胎儿右桡骨拇指缺如并左侧多囊性发育不良肾超声表现一例
·病例报告·胎儿右桡骨拇指缺如并左侧多囊性发育不良肾
超声表现一例
李广友杜峰曹彤
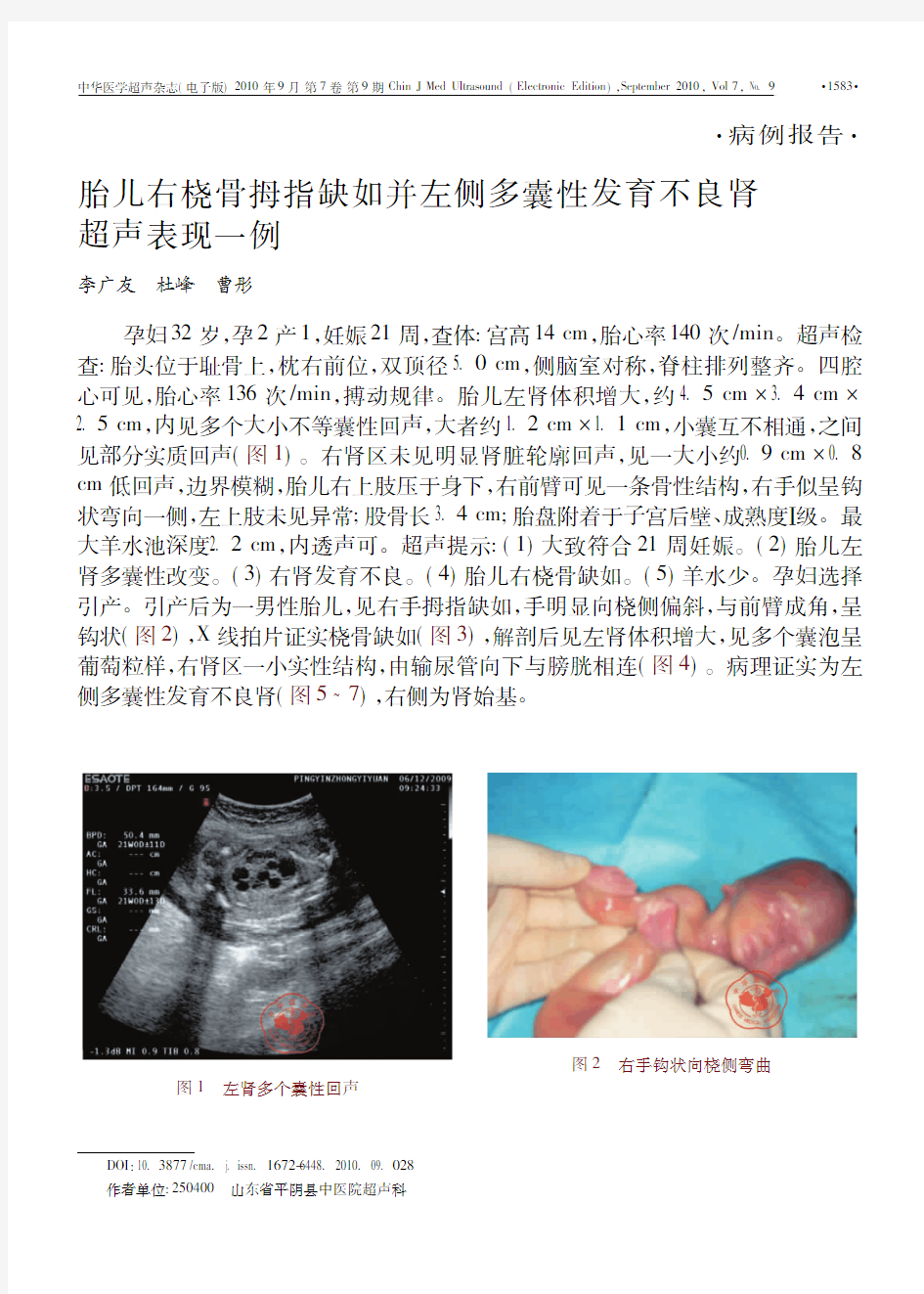
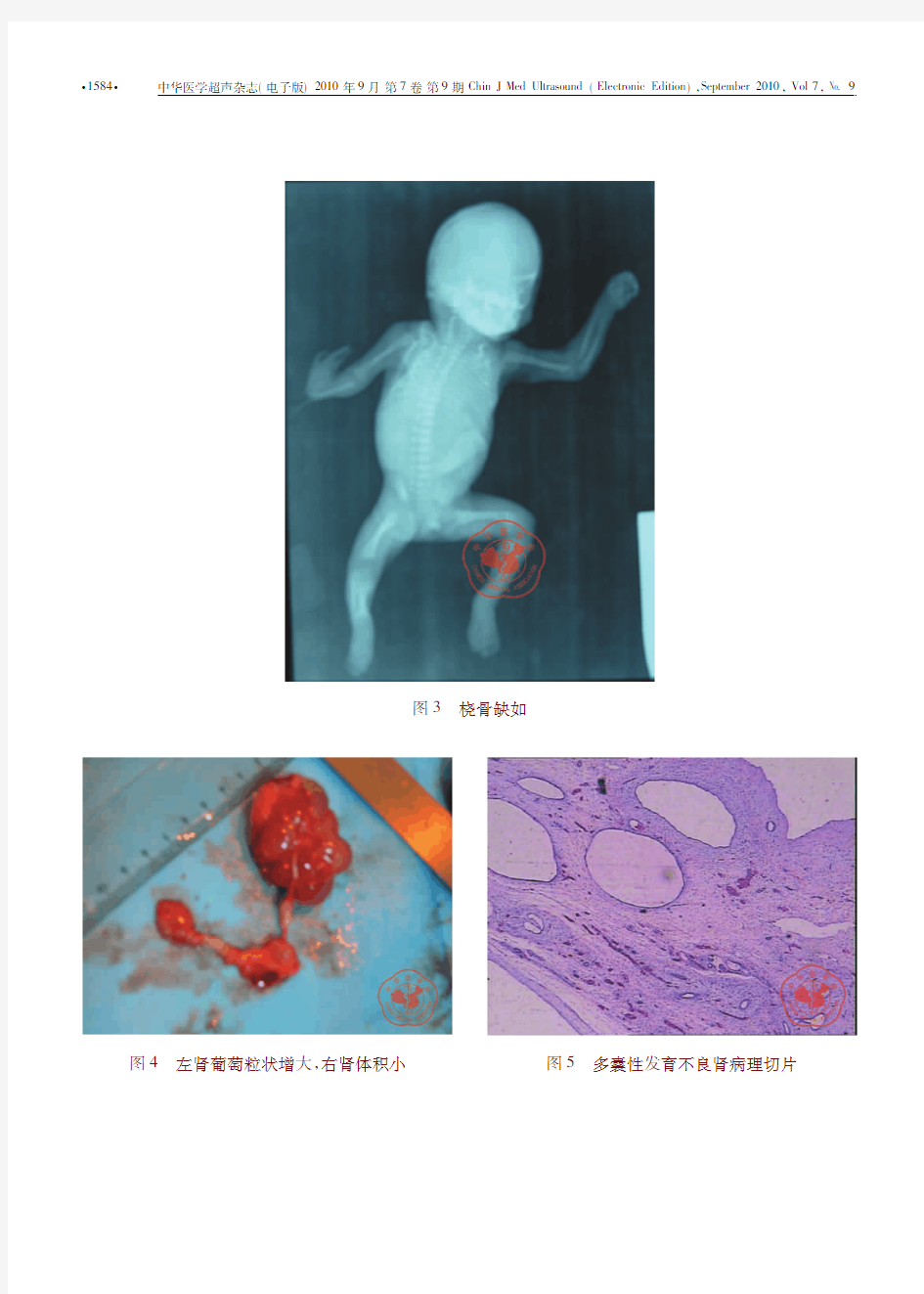
孕妇32岁,孕2产1,妊娠21周,查体:宫高14cm,胎心率140次/min。超声检查:胎头位于耻骨上,枕右前位,双顶径5.0cm,侧脑室对称,脊柱排列整齐。四腔心可见,胎心率136次/min,搏动规律。胎儿左肾体积增大,约4.5cm?3.4cm?2.5cm,内见多个大小不等囊性回声,大者约1.2cm?1.1cm,小囊互不相通,之间见部分实质回声(图1)。右肾区未见明显肾脏轮廓回声,见一大小约0.9cm?0.8 cm低回声,边界模糊,胎儿右上肢压于身下,右前臂可见一条骨性结构,右手似呈钩状弯向一侧,左上肢未见异常;股骨长3.4cm;胎盘附着于子宫后壁、成熟度Ⅰ级。最大羊水池深度2.2cm,内透声可。超声提示:(1)大致符合21周妊娠。(2)胎儿左肾多囊性改变。(3)右肾发育不良。(4)胎儿右桡骨缺如。(5)羊水少。孕妇选择引产。引产后为一男性胎儿,见右手拇指缺如,手明显向桡侧偏斜,与前臂成角,呈钩状(图2),X线拍片证实桡骨缺如(图3),解剖后见左肾体积增大,见多个囊泡呈葡萄粒样,右肾区一小实性结构,由输尿管向下与膀胱相连(图4)。病理证实为左侧多囊性发育不良肾(图5 7),右侧为肾始基。
图2右手钩状向桡侧弯曲图1左肾多个囊性回声
DOI:10.3877/cma.j.issn.1672-6448.2010.09.028
作者单位:250400山东省平阴县中医院超声科
图3桡骨缺如
图4左肾葡萄粒状增大,右肾体积小图5多囊性发育不良肾病理切片
