jade分析物相及晶胞参数和晶粒尺寸计算过程
《无极材料测试技术》课程作业
专业:2011级材料物理与化学姓名:王洪达学号:2011020204
作业要求:
对编号01N2009534的样品XRD测试数据进行物相分析,并计算其平均晶粒尺寸大小与晶胞参数。
1.物相分析过程
使用MDI Jade5.0软件对样品XRD测试数据进行分析,以定性分析样品的物相。
1.1.数据的导入
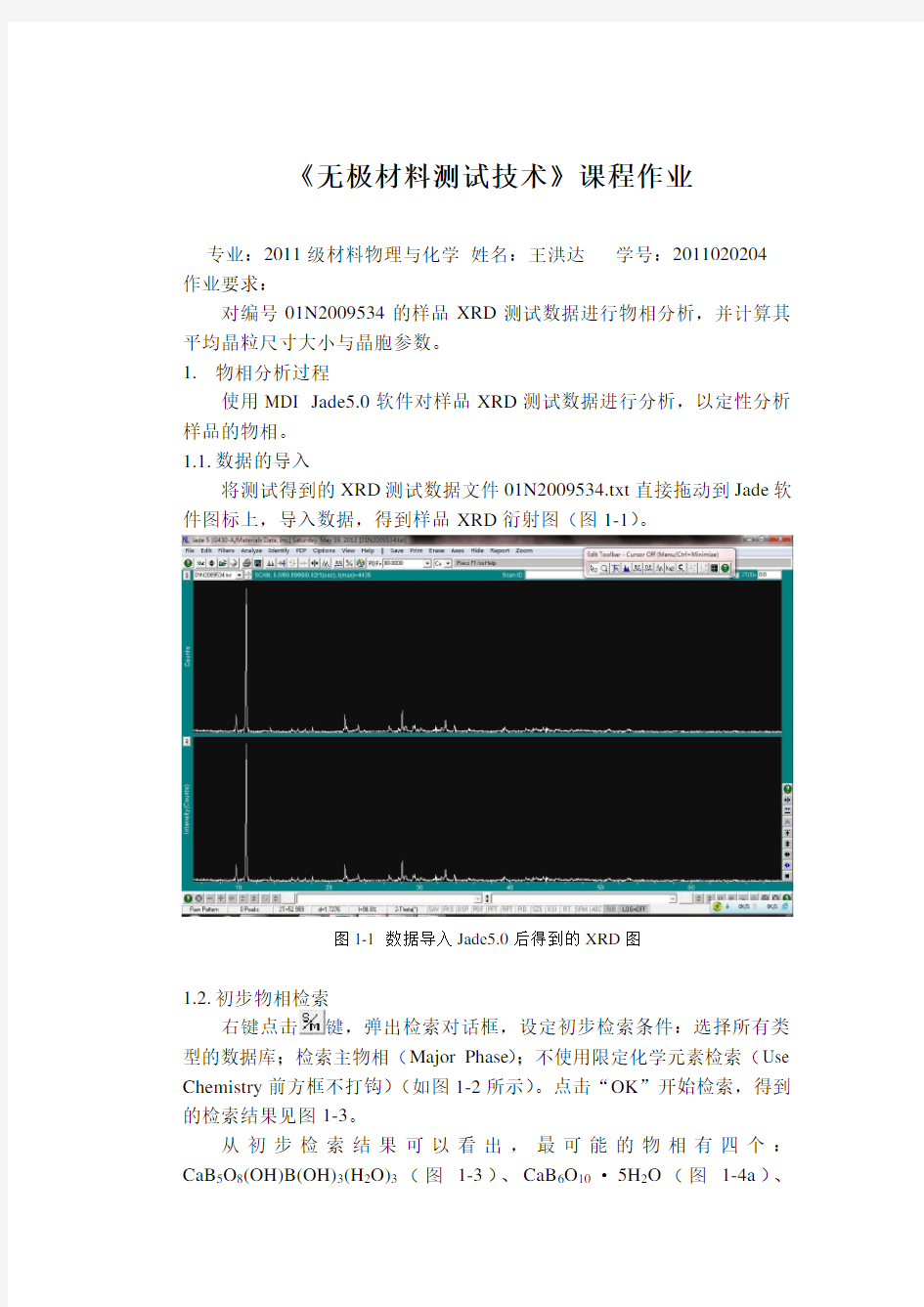
将测试得到的XRD测试数据文件01N2009534.txt直接拖动到Jade软件图标上,导入数据,得到样品XRD衍射图(图1-1)。
图1-1 数据导入Jade5.0后得到的XRD图
1.2.初步物相检索
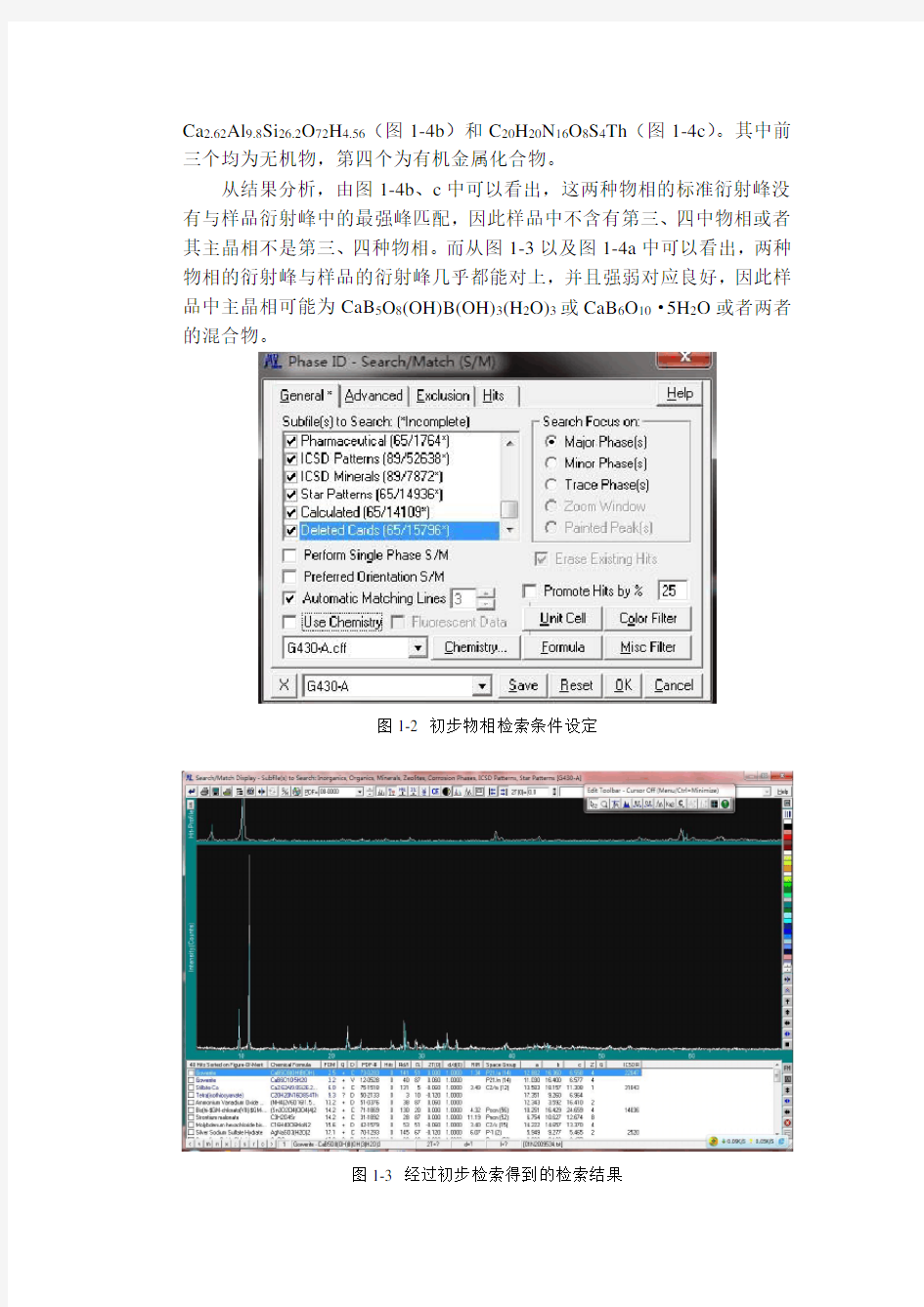
右键点击键,弹出检索对话框,设定初步检索条件:选择所有类型的数据库;检索主物相(Major Phase);不使用限定化学元素检索(Use Chemistry前方框不打钩)(如图1-2所示)。点击“OK”开始检索,得到的检索结果见图1-3。
从初步检索结果可以看出,最可能的物相有四个:CaB5O8(OH)B(OH)3(H2O)3(图1-3)、CaB6O10·5H2O(图1-4a)、
Ca2.62Al9.8Si26.2O72H4.56(图1-4b)和C20H20N16O8S4Th(图1-4c)。其中前三个均为无机物,第四个为有机金属化合物。
从结果分析,由图1-4b、c中可以看出,这两种物相的标准衍射峰没有与样品衍射峰中的最强峰匹配,因此样品中不含有第三、四中物相或者其主晶相不是第三、四种物相。而从图1-3以及图1-4a中可以看出,两种物相的衍射峰与样品的衍射峰几乎都能对上,并且强弱对应良好,因此样品中主晶相可能为CaB5O8(OH)B(OH)3(H2O)3或CaB6O10·5H2O或者两者的混合物。
图1-2 初步物相检索条件设定
图1-3 经过初步检索得到的检索结果
图1-4 初步检索结果
1.3. 限定条件的物相检索
初步分析结果,现对样品进行限定条件检索,检索条件设定如图1-5所示。检索结果见图1-6。
通过限定条件检索,发现CaB 5O 8(OH)B(OH)3(H 2O)3与CaB 6O 10·5H 2O 两物相的衍射峰与样品衍射峰均能对应。虽然CaB 5O 8(OH)B(OH)3(H 2O)3的FOM 值较小,但是从图上可以看出其标准衍射峰与样品峰(包括最强峰)有很小偏离,而CaB 6O 10·5H 2O 的衍射峰与样品峰能够更好的对应
a
(尤其是较强的衍射峰)。由于没有被告知样品的来历(合成或是天然矿物),因此,样品主晶相中一定含有CaB6O10·5H2O,可能有CaB5O8(OH)B(OH)3(H2O)3以及Ca2.62Al9.8Si26.2O72H4.56和C20H20N16O8S4Th。
如果样品为人工合成,考虑到Th元素的稀少性以及第四种物相元素与前三种差别较大,可以排除样品中含有此物相的可能性;但是若为天然矿物,则无法做出类似判断。CaB6O10·5H2O物相标准PDF卡号12-0528,卡片在附件中。
图1-5 限定条件物相检索前的条件设定
图1-6 经过限定元素后得到的分析结果
2.平均晶粒尺寸计算
Jade计算平均晶粒尺寸的基本原理就是谢乐公式,以衍射峰半高宽来计算。由于没有标准样品的衍射数据来制作仪器半高宽补正曲线,故计算
过程中选择Constant FWHM选项作为半高宽补正。
2.1.数据导入
