PCR实验步骤
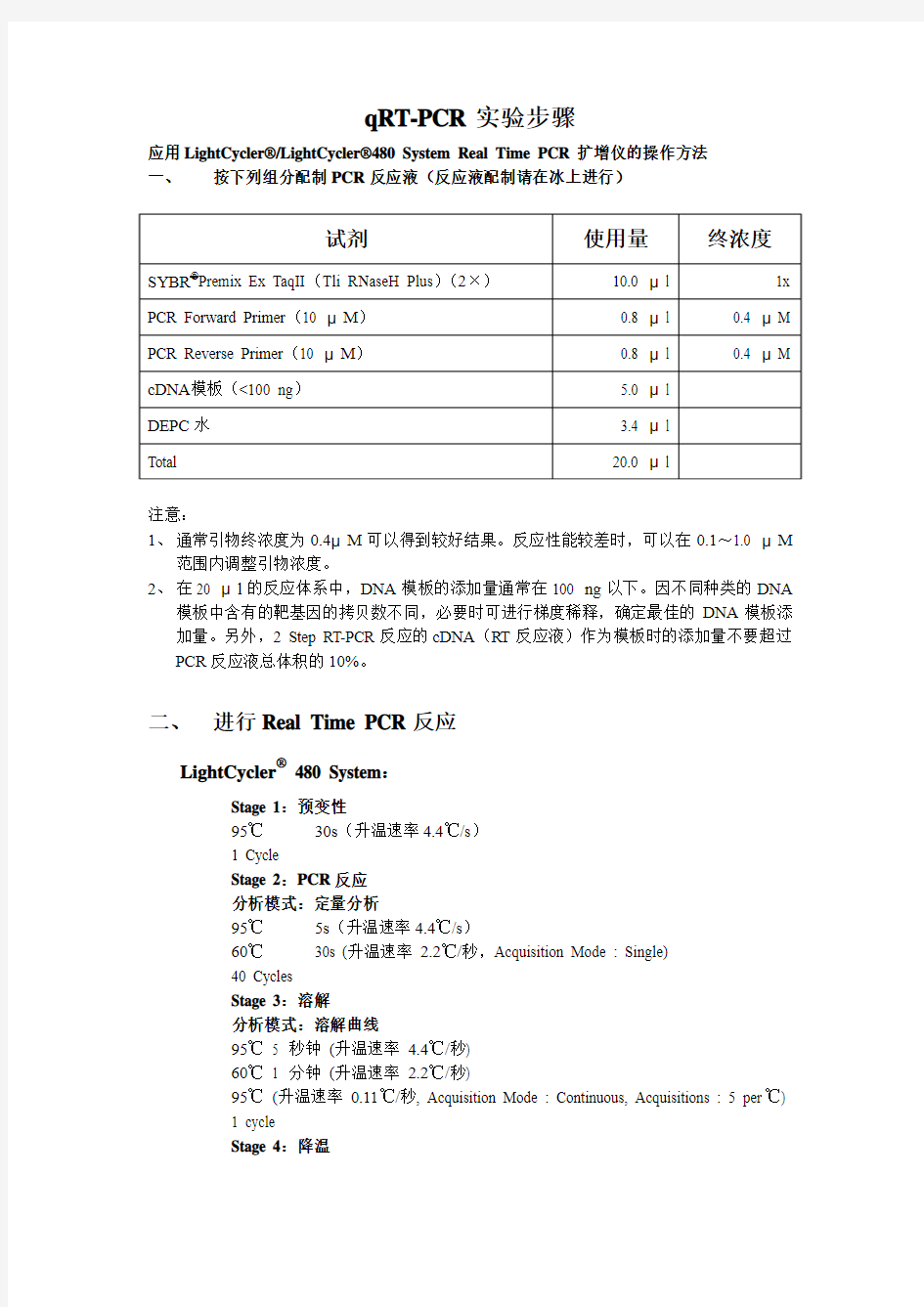

qRT-PCR实验步骤
应用LightCycler?/LightCycler?480 System Real Time PCR扩增仪的操作方法
一、按下列组分配制PCR反应液(反应液配制请在冰上进行)
注意:
1、通常引物终浓度为0.4μM可以得到较好结果。反应性能较差时,可以在0.1~1.0 μM
范围内调整引物浓度。
2、在20 μl的反应体系中,DNA模板的添加量通常在100 ng以下。因不同种类的DNA
模板中含有的靶基因的拷贝数不同,必要时可进行梯度稀释,确定最佳的DNA模板添加量。另外,2 Step RT-PCR反应的cDNA(RT反应液)作为模板时的添加量不要超过PCR反应液总体积的10%。
二、进行Real Time PCR反应
LightCycler? 480 System:
Stage 1:预变性
95℃30s(升温速率4.4℃/s)
1 Cycle
Stage 2:PCR反应
分析模式:定量分析
95℃5s(升温速率4.4℃/s)
60℃30s (升温速率2.2℃/秒,Acquisition Mode : Single)
40 Cycles
Stage 3:溶解
分析模式:溶解曲线
95℃5 秒钟(升温速率4.4℃/秒)
60℃1 分钟(升温速率2.2℃/秒)
95℃(升温速率0.11℃/秒, Acquisition Mode : Continuous, Acquisitions : 5 per℃)
1 cycle
Stage 4:降温
40℃30 秒钟(升温速率2.2℃/秒)
1 cycle
三、实验结果分析
反应结束后确认Real Time PCR的扩增曲线和融解曲线,进行PCR定量时制作标准曲线等。分析方法参见仪器操作手册。
RT-PCR的实验原理与操作步骤复习课程
R T-P C R的实验原理与 操作步骤
提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA。再以cDNA为模板进行PCR 扩增,而获得目的基因或检测基因表达。RT-PCR使测的灵敏性提高了几个数量级,使一些极为微量RNA样品分析成为可能。该技术主要用于:分析基因的转录产物、获取目的基因、合成cDNA探针、构建RNA高效转录系统。 (一) 反转录酶的选择 1. Moloney鼠白血病病毒(MMLV)反转录酶:有强的聚合酶活性,RNA酶H活性相对较弱。最适作用温度为37℃。 2. 禽成髓细胞瘤病毒(AMV)反转录酶:有强的聚合酶活性和RNA酶H活性。最适作用温度为42℃。 3. Thermus thermophilus、Thermus flavus等嗜热微生物的热稳定性反转录酶:在Mn2 存在下,允许高温反转录RNA,以消除RNA模板的二级结构。 4. MMLV反转录酶的RNase H-突变体:商品名为SuperScript 和 SuperScriptⅡ。此种酶较其它酶能多将更大部分的RNA转换成cDNA,这一特性允许从含二级结构的、低温反转录很困难的m模板合成较长cDNA。 (二) 合成cDNA引物的选择 1. 随机六聚体引物:当特定mRNA由于含有使反转录酶终止的序列而难于拷贝其全长序列时,可采用随机六聚体引物这一不特异的引物来拷贝全长mRNA。用此种方法时,体系中所有RNA分子全部充当了cDNA第一链模板,PCR引物在扩增过程中赋予所需要的特异性。通常用此引物合成的cDNA中96%来源于rRNA。 2. Oligo(dT):是一种对mRNA特异的方法。因绝大多数真核细胞mRNA具有3’端Poly(A )尾,此引物与其配对,仅mRNA被转录。由于Poly(A )RNA仅占总RNA的1-4%,故此种引物合成的cDNA比随机六聚体作为引物和得到的cDNA 在数量和复杂性方面均要小。 3. 特异性引物:最特异的引发方法是用含目标RNA的互补序列的寡核苷酸作为引物,若PCR反应用二种特异性引物,第一条链的合成可由与mRNA 3’端最靠近的配对引物起始。用此类引物仅产生所需要的cDNA,导致更为特异的PCR扩增。生物科研https://www.360docs.net/doc/3e13830474.html, 二、实验耗材与试剂 1.RNA提取" target="_blank" >RNA取试剂 2.第一链cDNA合成试剂盒
qPCR实验操作流程
Q-PCR实验流程 一、①实验前准备,每天早上到实验室后,先把超净工作台的紫外灯打 开15-20分钟。②超净台前做实验,需佩戴干净的橡胶手套/一次性薄膜手套,RNA抽提需带口罩。③取EP管/枪头时需用镊子,不可以用使用过的手套直接取用。取完EP管/枪头后,袋子及时封好。④橡胶手套须放入超净台照射紫外,实验操作过程中不得带出超净台,移液器在一天工作结束后调至最大量程,并用75%乙醇清洁移液器,枪头盒及超净台面。⑤实验进行的过程中或观看实验时,没有带口罩不要在超净台前讲话。 二、总RNA抽提 1)细胞培养皿中细胞样品用1*PBS洗两次后,用1ml枪将PBS吸干净,加入1ml Trizol (Invitrogen)溶液,吹打混匀,并吸至1.5ml RNase free EP管中使细胞充分裂解,室温静置5min; 组织样品用液氮充分研磨,加入1ml Trizol (Invitrogen)溶液,混匀,室温放置5min使其充分裂解;(管盖与管壁都需标记样品名称) 2)加入200μl氯仿,剧烈振荡混匀30s,使水相和有机相充分接触,室温静置3-5min;(离心时离心管按顺序排放,离心完毕,离心管的顺序也按顺序排好,与第一步的顺序一致) 3)4℃下,14,000g离心15min,可见分为三层,RNA在上层水相,移至另一个新的RNase free EP管;(用20-200ul的枪吸取上清,吸上清时,枪头应沿着液面上层吸取上清,枪头不可碰到、吸到中间层) 4)沉淀RNA:加入等体积异丙醇,轻柔地充分混匀(颠倒6-8次)(不应用振荡器混匀),室温静置10min; 5)4℃下,14,000g离心10min,收集RNA沉淀(如离心后仍不见EP管底部有沉淀,应将EP管放置在-80度冰箱过夜,继续在4℃下,14,000g 离心10min,收集RNA沉淀),去上清; 6)用75%乙醇洗涤两次(12,000g离心5min)(加入乙醇后只需轻轻颠倒EP管即可,不用振荡器震荡或枪头吸打沉淀),超净台风干;沉淀不