图6多囊性发育不良肾病理切片图7多囊性发育不良肾病理切片
讨论多囊性发育不良肾(Ⅱ型)是较常见的一种肾脏囊性疾病,其发生率约为0.0333%。本病无遗传性,以男性多见,常为单侧发病,对侧肾脏多发育正常[1],但15% 30%对侧肾脏有异常,左侧多囊性发育不良性肾的发生率约为右侧的2倍。本例胎儿对侧肾脏亦发育异常,为一始基肾。正常肾脏发育依赖于输尿管芽与后肾原基之间的相互作用。由于胚胎时输尿管及肾盂闭锁,即早期宫内梗阻干扰了这一过程,导致肾发育异常,常伴肾动脉发育不全。典型的多囊性发育不良肾受累肾脏形态明显异常,无肾脏基本形态,由大小不等数量不一的囊腔构成,像一串葡萄粒。由于本例胎儿双肾均有异常,伴羊水过少,提示发现羊水过少时应注意检查肾脏结构。
先天性桡骨发育不全或缺如又称轴旁性桡侧半肢畸形,发生率在围生儿中约为0.0033%[1],可单侧或双侧发病。可分为3型:Ⅰ型:桡骨完全缺如;Ⅱ型:桡骨部分缺如;Ⅲ型:桡骨发育不全。本例属Ⅰ型,最常见,占50%以上;桡骨完全未发育,腕部由于缺乏桡骨的支持而导致严重的桡偏畸形,手可呈直角或接近前壁桡侧表面。先天性桡骨缺如常伴有其他畸形综合征,如Holt-Oram综合征、TAR综合征、海豹肢畸形、VATER联合征等,但合并多囊性发育不良肾者少见报道。龚学德等[2]认为可能系某种临床综合征。陈琮瑛等[3]对7例桡骨畸形胎儿进行脐带血染色体检查,5例染色体为18-三体,表明桡骨畸形常合并的染色体异常为18-三体。本例胎儿肾脏畸形明显,拇指受胎位、姿势、羊水影响检出困难。桡骨发育畸形合并其他畸形的发生率较高,系统顺序超声检查可避免漏诊,对评价胎儿预后及降低围产期死亡率非常重要。
参考文献
1李胜利.胎儿畸形产前超声诊断学.北京:人民军医出版社,2007:271,352.