将编号01N2009534的文本数据拖动到Jade程序中,得到样品衍射图(图2-1)。
图2-1 数据导入Jade5.0后得到的XRD图
2.2.物相检索
不对数据做任何处理,直接进行物相检索,根据1中的物相分析结果,认为主晶相为CaB6O10·5H2O,不考虑其他物相。检索结果如图2-2所示。
图2-2 初步检索得到的检索结果
2.3.扣除背底、Kα2
点击键显示已有的背底(图2-3),然后再次点击键,去除背底以及Kα2(图2-4)。
图2-3 显示已有的背底
图2-4 扣除背底及Kα2后的XRD图
2.4.平滑曲线
点击键对衍射图进行一次平滑。平滑后的衍射图见图2-5。
图2-5 平滑后的XRD图
2.5.全谱拟合
点击键对XRD进行全谱拟合,系统提示衍射峰过多(如图2-6所示),需要对XRD进行选区拟合。从XRD图可以看出,样品的主要衍射峰都在40°以前。因此,选择40°区域进行拟合(图2-6)。
图2-6 系统提示全谱拟合峰数过多
图2-7 选区拟合结果
2.6.计算平均晶粒尺寸
在菜单栏,点击Report/Size&Strain Plot,弹出对话框,选择Constant FWHM为样品半高宽补正曲线,得到结果如图2-8所示,晶粒平均尺寸为1888?,即188.8nm
图2-8 平均晶粒尺寸计算结果
3.晶胞参数计算
由物相分析可知,样品中CaB6O10·5H2O为一定存在的主晶相,CaB5O8(OH)B(OH)3(H2O)3可能存在。因此计算晶胞参数仅计算CaB6O10·5H2O物相的。
3.1.数据导入
将编号01N2009534的文本数据拖动到Jade程序中,得到样品衍射图(图3-1)。
图3-1 数据导入Jade5.0后得到的XRD图
3.2.物相检索
点击进行物相检索,在得到检索结果中,在CaB6O10·5H2O相前方框里打钩(如图3-2)。
图3-2 初步检索得到的检索结果
3.3.扣除背底、Kα2
点击检索结果界面左上角键,退回主界面,点击键显示已有背
底(图3-3),再次点击键扣除背底与Kα2(图3-4)。
图3-3 显示已有的背底
图3-4 扣除背底及Kα2后的XRD图
3.4.平滑曲线
点击键对衍射图进行一次平滑。平滑后的衍射图见图2-5。
图3-5 平滑后的XRD图
3.5.标注衍射指数,选区拟合
点击主界面右下角的键,在途中标注个衍射峰的衍射指数(图3-6),然后对XRD图进行选区拟合,结果如图3-7所示。
图3-6 标注晶面指数的XRD图
图3-7 选区拟合结果
3.6.计算晶胞参数
点击工具栏中Option/Calculate Lattice,弹出对话框,直接得到计算
结果(如图3-8所示)。
图3-8 晶胞参数计算结果
计算结果表明:CaB6O10·5H2O的平均晶格参数为a=10.9769?,b=16.5742,c=6.5742;α=90°,β=91°,γ=90°。检索得到的CaB6O10·5H2O 标准PDF卡片中的晶胞参数为a=11.03?,b=16.4,c=6.577;α=90°,β=91.33°,γ=90°。两者相比,相差不大。
通过XRD测试数据以Origin软件绘制的CaB6O10·5H2O X射线衍射
图如图3-9所示,由于峰位密集程度较高,且部分衍射峰很低,图中只标注了40°以前的部分主要衍射峰的衍射指数。
10
20
30
40
50
60
70
1000
2000
3000
4000
5000
101
170
160
060250330131
311140
040
130
120110
020
I n t e n s i t y (a .u .)
