300-500bp范围内。 末端修复 使用Covaris剪切的DNA片段都会形成一些杂合的末端,其中包括了3’ 端悬垂结构、