2龚学德,曾莉,黄鲁刚.先天性桡骨缺如伴泌尿系多发畸形三例报道.中华小儿外科杂志,2007,28(12):667-668.
3陈琮瑛,李胜利,刘菊玲.连续顺序追踪超声法诊断胎儿桡骨畸形.中华超声影像学杂志,2004,13(8):594-596.
(收稿日期:2010-01-25)
(本文编辑:安京媛)
李广友,杜峰,曹彤.胎儿右桡骨拇指缺如并左侧多囊性发育不良肾超声表现一例[J/CD].中华医学超声杂志:电子版,2010,7(9):1583-1586.
正常人体运动学第四章腕关节运动学
三、腕关节运动学 (一)腕关节的组成和运动方向 (二)腕关节的功能解剖 (三)腕关节的生物力学 (一)腕关节的组成和运动方向 1.腕关节的组成 ●桡腕关节 ●腕骨间关节属于联合关节 ●腕掌关节 1.腕关节的组成(主要结构) (1)桡腕关节:桡骨腕关节面及尺骨头下方的关节盘组成关节窝与手舟骨、月骨及三角骨的近侧面组成的关节头构成,属于简单关节、椭圆关节。 (2)腕骨间关节:近侧的手舟骨、 ●月骨及三角骨和远侧的大多角骨、 ●小多角骨、头状骨、钩骨组成。 ●近侧腕骨间关节(平面关节) ●远侧腕骨间关节(平面关节) ●腕横关节或腕中关节(简单、球窝关节) (3)腕掌关节:由远侧列腕骨与5个掌骨底组成。 ●拇指腕掌关节:由大多角骨与第1掌骨底构成(鞍状关节) ●辅助结构:关节盘、腕桡侧副韧带、腕尺侧副韧带、桡腕掌侧韧带、桡腕背侧韧带。 ●关节特点:关节腔宽广,关节囊松弛,关节囊前、后、桡、尺侧都有韧带加固,腕掌侧韧带比桡腕背侧韧带坚 韧、限制桡腕关节后伸运动。 ●运动:屈伸、收展、环转(桡腕关节、腕横关节、拇指腕掌关节)。 (二)腕关节的功能解剖 1.运动腕关节的主要肌群 ●屈:桡侧腕屈肌、掌长肌、尺侧腕屈肌、指浅屈肌、指深屈肌 ●伸:桡侧腕长伸肌、桡侧腕短伸肌、指伸肌、示指伸肌、尺侧腕伸肌 ●外展:桡侧腕长伸肌、桡侧腕屈肌 ●内收:尺侧腕屈肌、尺侧腕伸肌 屈:桡侧腕屈肌(第2掌骨底)、掌长肌(掌腱膜)、尺侧腕屈肌(豌豆骨)、指浅屈肌(2-5指中节指骨两侧)、指深屈肌(第2至5指远节指骨底前面) 伸:桡侧腕长伸肌(第2掌骨底)、桡侧腕短伸肌(第3掌骨底)、指伸肌(2-5指中节远节指骨底背面)、示指伸肌(示指指背腱膜)、尺侧腕伸肌(第5掌骨底) 外展:桡侧腕长伸肌、桡侧腕屈肌(第2掌骨底) 内收:尺侧腕屈肌(豌豆骨)、尺侧腕伸肌(第5掌骨底) 2.运动拇指腕掌关节的主要肌群 ●屈:拇长屈肌、拇短屈肌 ●伸:拇长伸肌、拇短伸肌 ●外展:拇长展肌、拇短展肌 ●内收:拇收肌 ●对掌:拇对掌肌、小指对掌肌 (1)拇长屈肌 ●起点:前臂骨间膜 ●止点:拇指远节指骨底掌侧 ●作用:屈拇指腕掌、掌指和指骨间关节 (2)拇短屈肌 ●起点:屈肌支持带 ●止点:拇指近节指骨底
多囊肾的外科治疗
多囊肾的外科治疗 作者:董秉州,唐秀泉,谭斌,陈国强,李智 【关键词】多囊肾 成人型多囊肾是一种常染色体显性遗传性疾病,病程进展最终致高血压及肾功能严重损害。随着B超、CT等影像学检查的广泛应用,发现病例逐渐增多,目前该病尚缺乏有效的治疗方法,现将我院近3年收治的多囊肾病人行囊肿去顶减压术12例报告如下。 1 资料与方法 1.1 一般资料本组12例中男5例,女7例,年龄40~63岁,病变均累及双侧肾脏,临床上均表现为程度不同的腰痛,伴肉眼血尿者4例,伴高血压者4例,腹部可触及包块者11例,合并多囊肝3例。肾功能检查尿素氮和肌酐在正常范围内10例,超过正常者2例,其中肌酐最高值196 mmol/L,均经B超、CT检查明确诊断。 1.2 手术方法在连续硬膜外麻醉下经腰背部第十二肋缘下切口,直视下锐性游离肾脏充分显露。以电刀或组织钳将囊肿顶部透明部分切除,深层显露的透明囊肿顶部予以同样方法处理,尽可能切除所见全部囊肿的顶部,尤其是小囊肿和深层囊肿的减压处理,术毕肾
周放置引流管48~72 h。