2
CaB 6O 10·5H 2O
图3-9 CaB 6O 10·5H 2O 样品XRD 衍射图
XRD晶粒尺寸计算
XRD晶粒尺寸分析 很多人都想算算粒径有多大。 其实,我们专业的术语不叫粒径,而叫“亚晶尺寸”,它表征的并不是一个颗粒的直径。 A。这么说吧,粉末由很多“颗粒”组成,每个颗粒由很多个“晶粒”聚集而成,一个晶粒由很多个“单胞”拼接组成。X射线测得的晶块尺寸是指衍射面指数方向上的尺寸,如果这个方向上有M个单胞,而且这个方向上的晶面间距为d,则测得的尺寸就是Md。如果某个方向(HKL)的单胞数为N,晶面间距为d1,那么这个方向的尺寸就是Nd1。由此可见,通过不同的衍射面测得的晶块尺寸是不一定相同的。 B 如果这个晶粒是一个完整的,没有缺陷的晶粒,可以将其视为一个测试单位,但是,如果这个晶粒有缺陷,那它就不是一个测试单位了,由缺陷分开的各个单位称为“亚晶”。比如说吧,如果一个晶粒由两个通过亚晶界的小晶粒组成(称为亚晶),那么,测得的就不是这个晶粒的尺寸而是亚晶的尺寸了。 C 为什么那么多人喜欢抛开专业的解释而用“粒径”这个词呢?都是“纳米材料”惹的祸。纳米晶粒本来就很小,一般可以认为一个纳米晶粒中不再存在亚晶,而是一个完整的晶粒,因此,亚晶尺寸这个术语就被套用到纳米晶粒的“粒径”上来了。实际上,国家对于纳米材料的粒径及粒径分布的表征是有标准的,需要用“小角散射”方法来测量。比如,北京钢铁研究总院做这个就做了很长时间。但是呢,一则,做小角散射的地方还不多,做起来也特别麻烦(现在好一些了,特别是对光能自动一些了),所以,很少有人去做,而且,用衍射峰宽计算出来的“粒径”总是那么小,何乐而不为呢?我私下地觉得吧,这些人在偷换概念。久而久之,大家也就接受了。 为了这个事吧,有些人就问了,既然做出来的纳米材料的“粒径”是这么小,那么有没有办法在做SEM或TEM时将团聚在一起的小晶粒分开呢?确实分不开,分得开的是一个个的晶粒,分不开的是亚晶。 D 至于为什么通过衍射峰宽测出来的“粒径”为什么总是那么小,还有一个原因。实际上吧,使衍射峰变宽的原因可能有两个,一是晶粒变小了,另一个原因是晶粒内部存在“微观应变”。打个比方吧,甲乙两个人同时做一件事,结果把功劳算到甲一个人头上,当然这个人的功劳就大了(功能劳大就峰宽,峰越宽晶粒就越细)。有时候发现,有个别人在有意无意地避口不谈乙的功劳。 E 为什么允许将亚晶尺寸称为“粒径”呢?称为径,必假定晶粒为“球形”,从而假定了不论从哪个晶面去测都会是相同的,即忽略了A 所说的那种差别。事实上,这种不同方向的尺寸差异在很多情况下确实可以忽略。但是,也有一些特殊情况是不可以的。下面我们再谈。 注意这两个假定,这就是为什么很多人都说,XRD测出来的粒径不可靠,总是小于SEM和TEM量出来的值。因为概念都不相同,它们怎么可能相同呢? 既然大家都说是粒径,那么要怎么样来算粒径呢? 我们先来看一个简单的问题。 怎么做拟合?
xRD晶粒尺寸分析
xRD晶粒尺寸分析
XRD晶粒尺寸分析 注:晶粒尺寸和晶面间距不同 计算晶粒大小:谢乐公式:D=kλ/βcosθ D—垂直于反射晶面(hkl)的晶粒平均粒度D是晶粒大小 β--(弧度)为该晶面衍射峰值半高宽的宽化程度 K—谢乐常数,取决于结晶形状,常取0.89 θ--衍射角 λ---入射X射线波长(?) 计算晶面间距:布拉格方程:2dsinθ=nλd是晶面间距。 此文档是用XRD软件来分析晶粒尺寸,用拟合的办法,而不是用谢乐公式 很多人都想算算粒径有多大。 其实,我们专业的术语不叫粒径,而叫“亚晶尺寸”,它表征的并不是一个颗粒的直径。 A 这么说吧,粉末由很多“颗粒”组成,每个颗粒由很多个“晶粒”聚集而成,一个晶粒由很多个“单胞”拼接组成。X射线测得的晶块尺寸是指衍射面指数方向上的尺寸,
如果这个方向上有M 个单胞,而且这个方向上的晶面间距为d ,则测得的尺寸就是Md 。如果某个方向(HKL )的单胞数为N ,晶面间距为d 1,那么这个方向的尺寸就是Nd 1。由 此可见,通过不同的衍射面测得的晶块尺寸是不一定相同的。 B 如果这个晶粒是一个完整的,没有缺陷的晶粒,可以将其视为一个测试单位,但是,如果这个晶粒有缺陷,那它就不是一个测试单位了,由缺陷分开的各个单位称为“亚晶”。比如说吧,如果一个晶粒由两个通过亚晶界的小晶粒组成(称为亚晶),那么,测得的就不是这个晶粒的尺寸而是亚晶的尺寸了。 C 为什么那么多人喜欢抛开专业的解释而用“粒径”这个词呢?都是“纳米材料”惹的祸。纳米晶粒本来就很小,一般可以认为一个纳米晶粒中不再存在亚晶,而是一个完整的晶粒,因此,亚晶尺寸这个术语就被套用到纳米晶粒的“粒径”上来了。实际上,国家对于纳米材料的粒径及粒径分布的表征是有标准的,需要用“小角散射”方法来测量。比如,北京钢铁研究总院做这个就做了很长时间。但是呢,一则,做小角散射的地方还不多,做起来也特别麻烦(现在好一些了,特别是对光能自动一些了),所以,很少有人去做,而且,用衍射峰宽计算出来的“粒径”总
jade分析报告物相及晶胞全参数和晶粒尺寸计算过程
《无极材料测试技术》课程作业 专业:2011级材料物理与化学:王洪达学号:2011020204 作业要求: 对编号01N2009534的样品XRD测试数据进行物相分析,并计算其平均晶粒尺寸大小与晶胞参数。 1.物相分析过程 使用MDI Jade5.0软件对样品XRD测试数据进行分析,以定性分析样品的物相。 1.1.数据的导入 将测试得到的XRD测试数据文件01N2009534.txt直接拖动到Jade软件图标上,导入数据,得到样品XRD衍射图(图1-1)。 图1-1 数据导入Jade5.0后得到的XRD图 1.2.初步物相检索 右键点击键,弹出检索对话框,设定初步检索条件:选择所有类型的数据库;检索主物相(Major Phase);不使用限定化学元素检索(Use Chemistry前方框不打钩)(如图1-2所示)。点击“OK”开始检索,得到的检索结果见图1-3。 从初步检索结果可以看出,最可能的物相有四个: CaB 5O 8 (OH)B(OH) 3 (H 2 O) 3 (图1-3)、CaB 6 O 10 ·5H 2 O(图1-4a)、
Ca 2.62Al 9.8 Si 26.2 O 72 H 4.56 (图1-4b)和C 20 H 20 N 16 O 8 S 4 Th(图1-4c)。其中前三个 均为无机物,第四个为有机金属化合物。 从结果分析,由图1-4b、c中可以看出,这两种物相的标准衍射峰没有与样品衍射峰中的最强峰匹配,因此样品中不含有第三、四中物相或者其主晶相不是第三、四种物相。而从图1-3以及图1-4a中可以看出,两种物相的衍射峰与样品的衍射峰几乎都能对上,并且强弱对应良好,因此 样品中主晶相可能为CaB 5O 8 (OH)B(OH) 3 (H 2 O) 3 或CaB 6 O 10 ·5H 2 O或者两者的 混合物。 图1-2 初步物相检索条件设定 图1-3 经过初步检索得到的检索结果
jade分析物相及晶胞参数和晶粒尺寸计算过程