RT-PCR实验方法总结大全.
RT-PCR实验方法总结大全 RT-PCR实验有三步:抽提RNA,RT,PCR。 要求: 1.做RT前必需测RNA浓度,逆转录体系对RNA量还是有一些要求,常用500ng 或1ug。 2. RT按要求做,一般不会出太大问题。 3. PCR,按常规。但如需扩长片段,则对前两步要求较高,需要有完整的cDNA 存在,不是单改变Mg2+浓度、退火温度能解决的。 1RT和PCR时的引物设计是不是一定要先知道目的基因的序列?必须 在RT时,引物设计有3种方法即a:Random 9mers;b:Oligo dT-Adaptor Primer;和c:特异的下游引物。如果用a和b方法,是扩增的所有的cDNA(理论上,还要用此产物做PCR 的模板继续扩增。 如果用c方法,那么要去那里查它的序列呢?https://www.360docs.net/doc/3e13830474.html,问题: 在做RT-PCR遇到一怪现象,即对同一动物不同组织扩增同一段基因,结果从一种组织中可以扩出我的目的基因,条带非常的好,而另一组织在同样的条件下却得到许多非特异性的条带,尝试其他条件同样无法得到满意的结果,百思不得其解!(注:已肯定该基因在两种组织中都表达,且内参照在两种组织都可扩增出来 从这两种组织中提取的RNA的量是不一样的,我测过吸光度,差异还很大,会不会和这有关呢?请高手指教! 解答: 1.RT-PCR有两种做法:
条件具备的话可用kit进行一步法进行;若条件不太好的话可分两步进行逆转录再PCR。但后来发现两步法的结果更加理想,条带特异性强且无拖尾现象,我推测是体系更加单一比较利于PCR的进行,当然也可能是我买的kit不太好。(promega。 2.RT-PCR应具备的条件 高质量的RNA(保留后可做5‘,3’RACE;引物的(最好产物短点;若涉及粗略定量的话还应考虑RNA的浓度或是cDNA的浓度(如果由内标分子更好,但我发现其实很不容易将RNA的浓度以及内标分子的表达量调整的完全一样;体系的均一性等。 3.RACE 我做过RACE(3’RACE是宝生物的Kit;5‘RACE是Gibico,但现在再进行另一个同源基因的3‘RACE时却怎么也P不出来,这两个基因是由同一对引物扩增出来的,其中一个已经获得了全序列(RACE的方法,而另一个基因的3’UTR 却增么也扩不出来,我推测是不是该基因的3‘UTR太长的缘故,我都快绿了,有无 RT-PCR的常用内标b-actin 和GAPDH的使用有选择性吗?比如不同的细胞,不同的刺激。 有关内参: RT-PCR内参照可以在一个管子里做(那样也是图好看一些,最好分开两管,把除了引物之外的mixture统一配,拍照后,算目的基因和内参的比值,这就是基因表达的相对浓度。 问:我曾经作过同一管的PCR,内有actin 和目的基因引物。虽然可见到两条均一条带但图片质量不理想(而且酶量、Mg2+加倍。请教mxbdna2003 ,你是如何处理同一管的PCR的各成分的浓度? 答: it should determined the amount of RNA. but it not for the quantitity of the PCR. it just was convienent to guess the amount ot the template<(for RT and PCR and bettrer for publication and editor if he don not know the preocedure much. but the amount just
QPCR原理及应用
QPCR原理及应用 由于Real-time qPCR的众多优点,现在已经是生命科学领域的一项常规技术。越来越多的研究文章中涉及RT-PCR的实验,也基本上被real-time qPCR 所代替。由于real-time aPCR 输出的数据不同于常规的PCR 电泳检测,很多没有做过real-time qPCR的研究者常常感到高深莫测,不知从何入手;甚至一些做过次实验的研究者也会对数据处理分析感到迷惑,不知所措。 本文就从real-time qPCR的发展史说起,包括real-time qPCR的原理,实验设计,实际操作,数据分析,常见问题解答五个方面,手把手教你从各个方面了解real-time qPCR,彻底的从菜鸟到高手! 一、Real-time qPCR发展史 Real-time qPCR就是在PCR扩增过程中,通过荧光信号,对PCR进程进行实时检测。由于在PCR扩增的指数时期,模板的Ct 值和该模板的起始拷贝数存在线性关系,所以成为定量的依据。由于常规的PCR的缺点,real-time qPCR 由于其操作简便,灵敏度高,重复性好等优点发展非常迅速。现在已经涉及到生命科学研究的各个领域,比如基因的差异表达分析,SNP检测,等位基因的检测,药物开发,临床诊断,转基因研究等。 