2 结果 多囊肾体积随囊肿生长而进行性增大,通常可达正常肾脏的2~6倍,囊肿去顶减压后,肾脏体积可明显缩小,尤其伴有巨大囊肿者。本组12例中有10例术后腰背部酸胀痛立即减轻或消失,血尿停止。随访2年仍有大多数病人术侧无腰背部酸胀痛,肾功能的持续观察发现手术对肾功能无明显损害,并且肾功能略有改善,有高血压病人,术后随访病人有改善,本组随访病人中,不但症状缓解或消失,而且生活质量也得到提高。 3 讨论 多囊肾是一种先天性肾脏囊性疾患,有遗传和家族倾向,成人型多囊肾是一种常染色体的显性遗传,常在40岁左右出现临床症状,约1/3病人合并多囊肝,约10%病人末期肾功能衰竭和9%的肾透析者是由于多囊肾引起,是肾功能衰竭的第三位原因[1]。本病的发病机制与常染色体发育异常有关,主要病变是肾小管和集合管的囊性病变致泌尿引流不畅或集合管闭锁而引起的尿潴留所致。本病为双肾病变,但双肾轻重程度并不一致,其病理特征为双肾布满大小不等的囊肿,囊肿呈进行性增大,囊肿增大必然加重对肾实质的压迫并产生一系列并发症,从而使肾脏损伤进一步加重,逐渐出现肾功能不全,
肾发育不全常见类型
肾发育不全常见类型 *导读:临床上常见的先天性肾发育不良包括多囊性、梗阻性肾发育不良以及与基因有关的肾发育异常。…… 肾发育不良是肾脏未能进行正常生长发育形成的先天性疾病,过去对其发病机理了解甚少,随着分子生物技术的发展和应用,从分子学机理来阐明肾脏的发生,从分子生物学水平对肾发育不良的发生有了较深入的认识。本文就近期对此问题的研究进展作一介绍。 1 肾发生与肾发育不良 正常哺乳类肾脏位于间介中胚层,中胚层分化形成前肾导管,经进一步诱导形成中肾导管至输尿管芽,在输尿管芽诱导下,胚体尾端两侧的生肾素分化为后肾胚基,肾脏的胚胎发育正是由输尿管芽和后肾胚基二部分完成的,前者逐步发育成肾盂、肾盏和集合管,后者发育成肾小管和肾小球,最后肾小管和集合管对接,构成正常的肾单位。如果输尿管芽和后肾胚基二部分不能按正常程度发育和实行对接即造成肾发育不良。肾发育不良可以是部分性的,也可以是完全性的。多数类型的肾发育不良伴有囊肿,提示发育不良的各种形式在形成中有共同机制。 临床上常见的先天性肾发育不良包括多囊性、梗阻性肾发育不良以及与基因有关的肾发育异常。病理组织学重要特征是出现原始肾小管和化生软骨。完全性单侧肾发育不良,可表现为无症状。
多数发育不良病例中,肾缺陷是双侧性的,提示基因突变在正常肾发育中起重要作用。单侧性疾病则可能是一种获得性损伤所致,该损伤破坏了基因的正常表达,进而影响了对肾成熟有重要意义的蛋白质的产生。 2 肾发育不良常见类型 2.1 先天多囊性肾发育不良 多囊性肾发育不良(multiple cystic hypoplastic)是一种常见 的完全性肾发育不良,多为单侧病变(14-20%为双侧性),患肾失去正常形态,被不规则的大小囊肿所代替,肾脏功能丧失并常伴有输尿管梗阻,是新生儿腹部包块最常见的原因之一。 多囊性发育不良肾外型呈肾形结构,多数病例伴有一个闭锁的输尿管。妊娠早期的多囊肾含有正常发育所必须的成份,包括未诱导的后肾胚基岛和分支的输尿管导管,在此阶段肾单位各段已均可鉴别出囊性改变[1]。生后多囊性发育不良肾的病理组织学变 异包括原始肾小管的囊性改变、膨大且结构破坏、具有明显管周围反应的间质、纤维肌环的形成、软骨成分为标志的组织转化等。 2.2 先天梗阻性肾发育不良 先天性尿路梗阻在解剖位置上常发生于输尿管和膀胱的连接处,先天性后尿道瓣膜是婴幼儿泌尿系统梗阻的重要原因。先天梗阻性肾的组织学特征与多囊性肾发育不良相似,包括肾单位各段如肾小球的囊性转化、间质膨大且结构破坏、髓质和直小血管显著发育不全、发生管周围纤维肌环、多种形式的肾小球和发育的肾
常染色体显性多囊肾
常染色体显性多囊肾 常染色体显性多囊肾(ADPKU)是人类最常见的单基因遗传病之一,人群中的发病率为1:400—1:1000,是发病率最高的遗传性肾病,居我国终末期肾衰竭病因的第4位。引起ADPKU的突变基因有PKD1、PKD2和PKD3。PKD1是最常见的致病基因。ADPKU具有显性延迟性发病的特点,平均发病年龄在40岁左右。此时大多已将致病基因遗传给后代。因此,有必要对ADPKU家系进行产前诊断。该病主要表现为双侧肾脏形成多个液性囊泡,囊肿进行性增大,造成肾脏结构和功能的损害,最终引起终末期肾病(end-stage renal disease, ESRD),除了肾脏病变外,还累及全身多个器官,如肝囊肿、高血压、颅内动脉瘤、心瓣膜畸形等。 多囊肾病治疗一直是世界性的难题,目前尚无特效药,只是一些对症治疗和透析治疗。女性较男性患者进入终末期肾病的比例为高[(1.2-1.3)/1]。 临床表现 1.肾脏表现 (1)肾囊肿:多囊肾肾功能异常表现包括高血压,肾痛,肾功能不全等,这些表现与肾囊肿的进展直接关系。 (2)肾功能异常:较早发生尿浓缩能力和氨排泄减少,在疾病的早期阶段,这些异常较轻,对患者的生活影响不大。 (3)高血压等 产前基因诊断 两个基因已知与多囊肾相关:PKD1,占大约85%的患者;PKD2,占大约15%的患者。 一.突变扫描 PKD1:序列分折/突变扫描:阳性率约88%。 PKD2:序列分折/突变扫描:阳性率约88%。 二.连锁分析 在人口较多的家系进行症状前检测可运用PKD1和PKD2微卫星标记连锁分析。三.缺失/重复分析 适用于PKD2基因内或全基因缺失临床检测,检测频率估计不到1%。 产前诊断技术 多重连接依赖的探针扩增(MLPA)检测已发展可以筛查PKD1和PKD2重排。开发的PKD1位点特异性探针可以分析鉴别PKD1假基因。 遗传咨询 多囊肾是一种常染色体显性遗传性疾病。对多囊肾家庭调查表明,只有4%一8%的家庭成员将终止多囊肾胎儿的妊娠。 一.先证者的父母 1.大多数患者的父亲患有多囊肾。 2.从头突变的发生率显著,约有10%的家庭发生。 3.对于从头突变先证者的父母需进行影像筛查并评估建议,特别是在PKD2的情况下。 二.先证者的家系成员 1.先证者的家系成员的患病风险取决于父母的遗传状况。 2.如父母患病,子女的风险是50%。
第1腕掌关节炎教学
桡侧腕屈肌腱动力性悬吊结合掌骨间韧带重建 治疗第一腕掌关节骨性关节炎 赵建勇阚世廉宿晓雷张远林张植生刘振利陈广先 河北医科大学沧州中西医结合临床医学院手外科 天津医科大学天津医院手外科 【摘要】目的评价大多角骨切除,桡侧腕屈肌腱动力性悬吊结合掌骨基底间韧带重建治疗第一腕掌关节骨性关节炎的疗效。方法采用Scheker技术,应用大多角骨切除,桡侧半桡侧腕屈肌腱重建第一二掌骨基底间韧带,并与剩余肌腱自身悬吊控制掌骨基底背侧半脱位,并形成肌腱填塞物内置大多角骨切除遗留空间控制掌骨下沉等手术步骤,治疗第一腕掌关节骨性关节炎6例,按手部功能评价指标包括握力(Grip strength),捏力(Key-pinch),第一腕掌关节直观模拟疼痛标尺法(Visual analogue scales V AS)及第一腕掌关节有效活动度评分(Kapandji Score)以及随访12月时前后位X光片第一掌骨基底—舟骨远关节面间距评价手术疗效。结果本组随访时间12~26个月,平均15个月。手术前后疼痛(Visual analogue scales,V AS)平均分值为7/1.6;握力平均值11/22kg; Key-pinch捏力1.8/3.4kg; Kapandji Score 6/8.7; 12月时前后位X光第一掌骨基底—舟骨远关节面间距平均值8.8mm。患者术后疼痛的缓解程度最为突出,握力及捏力均有不同程度改善。结论大多角骨切除,桡侧腕屈肌腱动力性悬吊结合掌骨基底间韧带重建最大程度的接近了该部位韧带解剖及生物力学方面的结构,该项技术可以有效治疗第一腕掌关节骨性关节炎。 【关键词】成形术腕掌关节骨性关节炎动力悬吊 Treatment of the first trapeziometacarpal osteoarthritis with dynamic suspension-sling arthroplasty and intermetacarpal ligament reconstruction ZHAO Jian-yon g KAN Shi-lian ﹡XIU Xiao-lei ZHANG Yuan-lin ZHANG Zhi-sheng LIU Zhen-li CHEN Guang-xian ﹡Department of Hand Surgery Tianjin Medical University affiliated Tianjin Hospital, Tianjin,300211, China; Department of Hand Surgery Hebei Medical University affiliated Cangzhou Hospital of TCM and WM, Cangzhou,061001, China Corresponding author: ZHAO Jian-yon g, Email:zhaojyvip@https://www.360docs.net/doc/5515512137.html, 【Abstract】Objective To investigate the treatment outcomes of the new dynamic suspention sling