《无极材料测试技术》课程作业 对编号01N2009534的样品XRD测试数据进行物相分析,并计算其平均晶粒尺寸大小与晶胞参数。 1.物相分析过程 使用软件对样品XRD测试数据进行分析,以定性分析样品的物相。 1.1.数据的导入 将测试得到的XRD测试数据文件直接拖动到Jade软件图标上,导入数据,得到样品XRD衍射图(图1-1)。 图1-1数据导入后得到的XRD图 1.2.初步物相检索 右键点击键,弹出检索对话框,设定初步检索条件:选择所有类型的数据库;检索主物相(MajorPhase);不使用限定化学元素检索(UseChemistry前方框不打钩)(如图1-2所示)。点击“OK”开始检索,得到的检索结果见图1-3。 从初步检索结果可以看出,最可能的物相有四个:CaB5O8(OH)B(OH)3(H2O)3(图1-3)、CaB6O10·5H2O(图1-4a)、(图1-4b)和C20H20N16O8S4Th(图1-4c)。其中前三个均为无机物,第四个为有机金属化合物。 从结果分析,由图1-4b、c中可以看出,这两种物相的标准衍射峰没有与样品衍射峰中的最强峰匹配,因此样品中不含有第三、四中物相或者其主晶相不是第三、四种物相。而从图1-3以及图1-4a中可以看出,两种物相的衍射峰与样品的衍射峰几乎都能对上,并且强弱对应良好,因此样品中主晶相可能为CaB5O8(OH)B(OH)3(H2O)3或CaB6O10·5H2O 或者两者的混合物。 图1-2初步物相检索条件设定 图1-3经过初步检索得到的检索结果 a
b c 图1-4初步检索结果 1.3.限定条件的物相检索 初步分析结果,现对样品进行限定条件检索,检索条件设定如图1-5所示。检索结果见图1-6。 通过限定条件检索,发现CaB5O8(OH)B(OH)3(H2O)3与CaB6O10·5H2O两物相的衍射峰与样品衍射峰均能对应。虽然CaB5O8(OH)B(OH)3(H2O)3的FOM值较小,但是从图上可以看出其标准衍射峰与样品峰(包括最强峰)有很小偏离,而CaB6O10·5H2O的衍射峰与样品峰能够更好的对应(尤其是较强的衍射峰)。由于没有被告知样品的来历(合成或是天然矿物),因此,样品主晶相中一定含有CaB6O10·5H2O,可能有 CaB5O8(OH)B(OH)3(H2O)3以及和C20H20N16O8S4Th。 如果样品为人工合成,考虑到Th元素的稀少性以及第四种物相元素与前三种差别较大,可以排除样品中含有此物相的可能性;但是若为天然矿物,则无法做出类似判断。 CaB6O10·5H2O物相标准PDF卡号12-0528,卡片在附件中。 图1-5限定条件物相检索前的条件设定 图1-6经过限定元素后得到的分析结果 2.平均晶粒尺寸计算 Jade计算平均晶粒尺寸的基本原理就是谢乐公式,以衍射峰半高宽来计算。由于没有标准样品的衍射数据来制作仪器半高宽补正曲线,故计算过程中选择ConstantFWHM 选项作为半高宽补正。 2.1.数据导入 将编号01N2009534的文本数据拖动到Jade程序中,得到样品衍射图(图2-1)。 图2-1数据导入后得到的XRD图 2.2.物相检索 不对数据做任何处理,直接进行物相检索,根据1中的物相分析结果,认为主晶相为CaB6O10·5H2O,不考虑其他物相。检索结果如图2-2所示。 图2-2初步检索得到的检索结果 2.3.扣除背底、Kα2 点击键显示已有的背底(图2-3),然后再次点击键,去除背底以及Kα2(图2-4)。
jade分析物相与晶胞参数和晶粒尺寸计算过程
《无极材料测试技术》课程作业 对编号 01N2009534 的样品 XRD 测试数据进行物相分析,并计算其平 均晶粒尺寸大小与晶胞参数。 1. 物相分析过程 使用 MDI Jade5.0 软件对样品 XRD 测试数据进行分析,以定性分析样品的物相。 1.1. 数据的导入 将测试得到的 XRD 测试数据文件 01N2009534.txt 直接拖动到 Jade 软 件图标上,导入数据,得到样品 XRD 衍射图(图 1-1)。 图 1-1 数据导入 Jade5.0 后得到的 XRD 图 1.2. 初步物相检索 右键点击 键,弹出检索对话框,设定初步检索条件:选择所有类 型的数据库;检索主物相( Major Phase );不使用限定化学元素检索( Use Chemistry 前方框不打钩)(如图 1-2 所示)。点击“ OK ”开始检索,得到的检索结果见图 1-3。 从初步检索结果可以看出,最可能的物相有四个: 5 8 323(图 1-3 )、 CaB 6 O 10 · 5H 2 O ( 图 1-4a )、 CaB O (OH)B(OH) (H O) 2.62 Al 9.8 Si 26.2 O 72 H 4.56(图 1-4b )和 C 20 20 16 8 4(图 1-4c )。其中前 Ca H N O S Th 三个均为无机物,第四个为有机金属化合物。
从结果分析,由图 1-4b、c 中可以看出,这两种物相的标准衍射峰没有与样品衍射峰中的最强峰匹配,因此样品中不含有第三、四中物相或者其主晶相不是第三、四种物相。而从图 1-3 以及图 1-4a 中可以看出,两种 物相的衍射峰与样品的衍射峰几乎都能对上,并且强弱对应良好,因此样品中主晶相可能为 CaB5O8(OH)B(OH) 3(H 2O) 3或 CaB6 O10·5H2O 或者两者的混合物。 图 1-2 初步物相检索条件设定 图 1-3 经过初步检索得到的检索结果
晶粒尺寸的测定
用X射线粉末衍射法测定超细晶的粒径及微观应力 适用于1~100纳米的超细晶
用X射线粉末衍射法测定超细 晶的粒径及微观应力 问题-1 ?粉末衍射法测定超细晶粒径的原理是什么? ?用粉末衍射法测定超细晶粒径是怎样做的? ?用粉末衍射法测定超细晶粒径要注意什么? 问题-2 ?粉末衍射法测定超细晶微观应力的原理是什么? ?用粉末衍射法测定超细晶微观应力是怎样做的? ?用粉末衍射法测定超细晶微观应力要注意什么? 2
用X射线粉末衍射法测定 超细晶的粒径及微观应力 多晶衍射测定微晶粒径 ?衍射峰的基本要素; ?微晶宽化效应(粉末衍射法测定超细晶粒径的 原理与方法); ?粉末衍射法测定超细晶粒径应用举例; 多晶衍射测定微观应力 ?微观应力的测定方法; ?微观应力与微晶宽化的分离; ?微观应力计算应用实例; 3
4任何一个衍射峰都是由五个基本要素组成的。 ?衍射峰的位置,最大衍射强度(I max ),半高宽,形态(通常衍射峰的峰形态,可具有Gauss, Cauchy, Voigt 或Pearson VII 分布)及对称性或不对称性。 ?不对称有为左右半高宽不对称;B 为左右形态不对称;C 为左右半高宽与形态不对称;D 为上下不对称;以及任意不对称;完 全对称(图1)。 衍射峰5要素
衍射峰5要素 五个基本要素的物理学意义 ?衍射峰位置是衍射面网间距的反映(即Bragg定理); ?最大衍射强度是物相自身衍射能力强弱的衡量指标及在混合物当中百分含量的函数(Moore and Reynolds,1989); ?半高宽及形态是晶体大小与应变的函数(Stokes and Wilson,1944); ?衍射峰的对称性是光源聚敛性(Alexander,1948)、样品吸收性(Robert and Johnson,1995)、仪器机械装置等因素及其他衍射峰或物相存在的函数(Moore and Reynolds,1989;Stern et al.,1991)。 5
Jade是如何计算晶块尺寸的
Jade是如何计算晶块尺寸的? XRD 2008-07-01 23:15:43 阅读162 评论0 字号:大中小 Jade是如何计算晶块尺寸的?