在Real-time qPCR技术的发展过程中,定量PCR仪的发展起了至关重要的作用。1995年,美国PE公司(已经并入Invitrogen公司)成功研制了Taqman 技术,1996年推出了首台荧光定量PCR检测系统,通过检测每个循环的荧光强度,通过Ct值进行数据分析。从而荧光定量PCR获得广泛应用。现在的定量PCR 仪有ABI7000、7300、7500,7700、7900HT、StepOnePlusTM、StepOneTM、PRISM@StepOneTM系列;BIO-RAD的CFX96、iCycler iQ5@、MyiQ@、MJ Research Chromo4TM Opticon 系列;Stratagene MxTM系列;Roche LightCycler@系列;Eppendorf Masercycler@;Corbett Rotor-GeneTM;Cepheid SmartCycler@和BIOER的LineGene系列。 随国内生命科学的快速发展,科研水平不断提高,发高水平文章已不再是新鲜事。与其同时,国内公司经过长期不懈的努力,也有自主研发的real-time PCR
PCR扩增实验操作步骤
PCR扩增反应 一、实验原理 PCR:是一种选择性扩增DNA或RNA的方法,其基本原理是依据体内细胞分裂中的DNA半保留复制机理,以及在体外dNTP分子于不同温度下双链和单链可以互相转变的性质,人为地控制体外合成系统的温度,以促使双链DNA变成单链DNA;单链DNA与人工合成的引物退火,以及在dNTP存在下,耐高温的DNA聚合酶使引物沿单链模板延伸成为双链DNA。 PCR反应分3步:①变性:通过加热使DNA 双螺旋的氢键断裂,双链解离形成单链DNA;②退火:当温度突然降低时,由于模板分子结构较引物要复杂得多,而且反应体系中引物DNA量大大多于模板DNA,使引物和其互补的模板在局部形成杂交链,而模板DNA 双链之间互补的机会较少。③延伸:在DNA聚合酶和4 种dNTP底物及Mg2+存在的条件下,5'→3'的聚合酶催化以引物为起始点的DNA链延伸反应,以上3步为一个循环,每一循环的产物可以作为下一个循环的模板,数小时之后,介于两个引物之间的特异性DNA片段得到了大量复制,数量可达2×106~7拷贝。 变性 退火 延伸 图反应历程
二、实验材料 1·模板:细菌DNA 2·TsgDNA聚合酶3·dNTP混合液 4·10倍浓度PCR缓冲液5·2.5mmol/LMgCl2 6·RAPD引物:S14 S15 S18 S66 S74 S88 S97 S103 S110 S115 7·提取细菌DNA的相关试剂 三、操作步骤 1.细菌染色体DNA的提取(见上一组) 3·反应程序: 将RAPD反应试剂加入EP管中 轻混后用100ul石蜡油覆盖于反应混合液之上,防止样品在反复加热-冷却的过程中蒸发,盖好盖子 打开PCR反应仪输入以下反应数据 ●94 摄氏度预变性5min ●94摄氏度变性40s ●40摄氏度退火40s ●72摄氏度延伸1min 将EP管放入仪器开始扩增,循环35次;72摄氏度延伸10min 仪器为Model MyGene 25 Plus 三、注意事项 1、PCR反应体系中DNA样品及各种试剂的用量都极少,必须严格注意吸样量的准确性 及全部放入反应体系中。 2、为避免污染凡是用在PCR反应中的Tip尖、离心管、蒸馏水都要灭菌;吸每种试剂 时都要换新的灭菌Tip尖。 3、加试剂时先加消毒三蒸水,最后加DNA模板和Taq DNA聚合酶。 4、置PCR仪进行PCR反应前,PCR管要盖紧,否则使液体蒸发影响PCR反应。 5.引物条件首先引物与模板的序列要紧密互补,其次引物与引物之间避免形成稳定二聚 体或发夹结构,再次引物不能在模板的非目的位点引发DNA聚合反应(即错配)。
实时荧光定量PCR(Real-Time-PCR)实验流程
实时荧光定量PCR(Real-Time PCR)实验流程 一、RNA的提取(详见RNA提取及反转录) 不同组织样本的RNA提取适用不同的提取方法,因为Real-Time PCR对RNA样品的质量要求较高,所以,正式实验前要选择一款适合自己样品的提取方法,在实验过程中要防止RNA的降解,保持RNA的完整性。 在总RNA的提取过程中,注意避免mRNA的断裂;取2ug进行RNA的甲醛变性胶电泳检测,如果存在DNA污染时,要用DNase I进行消化(因为在处理过程中RNA极易降解,建议体系中加入适量RNA酶抑制剂)。 二、DNase I 消化样品RNA 中的DNA 用DNase I 消化DNA 组份加量 模板(RNA) 10ug RNase Inhibitor 4ul DNase I buffer 10ul DNase I 10ul DEPC处理H2O 至100ul 混匀,37℃ 90min 三、RNA琼脂糖凝胶电泳 1.1%的琼脂糖凝胶电泳凝胶的配制: 1)称取琼脂糖0.45g放入三角瓶中,向其中加入4.5ml的10×MOPS缓冲液和39.5ml 的DEPC水,放微波炉里溶化。 2)待冷却到60摄氏度左右时,加入1ml甲醛,摇匀(避免产生气泡)。倒入凝胶板上凝固30min。 2.取各个RNA样品4μl,加入6×RNA电泳上样缓冲液2μl混匀,加入变性胶加样孔中。3.120V电压下电泳25min。用凝胶紫外分析仪观察,照相保存。 4.RNA电泳结果如下图所示。可见28S和18S两条明亮条带,无DNA条带污染。 四.RNA反转录为cDNA 反转录程序(以MBI的M-MLV为例) 组份加量(20ul体系) 加量(40ul体系) 模板(RNA) 0.1~2.5ug(根据条带的亮度适当调整) 3ug(根据条带的亮度适当调整) 引物T18(50uM)(或其他引物) 2.0ul 4.0ul DEPC处理H2O 至12.5ul 至25ul