arthroplasty of the 1ST trapeziometacarpal joint stabilizes the base of the first metacarpal after the removal of the trapezium in patients who have suffered the 1st trapeziometacarpal joint osteoarthritis, Methods By adopting Scheker’s new techniques, 6 cases have received treatments of the removal of the trapezium , reconstruction of the intermetacarpal ligment with half of flexor carpi radial (FCR) and wrapping around its intact half to form an pinecone in order to prevent the dorsal subluxation and proximal migration of the thumb metacarpal. Evaluation indexs involve grip strength, key-pinch strength, visual analogue scales (V AS) and Kapandji Score for pre and
百度百科多囊肾
多囊肾(polycystic kidney)又名Potter(Ⅰ)综合征、Perlmann综合征、先天性肾囊肿瘤病、囊胞肾、双侧肾发育不全综合征、多囊肾、肾脏良性多房性囊瘤、多囊病。我国1941年朱宪彝首先报道,本征临床并不少见。多囊肾有两种类型,常染色体隐性遗传型(婴儿型)多囊肾,发病于婴儿期,临床较罕见;常染色体显性遗传型(成年型)多囊肾,常于青中年时期被发现,也可在任何年龄发病。 多囊肾是肾脏的皮质和髓质出现多个囊肿的一种遗传性肾脏疾病。其发病具有家族聚集性男女均可发病。按遗传方式分为二型:(1)常染色体显性遗传型,此型一般到成年才出现症状;(2)常染色体隐性遗传型,一般在婴儿即表现明显。常染色体显性遗传型多囊肾临床常见,约占终末期肾脏病的5%-10%。临床表现主要有肾脏肿大、血尿、蛋白尿、高血压,晚期可发生肾功能衰竭。 本病目前治疗主要是积极控制高血压等并发症和防治感染,保护肾功能,延缓尿毒症到来。出现尿毒症时可作透析治疗和肾移植。多囊肾是遗传性疾病。根据遗传学特点,分为常染色体显性遗传性多囊肾(ADPKD)和常染色体隐性遗传性多囊肾(AR PKD)两类。常染色体显性遗传性多囊肾常见。ADPKD 为常染色体显性遗传,其特点为具有家族聚集性,男女均可发病,两性受累机会相等,连续几代均可出现患者。常染色体显性遗传性多囊肾又称成人型多囊肾,是常见的多囊肾病。由于对本病的认识日益深入,预后明显改善。ARPKD是常染色体隐性遗传。父母几乎都无同样病史。常染色体隐性遗传性多囊肾又称婴儿型多囊肾,为多囊肾中少见类型。常于出生后不久死亡,只有极少数较轻类型,可存活至儿童时代甚至成人。ADPKD 常见于成年时出现症状。囊肿在出生时即已存在,随时间推移逐渐长大,抑或在成年时发生和发展尚未完全阐明。但大多数患者的病变可能在胎儿时期即已存在。绝大多数为双肾异常。两侧病变程度不一致。其特征是:全肾布满大小不等的囊肿,直径由刚能分辨至数厘米不等。乳头和锥体常难以辨认。肾盂肾盏明显变形。囊内有尿样液体,出血或感染时呈不同外观。囊肿呈进行性长大,可能与细胞增殖的相关过程、细胞分泌功能的改变以及囊肿周围组织受损有关。ARPKD囊肿上皮细胞经培养后显示了与ADPKD不相同的性质:ADPKD囊液中有内毒素或Gram阴性细菌,而ARPKD囊液中则无。 病因 90%ADPKD患者的异常基因位于16号染色体的短臂,称为ADPKD1基因,基因产物尚不清楚。该区域的许多编码基因已被阐明并被克隆,可望在不久的将来,ADPKD1可被确认 。另有10%不到患者的异常基因位于4号染色体的短臂,称为ADPKD2基因,其编码产物也不清楚。两组在起病、高血压出现以及进入肾功能衰竭期的年龄有所不同。本症确切病因尚不清楚。尽管大多在成人以后才出现症状,但在胎儿期即开始形成。囊肿起源于肾小管,其液体性质随起源部位不同而不同,起源于近端小管,囊肿液内成分如Na+、K+、