XRD 2008-07-01 23:12:54 阅读90 评论0 字号:大中小Jade是如何计算晶块尺寸 的? XRD 2008-07-01 23:14:56 阅读118 评论0 字号:大中小
Jade的一些使用经验
XRD 2009-01-03 23:29:26 阅读697 评论0 字号:大中小 摘要:本文简单介绍了作者在使用X射线衍射数据处理软件Jade进行物相检索、物相定量分析、晶胞参数修正以及晶粒尺寸与微应变计算等方面的一些经验和技巧。 Jade是一个32位Windows程序,用于处理X射线衍射数据。除基本的如显示图谱、打印图谱、数据平滑等功能外,主要功能有物相检索、结构精修、晶粒大小和微观应变计算等许多 功能。 1 Jade的物相检索方法和技巧 Jade的物相检索功能是非常强大的,通过软件基本上能检索出样品中全部物相。物相检索 的步骤包括: (1) 给出检索条件:包括检索子库(有机还是无机、矿物还是金属等等)、样品中可能存 在的元素等; (2) 计算机按照给定的检索条件进行检索,将最可能存在的前100种物相列出一个表; (3) 从列表中检定出一定存在的物相(人工完成)。 一般来说,判断一个物相的存在与否有三个条件: (1) 标准卡片中的峰位与测量峰的峰位是否匹配; (2) 标准卡片的峰强比与样品峰的峰强比要大致相同; (3) 检索出来的物相包含的元素在样品中必须存在。 Jade物相检索的常用方法有:无限制检索法和限定条件检索法。其中可限定的条件包括:PDF卡片库、元素组合、设置检索焦点、单峰检索。另外,也可以对物相进行反查。 1.1 无限制检索 无限制检索就是对图谱不作任何处理、不规定检索卡片库、也不作元素限定、检索对象选 择为主相(S/M Focus on Major Phases)。 这种方法一般可检测出样品中的主要的物相。在对样品无任何已知信息的情况下可试着检索出样品中的主要物相,进而通过检索出来的主要物相了解样品中元素的组成。另外,在考虑样品受到污染、反应不完全的情况可试探样品中是否存在未知的元素。但是,这种方法不可能检索出全部物相,并且检索结果可能与实际存在的物相偏差较大,需要其它实验作进一步证实。 2.2 PDF卡片库的选择 一般人认为,通过X射线衍射方法就能了解样品中存在某些元素,其实这是一个误解。X 射线衍射是一种结构分析手段,而不是元素分析手段,有很多物相虽然结构上也存在微小的差别,但是X射线衍射物相分析并不能真正区分它们。X射线衍射物相分析的目的应当是在已知样品元素组成的情况下检测这些元素的赋存状态。 PDF卡片库中有4个主要的数据库子库,即:Inorganic, ICSD Patterns, Minerals和ICSD Minerals。对于一般的样品,通常只需要选择这4个数据库就完全可以检索出全部物相。特别是当样品为天然矿物时,应当只选择矿物库的两个子库:ICSD Patterns, Minerals。否则,多选的数据库会对矿物物相分析带来困难。还有一点值得注意:ICSD(国际晶体学数据库)在物相检索中非常重要,因为很多新的物相在其它三个库中很难找到。我们应当注意的是,选择不同的数据库可能会得出不同的结果,数据库选择不合适时,可能会导致某些物相检索不出。
xRD晶粒尺寸分析1
XRD晶粒尺寸分析 注:晶粒尺寸和晶面间距不同 计算晶粒大小:谢乐公式:D=kλ/βcosθ D—垂直于反射晶面(hkl)的晶粒平均粒度 D是晶粒大小 β--(弧度)为该晶面衍射峰值半高宽的宽化程度 K—谢乐常数,取决于结晶形状,常取0.89 θ--衍射角 λ---入射X射线波长(?) 计算晶面间距:布拉格方程:2dsinθ=nλ d是晶面间距。 此文档是用XRD软件来分析晶粒尺寸,用拟合的办法,而不是用谢乐公式 很多人都想算算粒径有多大。 其实,我们专业的术语不叫粒径,而叫“亚晶尺寸”,它表征的并不是一个颗粒的直径。 A 这么说吧,粉末由很多“颗粒”组成,每个颗粒由很多个“晶粒”聚集而成,一个晶粒由很多个“单胞”拼接组成。X射线测得的晶块尺寸是指衍射面指数方向上的尺寸,如果这个方向上有M个单胞,而且这个方向上的晶面间距为d,则测得的尺寸就是Md。如果某个方向(HKL)的单胞数为N,晶面间距为d1,那么这个方向的尺寸就是Nd1。由此可见,通过不同的衍射面测得的晶块尺寸是不一定相同的。 B 如果这个晶粒是一个完整的,没有缺陷的晶粒,可以将其视为一个测试单位,但是,如果这个晶粒有缺陷,那它就不是一个测试单位了,由缺陷分开的各个单位称为“亚晶”。比如说吧,如果一个晶粒由两个通过亚晶界的小晶粒组成(称为亚晶),那么,测得的就不是这个晶粒的尺寸而是亚晶的尺寸了。 C 为什么那么多人喜欢抛开专业的解释而用“粒径”这个词呢?都是“纳米材料”惹的祸。纳米晶粒本来就很小,一般可以认为一个纳米晶粒中不再存在亚晶,而是一个完整的晶粒,因此,亚晶尺寸这个术语就被套用到纳米晶粒的“粒径”上来了。实际上,国家对于纳米材料的粒径及粒径分布的表征是有标准的,需要用“小角散射”方法来测量。比如,北京钢铁研究总院做这个就做了很长时间。但是呢,一则,做小角散射的地方还不多,做起来也特别麻烦(现在好一些
jade分析物相及晶胞参数和晶粒尺寸计算过程教学教材
j a d e分析物相及晶胞参数和晶粒尺寸计算 过程
《无极材料测试技术》课程作业 专业:2011级材料物理与化学姓名:王洪达学号:2011020204 作业要求: 对编号01N2009534的样品XRD测试数据进行物相分析,并计算其平均晶粒尺寸大小与晶胞参数。 1.物相分析过程 使用MDI Jade5.0软件对样品XRD测试数据进行分析,以定性分析样品的物相。 1.1.数据的导入 将测试得到的XRD测试数据文件01N2009534.txt直接拖动到Jade 软件图标上,导入数据,得到样品XRD衍射图(图1-1)。 图1-1 数据导入Jade5.0后得到的XRD图 1.2.初步物相检索 右键点击键,弹出检索对话框,设定初步检索条件:选择所有类型的数据库;检索主物相(Major Phase);不使用限定化学元素检索(Use Chemistry前方框不打钩)(如图1-2所示)。点击“OK”开始检索,得到的检索结果见图1-3。 从初步检索结果可以看出,最可能的物相有四个: CaB5O8(OH)B(OH)3(H2O)3(图1-3)、CaB6O10·5H2O(图1-4a)、