(待分)rtpcr原理和实验步骤
原理与实验步骤 一、知识背景: 、基因表达: 单拷贝基因表达存在逐步放大机制,如一个蚕丝心蛋白基因个丝心蛋白(每个存活,可以合成个丝心蛋白)共合成个丝心蛋白。因此单拷贝基因的表达水平对于其功能水平的调控是非常重要的。 、技术( ):即聚合酶链式反应。 在模板、引物和四种脱氧核苷酸存在的条件下依赖于聚合酶的酶促反应,其特异性由两我工合成的引物序列决定。反应分三步: .变性:通过加热使双螺旋的氢键断裂,形成单链。 .退火:将反应混合液冷却至某一温度,使引物与模板结合。 .延伸:在聚合酶和及+存在下,退火引物沿’’方向延伸。 以上三步为一个循环,如此反复。 、逆转录酶和 逆转录酶()是存在于病毒体内的依赖的聚合酶,至少具有以下三种活性: 、依赖的聚合酶活性:以为模板合成第一条链。 、水解活性:水解杂合体中的。 、依赖的聚合酶活性:以第一条链为模板合成互补的双链. 二、的准备: .引物的设计及其原则: ) 引物的特异性决定反应特异性。因此引物设计是否合理对于整个实验有着至关重要的影响。在引物设计时要充分考虑到可能存在的同源序列,同种蛋白的不同亚型,不同的剪切方式以及可能存在的对引物的特异性的影响。尽量选择覆盖相连两个内含子的引物,或者在目的蛋白表达过程中特异存在而在其他亚型中不存在的内含子。 ) 引物设计原则的把握 引物设计原则包括 : 、引物长度:一般为~,引物太短会影响的特异性,引物太长的最适延伸温度会超过酶的最适温度,也影响反应的特异性。 、碱基分布:四种碱基最好应随机分布,避免嘌呤或嘧啶的聚集存在,特别是连续出现个以上的单一碱基。含量(值):%~%,扩增的复性温度一般是较低值减去~度。 、’端要求:’端必须与模板严格互补,不能进行任何修饰,也不能有形成任何二级结构的可能。末位碱基是时错配的引发效率最低,、居中间,因此引物的’端最好选用、、而尽可能避免连续出现两个以上的。 、引物自身二级结构:引物自身不应存在互补序列,否则会自身折叠成发夹状结构或引物自身复性。 、引物之间的二级结构:两引物之间不应有多于个连续碱基互补,’端不应超过个。、同源序列:引物与非特异扩增序列的同源性应小于连续个的互补碱基存在。 、’端无严格限制:’末端碱基可以游离,但最好是或,使产物的末端结合稳定。还可以进行特异修饰(标记、酶切位点等)等等。 根据实验目的选择适当的引物。常用引物设计软件如,等对于这些条件都可以自行设置。 、耗材:
RT-PCR实验步骤
RT-PCR实验步骤 一、总RNA提取 1. 取200mg组织,放到1.5mlEP管中,加入1mlTrizol剪碎。 2. 震荡30s。 3. 加0.2ml氯仿,剧烈摇动30s,室温3min。 4. 12000×g,4℃离心,15min。 5. 吸上层无色水相,移入另一EP管中(约0.5ml)。 6. 加等体积异丙醇,-20℃,30min。 7. 12000×g,4℃离心,10min。在管底部可见微量RNA沉淀 8. 弃上清,加75%乙醇1ml,振荡。 9. 7500×g,4℃离心,10min。 10. 弃上清,用滤纸小心吸取残留液体,室温干燥5-10min。 11. 沉淀溶于20μlDEPC水,取1μl加入79μlDEPC水测OD260/OD280 12. 计算浓度与纯度,-70℃保存。 二、逆转录合成cDNA第一链 反应体系如下
混匀快速离心一次反应条件如下 -20℃ 冰箱冻存三、PCR反应 混匀快速离心一次反应条件如下
PCR产物-20℃冰箱保存 取PCR产物8μl 加5×Loading Buffer 2μl 2%琼脂糖凝胶电泳120V。 100mA 30min 溴化乙锭染色,凝胶成象仪成象并保存结果。 RT-PCR实验方法总结1,2 RT-PCR实验有三步:抽提RNA,RT,PCR。 要求: 1.做RT前必需测RNA浓度,逆转录体系对RNA量还是有一些要求,常用500ng或1ug。 2. RT按要求做,一般不会出太大问题。 3. PCR,按常规。但如需扩长片段,则对前两步要求较高,需要有完整的cDNA存在,不是单改变Mg2+浓度、退火温度能解决的。 1)RT和PCR时的引物设计是不是一定要先知道目的基因的序列?必须 在RT时,引物设计有3种方法即a:Random 9mers;b:Oligo dT-Adaptor Primer;和c:特异的下游引物。如果用a和b方法,是扩增的所有的cDNA(理论上),还要用此产物做PCR 的模板继续扩增。 如果用c方法,那么要去那里查它的序列呢?https://www.360docs.net/doc/3e13830474.html, 问题: 在做RT-PCR遇到一怪现象,即对同一动物不同组织扩增同一段基因,结果从一种组织中可以扩出我的目的基因,条带非常的好,而另一组织在同样的条件下却得到许多非特异性的条带,尝试其他条件同样无法得到满意的结果,百思不得其解!(注:已肯定该基因在两种组织中都表达,且内参照在两种组织都可扩增出来) 从这两种组织中提取的RNA的量是不一样的,我测过吸光度,差异还很大,会不会和这有关呢?请高手指教! 解答: 1.RT-PCR有两种做法: 条件具备的话可用kit进行一步法进行;若条件不太好的话可分两步进行逆转录再PCR。但后来发现两步法的结果更加理想,条带特异性强且无拖尾现象,我推测是体系更加单一比较利于PCR 的进行,当然也可能是我买的kit不太好。(promega)。 2.RT-PCR应具备的条件 高质量的RNA(保留后可做5‘,3’RACE);引物的(最好产物短点);若涉及粗略定量的话还应