正常人体运动学腕关节运动学
正常人体运动学腕关节 运动学 SANY标准化小组 #QS8QHH-HHGX8Q8-GNHHJ8-HHMHGN#
三、腕关节运动学 (一)腕关节的组成和运动方向 (二)腕关节的功能解剖 (三)腕关节的生物力学??? (一)腕关节的组成和运动方向 1.腕关节的组成 桡腕关节 腕骨间关节属于联合关节 腕掌关节 1.腕关节的组成(主要结构) (1)桡腕关节:桡骨腕关节面及尺骨头下方的关节盘组成关节窝与手舟骨、月骨及三角骨的近侧面组成的关节头构成,属于简单关节、椭圆关节。 (2)腕骨间关节:近侧的手舟骨、 月骨及三角骨和远侧的大多角骨、 小多角骨、头状骨、钩骨组成。 近侧腕骨间关节(平面关节) 远侧腕骨间关节(平面关节) 腕横关节或腕中关节(简单、球窝关节) (3)腕掌关节:由远侧列腕骨与5个掌骨底组成。 拇指腕掌关节:由大多角骨与第1掌骨底构成(鞍状关节) 辅助结构:关节盘、腕桡侧副韧带、腕尺侧副韧带、桡腕掌侧韧带、桡腕背侧韧带。 关节特点:关节腔宽广,关节囊松弛,关节囊前、后、桡、尺侧都有韧带加固,腕掌侧韧带比桡腕背侧韧带坚韧、限制桡腕关节后伸运动。 运动:屈伸、收展、环转(桡腕关节、腕横关节、拇指腕掌关节)。 (二)腕关节的功能解剖 1.运动腕关节的主要肌群 屈:桡侧腕屈肌、掌长肌、尺侧腕屈肌、指浅屈肌、指深屈肌 伸:桡侧腕长伸肌、桡侧腕短伸肌、指伸肌、示指伸肌、尺侧腕伸肌 外展:桡侧腕长伸肌、桡侧腕屈肌 内收:尺侧腕屈肌、尺侧腕伸肌 屈:桡侧腕屈肌(第2掌骨底)、掌长肌(掌腱膜)、尺侧腕屈肌(豌豆骨)、指浅屈肌(2-5指中节指骨两侧)、指深屈肌(第2至5指远节指骨底前面) 伸:桡侧腕长伸肌(第2掌骨底)、桡侧腕短伸肌(第3掌骨底)、指伸肌(2-5指中节远节指骨底背面)、示指伸肌(示指指背腱膜)、尺侧腕伸肌(第5掌骨底) 外展:桡侧腕长伸肌、桡侧腕屈肌(第2掌骨底) 内收:尺侧腕屈肌(豌豆骨)、尺侧腕伸肌(第5掌骨底) 2.运动拇指腕掌关节的主要肌群 屈:拇长屈肌、拇短屈肌 伸:拇长伸肌、拇短伸肌 外展:拇长展肌、拇短展肌 内收:拇收肌 对掌:拇对掌肌、小指对掌肌 (1)拇长屈肌 起点:前臂骨间膜 止点:拇指远节指骨底掌侧
多囊肝多囊肾是怎么回事
多囊肝多囊肾是怎么回事 *导读:多囊肝多囊肾是怎么回事?生活中还有部分人对多囊肝多囊肾的认识不是十分清楚,常常容易忽视,甚至是误诊,耽误治疗,导致病情进一步发展恶化。那多囊肝多囊肾是怎么回事呢?下文对这个问题进行了解答,一起来了解。…… 多囊肝多囊肾是怎么回事?生活中还有部分人对多囊肝多 囊肾的认识不是十分清楚,常常容易忽视,甚至是误诊,耽误治疗,导致病情进一步发展恶化。那多囊肝多囊肾是怎么回事呢?下文对这个问题进行了解答,一起来了解。 多囊肝多囊肾是怎么回事?很多患者在检察过程中同时发 现有多囊肾和多囊肝,或相继检察发现多囊肾多囊肝。这和遗传因素有关,也时还可伴有其它脏器的囊肿,如多囊卵巢、多囊脾等实质性器官。多囊肝是由于胆管上皮和间质的过渡增生致肝脏纤维组织出现多发性囊肿,包括肝内和肝外胆管扩张,肝内胆管局限性护张,先天性肝纤维化,雌激素对肝囊肿的产生有一定作用。 多囊肝多囊肾是怎么回事?多囊肾常同时伴有多囊肝,多囊肝是指肝内存在3个以上的囊肿,并沿肝管分布存在。女性较男性多见,发病率自3岁时的20%左右到70岁的75%。有家族遗传倾向,在同种异体肾移植成功的患者中,存活10年以上者均合并有肝囊肿。PLD是由于胆管上皮和间质的过渡增生致肝脏纤维
组织出现多发性囊肿,包括肝内和肝外胆管扩张,肝内胆管局限性护张,先天性肝纤维化,雌激素对肝囊肿的产生有一定作用。 多囊肝的囊肿大小,小至需经显微镜才能发现,大到可占据整个腹腔,肝脏大小由正常至巨大型。囊中所致的肝肿大引起未受侵害的肝实质被压迫而萎缩,肝实质可以保存较好,肝功能可在正常范围。腹部B超和CT扫描是诊断囊内出血的可靠方法,MRI则是对鉴别出血性肝囊肿和非出血性肝囊肿及其他肝脏疾病最可靠的方法。 多囊肝多囊肾是怎么回事?通过上述讲解,是否对你深入认识疾病有所帮助呢。同时需要注意的是,多囊肝多囊肾的危害是相当大的,治疗越早越好,避免病情恶化,给患者带来更大的伤害。若你还有哪些疑问需要解答,不妨向39专家在线咨询。 更多精彩内容请用微信扫描下方二维码,关注39疾病百科官方微信获取,关注有惊喜哦!