Ca2.62Al9.8Si26.2O72H4.56(图1-4b)和C20H20N16O8S4Th(图1-4c)。其中前三个均为无机物,第四个为有机金属化合物。 从结果分析,由图1-4b、c中可以看出,这两种物相的标准衍射峰没有与样品衍射峰中的最强峰匹配,因此样品中不含有第三、四中物相或者其主晶相不是第三、四种物相。而从图1-3以及图1-4a中可以看出,两种物相的衍射峰与样品的衍射峰几乎都能对上,并且强弱对应良好,因此样品中主晶相可能为CaB5O8(OH)B(OH)3(H2O)3或 CaB6O10·5H2O或者两者的混合物。 图1-2 初步物相检索条件设定 图1-3 经过初步检索得到的检索结果
Jade 是如何计算晶粒尺寸的
Jade 是如何计算晶粒尺寸的? 不止10次有人问到这个问题,让我有兴趣去了解。看了看这个软件的帮助,也没有得到答案。只好一种一种方法去试,好象还真是得到了解答。今天,把它写出来供大家验证。 Jade 按照谢乐公式来计算。 θ βλcos k D = λ 是辐射的波长,按K α1的波长计算,如铜靶,则λ=0.154056nm 。 D 就是晶块尺寸,单位可以是纳米,与波长λ的单位相同。 k 是一个参数,可以取0.89,0.95或者1,一般人都愿意取1。但是,软件是按0.89计算的。 θ是半衍射角,单位可以是度或者弧度,只要你能正确计算出它的余弦就可以。 β是衍射峰的加宽。一般按两种方法来计算,即b B ?=β,22b B ?=β一般人愿意用b B ?=β。但是,Jade 却用后者。确实,一些教科书中都提到,后者更符合实际情况。 这里的B 就是FWHM ,即样品的衍射峰宽,b 则是仪器宽度。 好了。让大家来看看我的试验过程。 有这么一个衍射峰,我们先来做拟合:
通过Report----peak profile report菜单,查看到拟合的结果: 通过菜单Edit-----Preferences,可看到下面的窗口:
单击View FWHM Curve,你看到: 你可能看到的不一样,这是因为你没有做仪器校正,而使用了软件自带的某个“标样”,如Constant FWHM。这里看到的是我在07年12月19日做的硅标数据。 移动你的鼠标,并定位于116°处,你可看到FWHM=0.140°。这就是仪器宽度,即b。
在这个窗口中,你还看到了仪器波长是 1.54056埃,即0.145056nm。 怎么样?把这些数据代入到公式,得到14.40902nm。 这里讲的是单峰处理时的晶块尺寸。要注意,除非你的样品是分散单体纳米晶,否则,这个数据是不可信的。 关于晶块尺寸计算与微观应变更详细的解释,请访问我的QQ空间,也许会有些帮助。
Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸)
Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸) Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸) Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸) 根据X射线衍射理论,在晶粒尺寸小于100nm时,随晶粒尺寸的变小衍射峰宽化变得显著,考虑样品的吸收效应及结构对衍射线型的影响,样品晶粒尺寸可以用Debye-Scherrer公式计算。 Scherrer公式:Dhkl=kλ/βcosθ 其中,Dhkl为沿垂直于晶面(hkl)方向的晶粒直径,k为Scherrer 常数(通常为0.89),λ为入射X射线波长(Cuka 波长为0.15406nm,Cuka1 波长为0.15418nm。),θ为布拉格衍射角(°),β为衍射峰的半高峰宽(rad)。 但是在实际操作中如何从一张普通的XRD图谱中获得上述的参数来计算晶粒尺寸还存在以下问题: 1) 首先,用XRD计算晶粒尺寸必须扣除仪器宽化和应力宽化影响。如何扣除仪器宽化和应力宽化影响?在什么情况下,可以简化这一步骤? 答:在晶粒尺寸小于100nm时,应力引起的宽化与晶粒尺度引起的宽化相比,可以忽略。此时,Scherrer公式适用。但晶粒尺寸大到一定程度时,应力引起的宽化比较显著,此时必须考虑引力引起的宽化, Scherrer公式不再适用。
2) 通常获得的XRD数据是由Kα线计算得到的。此时,需要Kα1 和Kα2必须扣除一个,如果没扣除,肯定不准确。 3) 扫描速度也有影响,要尽可能慢。一般2°/min。 4)一个样品可能有很多衍射峰,是计算每个衍射峰对应晶粒尺寸后 平均?还是有其它处理原则? 答:通常应当计算每个衍射峰晶粒尺寸后进行平均。当然只有一两 峰的时候,就没有必要强求了! 5) 有的XRD数据中给出了width值,是不是半高宽度的值?能不能 直接代入上面公式吗?如果不能,如何根据XRD图谱获得半峰宽? TOP 20 β为衍射峰的半高峰宽时,k=0.89 β为衍射峰的积分宽度时,k=1.0。其中积分宽度=衍射峰面积积分/峰高 如何获得单色Kα1: 1)硬件滤掉Kβ:K系射线又可以细分为Kα(L层电子填充)和Kβ(M层电 子填充)两种波长略有差异的两种射线。而X射线衍射仪要求使用单色X射线,因此,需要在XRD实验时把后者除掉。 a). 传统的方法是在光路上加入一个滤波片(如Ni)。 b).现在一般使用铜靶,在光路上增加一个石墨晶体单色器来去除Kβ射线。通常的做法是在衍射线的光路上,安装弯曲晶体单色器。石墨单晶体单色器是一块磨成弯曲面的石墨单晶体。由试样衍射产生的衍射线(称为一次衍射)经单色器时,通过调整单晶体的方位使它的某个高反射本领晶面与一次衍射线的夹角刚好等于该晶面对一次衍射的Kα辐射的布拉格角。单色器可以去除衍射背底,也可以去除Kβ射线的干扰。这样,由单晶体衍射后发出的二次衍射线就是纯净的与试样衍射对应的Kα衍射线。 2) 软件分离Kα2:Kα辐射还可以细分为Kα1和Kα2两种波长差很小的辐射。由于它们的波长差很小,无法通过硬件的方法来消除其中任何一种,因此,只有
XRD晶粒尺寸分析