RT-PCR的实验原理与操作步骤
提取组织或细胞中的总RNA以其中的mRN作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA再以cDNA为模板进行PCR扩增,而获得目的基因或检测基因表达。RT-PCF使测的灵敏性提高了几个数量级,使一些极为微量 RNA羊品分析成为可能。该技术主要用于:分析基因的转录产物、获取目的基因、合成cDNA探针、构建RNA高效转录系统。 (一)反转录酶的选择 1. Moloney 鼠白血病病毒(MMLV)反转录酶:有强的聚合酶活性,RNA酶H活 性相对较弱。最适作用温度为37 C。 2. 禽成髓细胞瘤病毒(AMV)反转录酶:有强的聚合酶活性和 RNA 酶 H 活性。 最适作用温度为42 C。 3. Thermus thermophilus 、 Thermus flavus 等嗜热微生物的热稳定性反转录 酶:在Mn2存在下,允许高温反转录 RNA,以消除RNA模板的二级结构。 4. MMLV 反转录酶的 RNase H- 突变体:商品名为 SuperScript 和 SuperScript U。此种酶较其它酶能多将更大部分的RNA转换成cDNA,这一 特性允许从含二级结构的、低温反转录很困难的 m 模板合成较长 cDNA 。 (二)合成 cDNA 引物的选择 1. 随机六聚体引物:当特定 mRNA 由于含有使反转录酶终止的序列而难于拷贝 其全长序列时,可采用随机六聚体引物这一不特异的引物来拷贝全长 mRNA 。 用此种方法时,体系中所有RNA分子全部充当了 cDNA第一链模板,PCR引物在扩增过程中赋予所需要的特异性。通常用此引物合成的 cDNA 中 96%来源 于 rRNA。 2. Oligo(dT) :是一种对 mRNA 特异的方法。因绝大多数真核细胞 mRNA 具有
定量PCR的实验步骤。
定量PCR的实验步骤 一、标准曲线的标准DNA样品准备 1.将目的片段进行克隆,对克隆得到的阳性菌落进行质粒DNA的提取和纯化(试剂盒),纯化后的质粒DNA纯度用DO260/DO280的比值来鉴定,纯的DNA比值应该在1.8-1.9之间,如果比值大于1.9的时候表示有RNA污染,小于1.6的时候表示有蛋白质的污染(DO260的1OD值相当于50ng/ul的双链DNA 或33ng/ul的单链DNA或40ng/ul的RNA); 2.采用紫外分光光度法(DO260 nm)测定质粒DNA的吸光度DO260值为A,(B=1DO260=50 ng/ul),由于载体质粒序列和插入的目标片段的序列都是已知的,这样通过核苷酸的相对分子量和标准品序列信息估算出单个质粒DNA (阳性质粒)的分子量C (ng/mol),进而计算标准品的拷贝数D(copy/uL): 拷贝数D=A×B C ×6.02×1023(copy/ul) 3.将2步骤中计算出来的已知拷贝数的质粒DNA以10倍为梯度进行稀释,稀释7个系列(各个梯度的浓度一般为101~107),作为标准曲线的DNA模板; 二、未知样品的定量标定 模板质粒DNA每个梯度设置3个平行样。未知样品设置2个平行样,同时设置一个没有质粒DNA的阴性对照。(定量实验,误差是不可避免的,设立平行样,对数据进行统计处理,可以将误差降低到最小,以满足小样本统计的要求;确保数据的有效性,排除假阳性的干扰,必须设置阴性对照,阳性对照有标准样品就做,没有标准样品就可以省略不做); 2.按照下面的程序进行荧光定量PCR反应: 95℃ 3 min 95℃10 s 58℃20 s 40个循环 72℃20 s
RT-PCR实验步骤
RT-PCR实验步骤
RT-PCR实验有三步:抽提RNA,RT,PCR。 要求: 1.做RT前必需测RNA浓度,逆转录体系对RNA量还是有一些要求,常用500ng或1ug。 2. RT按要求做,一般不会出太大问题。 3. PCR,按常规。但如需扩长片段,则对前两步要求较高,需要有完整的cDNA存在,不是单改变Mg2+浓度、退火温度能解决的。 1)RT和PCR时的引物设计是不是一定要先知道目的基因的序列?必须 在RT时,引物设计有3种方法即a:Random 9mers;b:Oligo dT-Adaptor Primer;和c:特异的下游引物。如果用a和b方法,是扩增的所有的cDNA(理论上),还要用此产物做PCR 的模板继续扩增。 如果用c方法,那么要去那里查它的序列呢?https://www.360docs.net/doc/3e13830474.html, 问题: 在做RT-PCR遇到一怪现象,即对同一动物不同组织扩增同一段基因,结果从一种组织中可以扩出我的目的基因,条带非常的好,而另
一组织在同样的条件下却得到许多非特异性的条带,尝试其他条件同样无法得到满意的结果,百思不得其解!(注:已肯定该基因在两种组织中都表达,且内参照在两种组织都可扩增出来) 从这两种组织中提取的RNA的量是不一样的,我测过吸光度,差异还很大,会不会和这有关呢?请高手指教! 解答: 1.RT-PCR有两种做法: 条件具备的话可用kit进行一步法进行;若条件不太好的话可分两步进行逆转录再PCR。但后来发现两步法的结果更加理想,条带特异性强且无拖尾现象,我推测是体系更加单一比较利于PCR的进行,当然也可能是我买的kit不太好。(promega)。 2.RT-PCR应具备的条件 高质量的RNA(保留后可做5‘,3’RACE);引物的(最好产物短点);若涉及粗略定量的话还应考虑RNA的浓度或是cDNA的浓度(如果由内标分子更好,但我发现其实很不容易将RNA的浓度以及内标分子的表达量调整的完全一样);体系的均一性等。 3.RACE 我做过RACE(3’RACE是宝生物的Kit;5‘RACE是Gibico),但现在再进行另一个同源基因的3‘RACE时却怎么也P不出来,这两个基因是由同一对引物扩增出来的,其中一个已经获得了全序列(RACE的方法),而另一个基因的3’UTR却增么也扩不出来,我推测是不是该基因的3‘UTR太长的缘故,我都快绿了,有无
PCR实验步骤
qRT-PCR实验步骤 应用LightCycler?/LightCycler?480 System Real Time PCR扩增仪的操作方法 一、按下列组分配制PCR反应液(反应液配制请在冰上进行) 注意: 1、通常引物终浓度为0.4μM可以得到较好结果。反应性能较差时,可以在0.1~1.0 μM 范围内调整引物浓度。 2、在20 μl的反应体系中,DNA模板的添加量通常在100 ng以下。因不同种类的DNA 模板中含有的靶基因的拷贝数不同,必要时可进行梯度稀释,确定最佳的DNA模板添加量。另外,2 Step RT-PCR反应的cDNA(RT反应液)作为模板时的添加量不要超过PCR反应液总体积的10%。 二、进行Real Time PCR反应 LightCycler? 480 System: Stage 1:预变性 95℃30s(升温速率4.4℃/s) 1 Cycle Stage 2:PCR反应 分析模式:定量分析 95℃5s(升温速率4.4℃/s) 60℃30s (升温速率2.2℃/秒,Acquisition Mode : Single) 40 Cycles Stage 3:溶解 分析模式:溶解曲线 95℃5 秒钟(升温速率4.4℃/秒) 60℃1 分钟(升温速率2.2℃/秒) 95℃(升温速率0.11℃/秒, Acquisition Mode : Continuous, Acquisitions : 5 per℃) 1 cycle Stage 4:降温
40℃30 秒钟(升温速率2.2℃/秒) 1 cycle 三、实验结果分析 反应结束后确认Real Time PCR的扩增曲线和融解曲线,进行PCR定量时制作标准曲线等。分析方法参见仪器操作手册。
半定量RT-PCR的实验原理和方法步骤
半定量RT-PCR的实验原理和方法步骤 以下实验步骤仅供参考: 1 样品RNA的抽提 ①取冻存已裂解的细胞,室温放置5分钟使其完全溶解。 ②两相分离每1ml的TRIZOL试剂裂解的样品中加入0.2ml的氯仿,盖紧管盖。手动剧烈振荡管体15秒后,15到30℃孵育2到3分钟。4℃下12000rpm离心15分钟。离心后混合液体将分为下层的红色酚氯仿相,中间层以及无色水相上层。RNA全部被分配于水相中。水相上层的体积大约是匀浆时加入的TRIZOL试剂的60%。 ③RNA沉淀将水相上层转移到一干净无RNA酶的离心管中。加等体积异丙醇混合以沉淀其中的RNA,混匀后15到30℃孵育10分钟后,于4℃下12000rpm 离心10分钟。此时离心前不可见的RNA沉淀将在管底部和侧壁上形成胶状沉淀块。 ④RNA清洗移去上清液,每1mlTRIZOL试剂裂解的样品中加入至少1ml的75%乙醇(75%乙醇用DEPCH2O配制),清洗RNA沉淀。混匀后,4℃下7000rpm离心5分钟。 ⑤RNA干燥小心吸去大部分乙醇溶液,使RNA沉淀在室温空气中干燥5-10分钟。 ⑥溶解RNA沉淀溶解RNA时,先加入无RNA酶的水40μl用枪反复吹打几次,使其完全溶解,获得的RNA溶液保存于-80℃待用。 2 RNA质量检测 1)紫外吸收法测定 先用稀释用的TE溶液将分光光度计调零。然后取少量RNA溶液用TE稀释(1:100)后,读取其在分光光度计260nm和280nm处的吸收值,测定RNA溶液浓度和纯度。 ①浓度测定 A260下读值为1表示40 μg RNA/ml。样品RNA浓度(μg/ml)计算公式为:A260 ×稀释倍数× 40 μg/ml。具体计算如下: RNA溶于40 μl DEPC水中,取5ul,1:100稀释至495μl的TE中,测得A260 = 0.21 RNA 浓度= 0.21 ×100 ×40 μg/ml = 840 μg/ml 或0.84 μg/μl 取5ul用来测量以后,剩余样品RNA为35 μl,剩余RNA总量为: 35 μl × 0.84 μg/μl = 29.4 μg ②纯度检测 RNA溶液的A260/A280的比值即为RNA纯度,比值范围1.8到2.1。 2)变性琼脂糖凝胶电泳测定 ①制胶 1g琼脂糖溶于72ml水中,冷却至60℃,10 ml的10× MOPS电泳缓冲液和18 ml 的37% 甲醛溶液(12.3 M)。 10×MOPS电泳缓冲液 浓度成分 0.4M MOPS,pH 7.0 0.1M 乙酸钠 0.01M EDTA 灌制凝胶板,预留加样孔至少可以加入25 μl溶液。胶凝后取下梳子,将凝胶板
菌落PCR实验步骤.doc