很多人都想算算粒径有多大。 其实,我们专业的术语不叫粒径,而叫“亚晶尺寸”,它表征的并不是一个颗粒的直径。 A。这么说吧,粉末由很多“颗粒”组成,每个颗粒由很多个“晶粒”聚集而成,一个晶粒由很多个成。X射线测得的晶块尺寸是指衍射面指数方向上的尺寸,如果这个方向上有M个单胞,而且这个为d,则测得的尺寸就是Md。如果某个方向(HKL)的单胞数为N,晶面间距为d1,那么这个方向的此可见,通过不同的衍射面测得的晶块尺寸是不一定相同的。 B如果这个晶粒是一个完整的,没有缺陷的晶粒,可以将其视为一个测试单位,但是,如果这个晶不是一个测试单位了,由缺陷分开的各个单位称为“亚晶”。比如说吧,如果一个晶粒由两个通过成(称为亚晶),那么,测得的就不是这个晶粒的尺寸而是亚晶的尺寸了。 C为什么那么多人喜欢抛开专业的解释而用“粒径”这个词呢?都是“纳米材料”惹的祸。纳米晶般可以认为一个纳米晶粒中不再存在亚晶,而是一个完整的晶粒,因此,亚晶尺寸这个术语就被套用径”上来了。实际上,国家对于纳米材料的粒径及粒径分布的表征是有标准的,需要用“小角散射如,北京钢铁研究总院做这个就做了很长时间。但是呢,一则,做小角散射的地方还不多,做起来好一些了,特别是对光能自动一些了),所以,很少有人去做,而且,用衍射峰宽计算出来的“粒何乐而不为呢?我私下地觉得吧,这些人在偷换概念。久而久之,大家也就接受了。 ?为了这个事吧,有些人就问了,既然做出来的纳米材料的“粒径”是这么小,那么有没有办法在做团聚在一起的小晶粒分开呢?确实分不开,分得开的是一个个的晶粒,分不开的是亚晶。 D至于为什么通过衍射峰宽测出来的“粒径”为什么总是那么小,还有一个原因。实际上吧,使衍能有两个,一是晶粒变小了,另一个原因是晶粒内部存在“微观应变”。打个比方吧,甲乙两个人果把功劳算到甲一个人头上,当然这个人的功劳就大了(功能劳大就峰宽,峰越宽晶粒就越细)。别人在有意无意地避口不谈乙的功劳。 E为什么允许将亚晶尺寸称为“粒径”呢?称为径,必假定晶粒为“球形”,从而假定了不论从哪
Jade计算晶胞参数及晶粒尺寸、EXPGUI结构精修
2、操作题 (1)分析试样的相组成: (2)采用Jade计算主相的晶粒尺寸、晶胞参数,操作步骤及结果如下: 在完成(1)中的物相检索后,(已扣除背底和平滑),对图像进行全谱拟合,观察拟合效果,因为需要求的是主相即Li4Ti5O12相的晶粒 尺寸与晶胞参数。所以在TiO2的两个峰处按下右键,去除对TiO2的
468?。 a=
(3)利用Jade将.raw的数据文件导出并存为.txt格式→Excel转换另存为.csv格式→Cmpr转换为.gsas,进行峰的拟合并得出仪器参数→EXPGUI进行结构精修。(此步骤中的所有文件存在同一个文件下,且不要出现汉字命名。) ●Excel中的操作: 读取.txt文件,删除数据第一行。选择“数据”→“分列”→“文件另存为.csv” ●Cmpr中的操作: ○1Read(选择正确的数据格式,并读取.csv文件)→Write(选择 相应的数据格式,存储位置选在前述文件所在的文件夹),点击Write Selected Datasets存为.gsas文件→Fit,对选中的峰用“P”选中,可以在框中看到相应的峰的位置→Set Range to Fit, 进行峰的拟合,注意观察GOF值,使之最小。所有的峰都拟合完 成。→FitWidths,Select peak list选择peaklist 1,将蓝点与
红点交叉的位置去除掉(在相应的框中打勾)→Fit Profile,即可得到相应的仪器参数值。 只能选择8个峰。
EXPGUI中的操作: 设置文件名(禁止出现中文)→Read→Creat→Set→LS Contr ols中的Number of Cycles可设为3→Phase→Add Phase,加入两相的数据,为.cif格式的文件(Add→Continue→Add Atoms)→Histo gram
RD晶粒尺寸分析
R D晶粒尺寸分析 Revised final draft November 26, 2020
很多人都想算算粒径有多大。 其实,我们专业的术语不叫粒径,而叫“亚晶尺寸”,它表征的并不是一个颗粒的直径。 A。这么说吧,粉末由很多“颗粒”组成,每个颗粒由很多个“晶粒”聚集而成,一个晶粒由很多个“单胞”拼接组成。X 是指衍射面指数方向上的尺寸,如果这个方向上有M个单胞,而且这个方向上的晶面间距为d,则测得的尺寸就是Md。如单胞数为N,晶面间距为d1,那么这个方向的尺寸就是Nd1。由此可见,通过不同的衍射面测得的晶块尺寸是不一定相同B如果这个晶粒是一个完整的,没有缺陷的晶粒,可以将其视为一个测试单位,但是,如果这个晶粒有缺陷,那它就不是陷分开的各个单位称为“亚晶”。比如说吧,如果一个晶粒由两个通过亚晶界的小晶粒组成(称为亚晶),那么,测得的寸而是亚晶的尺寸了。 C为什么那么多人喜欢抛开专业的解释而用“粒径”这个词呢都是“纳米材料”惹的祸。纳米晶粒本来就很小,一般可以再存在亚晶,而是一个完整的晶粒,因此,亚晶尺寸这个术语就被套用到纳米晶粒的“粒径”上来了。实际上,国家对于径分布的表征是有标准的,需要用“小角散射”方法来测量。比如,北京钢铁研究总院做这个就做了很长时间。但是呢,地方还不多,做起来也特别麻烦(现在好一些了,特别是对光能自动一些了),所以,很少有人去做,而且,用衍射峰宽总是那么小,何乐而不为呢我私下地觉得吧,这些人在偷换概念。久而久之,大家也就接受了。 为了这个事吧,有些人就问了,既然做出来的纳米材料的“粒径”是这么小,那么有没有办法在做SEM或TEM时将团聚在确实分不开,分得开的是一个个的晶粒,分不开的是亚晶。 D至于为什么通过衍射峰宽测出来的“粒径”为什么总是那么小,还有一个原因。实际上吧,使衍射峰变宽的原因可能有了,另一个原因是晶粒内部存在“微观应变”。打个比方吧,甲乙两个人同时做一件事,结果把功劳算到甲一个人头上,大了(功能劳大就峰宽,峰越宽晶粒就越细)。有时候发现,有个别人在有意无意地避口不谈乙的功劳。 E为什么允许将亚晶尺寸称为“粒径”呢?称为径,必假定晶粒为“球形”,从而假定了不论从哪个晶面去测都会是相同那种差别。事实上,这种不同方向的尺寸差异在很多情况下确实可以忽略。但是,也有一些特殊情况是不可以的。下面我注意这两个假定,这就是为什么很多人都说,XRD测出来的粒径不可靠,总是小于SEM和TEM量出来的值。因为概念都不同呢? 既然大家都说是粒径,那么要怎么样来算粒径呢? 我们先来看一个简单的问题。 怎么做拟合? 我们并不需要对所有的峰都做拟合,也不能用“全谱自动拟合”,正确的方法是做单峰拟合。 今天有同学发过来一个数据,看看。 当晶粒细化时,衍射峰就会变宽,随之而来的是强度降低,峰就不那么好看了,做拟合时有三点要注意: 1)并不需要选择全部的峰来参与计算,如果某个峰长得不好,宁可不要这个峰的数据; 2)做单峰拟合,有的同学不管三七二十一,一个拟合按钮按下去,自动去算吧,结果当然是错误的; 3)如果有重叠峰,可以先将重叠峰分离,但最后最好将其去掉,软件自动分解重叠峰的效果可能并不是令人满意的。 这里,拟合分作两段,只用到六个峰。特别是一些背底不平的情况,尤其要如此。 看看拟合结果,每个晶面计算出来的晶粒尺寸差不了多少,说明: 1)确实不存在微应变; 2)晶粒基本上是球形。 