菌落PCR资料 PCR, 菌落, 资料 菌落PCR 菌落PCR与我们通常的普通DNA的PCR的不同在于,直接以单个菌作为模板。是一种可以快速鉴定菌落是否为含目的质粒的阳性菌落。操作简单,快接,阳性率较高,在转化鉴定中较常见。 操作方法: 1,挑取单个菌落,可以用高压灭菌的牙签。现在含抗性的平板上画线,做保种用,然后置于20ultriton-x100中搅和一下,将相应的菌和板子上的画线区做上对应的记号 2,将装有20ulTritonx-100的EP管100度煮2分钟 3,按照菌中预期包含的质粒加好PCR体系,建议做20ul体系,模板加煮过的菌的上清1ul 4,上机即可。退火温度可以稍低,虽然没有什么根据可言,但是个人经验,推荐比质粒PCR 时稍低 5,电泳,看结果,阳性的菌落对应的板子可以直接挑菌小摇,保种或者送测序都可以 我的做法是: 用枪头挑选单菌落到装有20uL水的pcr管中,然后悬浮菌液。 在做PCR时,吸取1uL做摸板,但电泳检测后,选有阳性克隆相对的菌液10uL与试管的液体培养基中培养。这样做,既初步检测了阳性克隆,又保存了所需要的菌落。 当然,在挑菌时也有用接种环挑的,有用牙签挑的,看个人的爱好了 我们都是直接拿牙签挑单菌落往PCR反应体系中搅拌一下,一般都能鉴定出来。想节省麻烦的话,还可以菌落鉴定同时挑单克隆,用1.5的EP管装1mL左右培养基,牙签挑出菌落后先在培养基上搅拌然后再放到PCR反应体系中。 把PCR体系先加好,用接种针直接往菌落上戳一下然后伸到体系里面晃晃就可以上机P了,我每次都是这样,可能是最方便的方法了。 取200ul水,牙签挑菌,煮沸5‘,冰浴5’,12000rpm,取上清5ul做模版。其他的和普通pcr一样。 我们都是用过夜培养的菌液0.3ul(不要超过0.5ul)做模板,很简单的,其它都不变。先94度5min 裂解菌,最好5min后再加入taq酶。 是的,菌落PCR不难。。但是如果发现结果出现非特异带,在目的带位置有很弱的条带出现,最好再用液体培养后再做一次PCR,我就因为这个阴性结果折腾了1个月。。扩培后再作菌落PCR就得到阳性结果了。。 我是这样做的,取菌液60ul至ep管中,沸水中煮5min,离心,取其中上清1ul作模板,屡试不爽 拿牙签点一下菌落然后再体系里面涮一下就可以了 如果觉得不可靠,就索性接个种,摇个小管,然后取1ul作模板 模板如果太多了,引物就不够用了,显然扩不出来,这种现象在质粒这种模板的PCR里面很多,你提出来的质粒不要忘记稀释
PCR实验室操作流程
乙型肝炎病毒(HBV DNA PCR ABI7300)检侧标准操作程序 飞INFORMATIONFORTE检测信息 二、ANALYTICALPRINCTP检测原理 聚台酶链式反应(PCR可对特定核苷酸片段进行指数级的扩增,而实时荧光定量PCF技术,是指在PCF反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。在实时荧光定量PCR反应中,引入了一种荧光化学物质,荧光物质有两种:荧光探针和荧光染料。我们目前是使用TaqMan荧光探针的检测原理:PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCRT增时,Taq 酶的5' —3'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,荧光信号强度也等比例增加(图一)。这样就可以通过荧光 强度变化监测产物量的变化,从而得到一条荧光扩增曲线图 3 ■- atwnch-ftr
三、操作步骤 (一)试剂配制 1、进入试剂准备区,开启冰箱冷冻层取出 HBV-DNA 佥测试剂盒。查 看外包装盒(包括生产批号、有效期),无误后拆开试剂盒,取出核酸 提取液、反应液及Taq 酶(核对核酸提取液、HBVPC 反应液及Taq 酶 管上批号与试剂外包装盒批号是否一致)(图1),不一致则不可使用, 需更换新的试剂。室温平衡20min ,完全融化后2000rpm 离心备用。 2、核酸提取液的分装 (1)取0.5ml 离心管架置于超净工作台内(开机前用紫外线消毒30 分钟),用右手拿起镊子逐个将离心管放入离心管架(注意应夹住离 心管外壁,不能碰到管内壁),摆放顺序为横列从左往右摆,竖列为 从上往下隔行排,离心管个数为n (n 二样本数+对照品+质控品)。完成 后将镊子放回原位(图 2)。 对 及 有 效 La i
RTPCR实验步骤
RT-PCR实验有三步:抽提RNA,RT,PCR。 要求: 1.做RT前必需测RNA浓度,逆转录体系对RNA量还是有一些要求,常用500ng或1ug。 2.RT按要求做,一般不会出太大问题。 3.PCR,按常规。但如需扩长片段,则对前两步要求较高,需要有完整的cDNA存在,不是单改变Mg2+浓度、退火温度能解决的。 1)RT和PCR时的引物设计是不是一定要先知道目的基因的序列?必须 在RT时,引物设计有3种方法即a:Random9mers;b:OligodT-AdaptorPrimer;和c:特异的下游引物。如果用a和b 方法,是扩增的所有的cDNA(理论上),还要用此产物做PCR的模板继续扩增。
问题: 在做RT-PCR遇到一怪现象,即对同一动物不同组织扩增同一段基因,结果从一种组织中可以扩出我的目的基因,条带非常的好,而另一组织在同样的条件下却得到许多非特异性的条带,尝试其他条件同样无法得到满意的结果,百思不得其解!(注:已肯定该基因在两种组织中都表达,且内参照在两种组织都可扩增出来) 从这两种组织中提取的RNA的量是不一样的,我测过吸光度,差异还很大,会不会和这有关呢?请高手指教! 解答: 1.RT-PCR有两种做法: 条件具备的话可用kit进行一步法进行;若条件不太好的话可分两步进行逆转录再PCR。但后来发现两步法的结果更加理想,条带特异性强且无拖尾现象,我推测是体系更加单一比较利于PCR的进行,当然也可能是我买的kit不太好。(promega)。 2.RT-PCR应具备的条件 高质量的RNA(保留后可做5‘,3’RACE);引物的(最好产物短点);若涉及粗略定量的话还应考虑RNA的浓度或是cDNA的浓
RTPCR原理和实验步骤
RT—PCR原理与实验步骤 一、知识背景: 1、基因表达:DNA RNA Protein 单拷贝基因表达存在逐步放大机制,如一个蚕丝心蛋白基因 104个丝心蛋白mRNA(每个mRNA存活4d,可以合成105个丝心蛋白) 共合成109个丝心蛋白。因此单拷贝基因的mRNA表达水平对于其功能水平的调控是非常重要的。 2、PCR技术(Polymerase chain reaction):即聚合酶链式反应。 在模板、引物和四种脱氧核苷酸存在的条件下依赖于DNA聚合酶的酶促反应,其特异性由两个人工合成的引物序列决定。反应分三步: A。变性:通过加热使DNA双螺旋的氢键断裂,形成单链DNA; B.退火:将反应混合液冷却至某一温度,使引物与模板结合. C。延伸:在DNA聚合酶和dNTPs及Mg2+存在下,退火引物沿5’3’方向延伸。以上三步为一个循环,如此反复。 3、逆转录酶和RT-PCR 逆转录酶(reverse transcriptase)是存在于RNA病毒体内的依赖RNA的DNA聚合酶,至少具有以下三种活性: 1、依赖RNA的DNA聚合酶活性:以RNA为模板合成cDNA第一条链; 2、Rnase水解活性:水解RNA:DNA杂合体中的RNA; 3、依赖DNA的DNA聚合酶活性:以第一条DNA链为模板合成互补的双链cDNA。 二、RT—PCR的准备: 1。引物的设计及其原则: 1)引物的特异性决定PCR反应特异性.因此引物设计是否合理对于整个实验有着至关重要的影响。在引物设计时要充分考虑到可能存在的同源序列,同种蛋白的不同亚型,不同的mRNA剪切方式以及可能存在的hnRNA对引物的特异性的影响。尽量选择覆盖相连两个内含子的引物,或者在目的蛋白表达过程中特异存在而在其他亚型中不存在的内含子。 2) 引物设计原则的把握 引物设计原则包括: a、引物长度:一般为15~30bp ,引物太短会影响PCR的特异性,引物太长PCR的最适延伸温度会超过Taq酶的最适温度,也影响反应的特异性。 b、碱基分布:四种碱基最好应随机分布,避免嘌呤或嘧啶的聚集存在,特别是连续出现3个以上的单一碱基。GC含量(Tm值):40%~60%,PCR扩增的复性温度一般是较低Tm值减去5~10度.
反转录PCR-步骤和方法
反转录P C R-步骤和方 法 本页仅作为文档封面,使用时可以删除 This document is for reference only-rar21year.March
反转录PCR 科技名词定义 中文名称:反转录PCR 英文名称:reverse transcription PCR;RT-PCR 定义:扩增mRNA的一种实验技术。先将mRNA反转录成cDNA,然后再以 cDNA为模板,用PCR方法加以扩增。 应用学科:遗传学(一级学科);基因组学(二级学科) 以上内容由全国科学技术名词审定委员会审定公布 目录 一、反转录酶的选择 二、合成cDNA引物的选择 三、操作步骤 四、注意事项 RT-PCR反应原理 [1] 反转录PCR(Reverse Transcription-Polymerase Chain Reaction,RT-PCR)又称为逆转录PCR。其原理是:提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA。再以cDNA为模板进行PCR扩增,而获得目的基因或检测基因表达。RT-PCR使RNA检测的灵敏性提高了几个数量级,使一些极为微量RNA样品分析成为可能。该技术主要用于:分析基因的转录产物、获取目的基因、合
成cDNA探针、构建RNA高效转录系统。 RT- PCR(Reverse Transcription-Polymerase
Chain Reaction)即逆转录PCR,是将RNA的逆转录(RT)和cDNA的聚合酶链式扩增反应(PCR)相结合的技术。RT-PCR技术灵敏而且用途广泛,可用于检测细胞/组织中基因表达水平,细胞中RNA病毒的含量和直接克隆特定基因的cDNA序列等。 编辑本段一、反转录酶的选择 1. Money 鼠白血病病毒(MMLV)反转录酶:有强的聚合酶活性,RNA酶H活性相对较弱。最适作用温度为37℃。 2.禽成髓细胞瘤病毒(AMV)反转录酶:有强的聚合酶活性和RNA酶H活性。最适作用温度为42℃。 3.Thermus thermophilus、Thermus flavus等嗜热微生物的热稳定性反转录酶:在Mn2 存在下,允许高温反转录RNA,以消除RNA模板的二级结构。 4.MMLV反转录酶的RNase H-突变体:商品名为SuperScript 和SuperScriptⅡ。此种酶较其它酶能多将更大部分的RNA转换成cDNA,这一特性允许从含二级结构的、低温反转录很困难的mRNA模板合成较长cDNA。 编辑本段二、合成cDNA引物的选择 1.随机六聚体引物:当特定mRNA由于含有使反转录酶终止的序列而难于拷贝其全长序列时,可采用随机六聚体引物这一不特异的引物来拷贝全长mRNA。用此种方法时,体系中所有RNA分子全部充当了cDNA第一链模板,PCR引物在扩增过程中赋予所需要的特异性。通常用此引物合成的cDNA 中96%来源于rRNA。 2. Oligo(dT):是一种对mRNA特异的方法。因绝大多数真核细胞mRNA具有3’端Poly(A )尾,此引物与其配对,仅mRNA可被转录。由于Poly(A )RNA仅占总RNA的1-4%,故此种引物合成的cDNA比随机六聚体作为引物和得到的cDNA在数量和复杂性方面均要小。 3.特异性引物:最特异的引发方法是用含目标RNA的互补序列的寡核苷酸作为引物,若PCR反应用二种特异性引物,第一条链的合成可由与mRNA 3’端最靠近的配对引物起始。用此类引物仅产生所需要的cDNA,导致更为特异的PCR扩增。