既然这样,那么计算粒径的时候只要一个峰不是也差不离吗确实是这样的,如果能假定样品中不存在微观应变,用一个低尺寸就可以了,没有必要用很多峰,很多峰算出来不就是“平均粒径”吗 按下上面那个窗口中的“Size&Strain”按钮,弹出上面的窗口,发现这些数据点基本上落在一条水平线上,那么选择“S “平均粒径”。 我们再来玩玩这个样品。将这个样品做一点处理,比如加热烧一会,会得到怎么样的衍射谱呢? 这个图比上面那个图好看多了,为什么呢?因为峰明显变窄了,窄了也就高了,高了也就掩盖误差了。 注意,尽管这个图长得这么好,我们还是做单峰拟合,不厌其烦地,任劳任怨地一个峰一个峰地做拟合。 有点意思了,看看XS下面的数据,从衍射角由低到高的顺序,XS值是由大到小(除了最后两个例外)。点下“Size&Str 据点落在一条斜线上。而且还不过原点。在纵坐标上的截距大于0,这就说明,这个峰变宽,有你的一半也有我的一半。计算的结果可以从图上看得清(吧) 那么,最后两个点,为什么会突然变大了呢?都是重叠峰惹的祸。注意,上面的谱图可以清楚看到,最后两个点的数据是怎么办呢?去掉呀,在图上的红点上用鼠标点一下,红点就消失了,这个数据点被舍弃。如果舍弃一个点还不满足,干脆什么大不了的。
XRD计算晶粒尺寸
Scherrer公式计算晶粒尺寸() Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸) 根据X射线衍射理论,在晶粒尺寸小于100nm时,随晶粒尺寸的变小衍射峰宽化变得显著,考虑样品的吸收效应及结构对衍射线型的影响,样品晶粒尺寸可以用Debye-Scherrer公式计算。 Scherrer公式:Dhkl=kλ/βcosθ 其中,Dhkl为沿垂直于晶面(hkl)方向的晶粒直径,k为Scherrer常数(通常为0.89),λ为入射X射线波长(Cuka 波长为0.15406nm,Cuka1 波长为0.15418nm。),θ为布拉格衍射角(°),β为衍射峰的半高峰宽(rad)。 但是在实际操作中如何从一张普通的XRD图谱中获得上述的参数来计算晶粒尺寸还存在以下问题: 1) 首先,用XRD计算晶粒尺寸必须扣除仪器宽化和应力宽化影响。如何扣除仪器宽化和应力宽化影响?在什么情况下,可以简化这一步骤? 答:在晶粒尺寸小于100nm时,应力引起的宽化与晶粒尺度引起的宽化相比,可以忽略。此时,Scherrer公式适用。但晶粒尺寸大到一定程度时,应力引起的宽化比较显著,此时必须考虑引力引起的宽化,Scherrer公式不再适用。 2) 通常获得的XRD数据是由Kα线计算得到的。此时,需要Kα1和Kα2必须扣除一个,如果没扣除,肯定不准确。 3) 扫描速度也有影响,要尽可能慢。一般2°/min。 4)一个样品可能有很多衍射峰,是计算每个衍射峰对应晶粒尺寸后平均?还是有其它处理原则? 答:通常应当计算每个衍射峰晶粒尺寸后进行平均。当然只有一两峰的时候,就没有必要强求了! 5) 有的XRD数据中给出了width值,是不是半高宽度的值?能不能直接代入上面公式吗?如果不能,如何根据XRD图谱获得半峰宽? β为衍射峰的半高峰宽时,k=0.89 β为衍射峰的积分宽度时,k=1.0。其中积分宽度=衍射峰面积积分/峰高 如何获得单色Kα1: 1)硬件滤掉Kβ:K系射线又可以细分为Kα(L层电子填充)和Kβ(M层电子填充)两种波长略有差异的两种射线。而X射线衍射仪要求使用单色X射线,因此,需要在XRD实验时把后者除掉。 a). 传统的方法是在光路上加入一个滤波片(如Ni)。 b).现在一般使用铜靶,在光路上增加一个石墨晶体单色器来去除Kβ射线。通常的做法是在衍射线的光路上,安装弯曲晶体单色器。石墨单晶体单色器是一块磨成弯曲面的石墨单晶体。由试样衍射产生的衍射线(称为一次衍射)经单色器时,通过调整单晶体的方位使它的某个高反射本领晶面与一次衍射线的夹角刚好等于该晶面对一次衍射的Kα辐射的布拉格角。单色器可以去除衍射背底,也可以去除Kβ射线的干扰。这样,由单晶体衍射后发出的二次衍射线就是纯净的与试样衍射对应的Kα衍射线。 2) 软件分离Kα2:Kα辐射还可以细分为Kα1和Kα2两种波长差很小的辐射。由于它们的波长差很小,无法通过硬件的方法来消除其中任何一种,因此,只有通过软件的方法来消除
14.谢乐公式计算XRD样品的晶粒尺寸的实例
谢乐公式计算XRD样品的晶粒尺寸的实例 我们常见的谢乐(Scherrer)公式表达式为D=Kλ /(βcos θ)(K为常数;λ 为X 射线波长;β 为为衍射峰半高宽;θ为衍射角)。在上式中常数K的取值与β的定义有关,当β为半宽高时,K取0.89。当β为积分宽度时,K取1.0。 我们在计算晶粒尺寸时,一般采用低角度的衍射线,如果晶粒尺寸较大,可用较高衍射角的衍射线来代替。谢乐公式适用范围为1-100nm,晶粒尺寸小于1nm大于100nm时,使用用谢乐公式不太准确,当晶粒尺寸在30nm时其计算的结果最准确。同时,谢乐公式只适合球形粒子,对立方体粒子常数K应改为0.943,半高宽应该转化为弧度制,即[(β÷180)×3.14]。 下面这个图是Jade5.0所读的晶粒尺寸为264(A°)即为26.4nm。
38.26)2 159.36(14.3180332.015405.0943.0=???=COS D 这边有的数据是X 射线波长λ=0.15405 nm , 半高宽β=0.332,2θ=36.159。 我是这样算的: 自己计算出来的值和用软件计算出来的值很接近。 我这里有2004的PDF 标准卡片,如果有哪位需要的话直接加我qq ,我发给你,我的qq 是425841088。 Scherrer 公式计算晶粒尺寸() Scherrer 公式计算晶粒尺寸(XRD 数据计算晶粒尺寸) 根据X 射线衍射理论,在晶粒尺寸小于100nm 时,随晶粒尺寸的变小衍射峰宽化变得显著,考虑样品的吸收效应及结构对衍射线型的影响,样品晶粒尺寸可以用Debye-Scherrer 公式计算。 Scherrer 公式:Dhkl=k λ/βcos θ 其中,Dhkl 为沿垂直于晶面(hkl )方向的晶粒直径,k 为Scherrer 常数(通常为0.89), λ为入射X 射线波长(Cuka 波长为0.15406nm ,Cuka1 波长为0.15418nm 。),θ为布拉格衍射角(°),β为衍射峰的半高峰宽(rad )。 但是在实际操作中如何从一张普通的XRD 图谱中获得上述的参数来计算晶粒尺寸还存在以下问题: 1) 首先,用XRD 计算晶粒尺寸必须扣除仪器宽化和应力宽化影响。如何扣除仪器宽化和应力宽化影响?在什么情况下,可以简化这一步骤? 答:在晶粒尺寸小于100nm 时,应力引起的宽化与晶粒尺度引起的宽化相比,