常染色体隐性遗传性多囊肾(ARPKD)
常染色体隐性遗传性多囊肾(ARPKD) 多囊肾是遗传性疾病。根据遗传学特点,分为常染色体显性遗传性多囊肾(ADPKD)和常染色体隐性遗传性多囊肾(ARPKD)两类。常染色体显性遗传性多囊肾常见。ADPKD为常染色体显性遗传,其特点为具有家族聚集性,男女均可发病,两性受累机会相等,连续几代均可出现患者。常染色体显性遗传性多囊肾又称成人型多囊肾,是常见的多囊肾病。由于对本病的认识日益深入,预后明显改善。ARPKD是常染色体隐性遗传。父母几乎都无同样病史。常染色体隐性遗传性多囊肾又称婴儿型多囊肾,为多囊肾中少见类型。常于出生后不久死亡,只有极少数较轻类型,可存活至儿童时代甚至成人。 囊肿上皮细胞经培养后显示了与ADPKD不相同的性质:ADPKD囊液中有内毒素或Gram阴性细菌,而ARPKD囊液中则无。 正确的遗传学诊断运用重组DNA技术已发现约85%的APD-KD家族中,被称为PKD1的基因突变定位于16号染色体短臂(P)上,它具有两个特异性标志:α球蛋白复合体及磷酸甘油酸激酶的基因.大多数其余的家族发现在4号染色体(PKD2)上有基因缺陷,但也有少数家族不与如何基因座相关. 多囊肾分为常染色体显性遗传性多囊肾(ADPKD)和常染色体隐性遗传性多囊肾(ARPKD)。多囊肾的病人,ARPKD罕见,一般出世不久后死亡只有极少数较轻类型,可存活至儿童或成人,故这里指的手术者多为ADPKD。ADPKD多表现为肾实质弥漫性进行性囊肿形成,可同时伴有肝囊肿、肺囊肿、胰腺囊肿、肺囊肿,最终会发展为尿毒症,需作透析治疗和肾移植,抽吸减压后,囊肿仍会增多,故对多囊肾并没太大价值。 单纯肾囊肿发展慢,不一定损害肾脏,且发现时多数患者年龄已较大,近年来在治疗方面趋于保守。一般以定期复查,门诊随诊为主。只有囊肿大于4cm时,才采取穿刺加硬化剂治疗。另外长期血液透析的患者中有部分获得性肾囊肿以单个或多个肾囊肿为表现。单一肾囊肿的唯一危害就是可能发展成恶性囊肿,如肾囊肿短时间内长大明显,抽吸不仅为了减压,也是为了排除肿瘤可能。 多囊肾的定义是什么 多囊肾系累及双侧肾脏的先天性疾病。其肾内布满大小不等的囊肿,有的相互间可沟通,使肾脏体积增大并压迫肾实质,使其萎缩造成功能损害,直至慢性肾功能衰竭。 多囊肾分为两型:婴儿型为染色体隐性遗传,常伴其它先天畸形,多于数月内死亡;成人型为染色体显性遗传,多中年发病,常伴有肝、脾、胰、卵巢、骨等器官的多囊性病变及颅内动脉瘤。 临床上多数病人可有家族史、男多于女,表现各异。常见的表现有: (1)腰腹部疼痛:为多数病人的首发症状,呈持续性或阵发性不等,劳累后加重。 (2)血尿:常为首发症状,约半数人出现,为间歇无痛性肉眼血尿。 (3)腹部肿物:多在双侧上腹部摸到,肿块大小不一。 (4)可以有高血压伴头晕、头痛。 (5)肾功能不全:肾功能检查明显异常,尿比重低而固定。 (6)约1/4病人有肾绞痛表现及尿频、尿急等不适。 X线平片、B超、核磁共振等检查有助于诊断。 治疗原则上以非手术疗法为主,外科手术治疗仅限于顽固性疼痛、肾动脉受压、肾盂输尿管梗阻及结石、积脓等并发症时的治疗。
2015年泌尿外科专业卫生高级职称考试模拟题1-(4)
泌尿外科分类模拟题1-(4) 一、单项选择题 151、经影像学检查未发现睾丸,最可有的诊断是 A.睾丸缺如 B.高位隐睾 C.睾丸萎缩 D.设备误差 E.睾丸坏死 152、隐睾对人体构成的危险是 A.不育 B.睾丸萎缩 C.睾丸恶变 D.睾丸炎 E.睾丸鞘膜积液 153、单侧隐睾对患者的生育能力有影响,其主要原因为 A.患侧睾丸发育不良 B.先天性不育 C.对侧睾丸发育不良 D.患侧睾丸产生“抗睾丸因子”影响对侧睾丸 E.双侧睾丸发育不良 154、包皮嵌顿三天,阴茎头青紫、包皮明显水肿、瘀血,紧急处理 A.用药物控制感染 B.试行包皮复位 C.包皮嵌顿环切开 D.包皮水肿部位穿刺抽水 E.包皮环切术 155、输尿管口囊肿常伴发什么疾病 A.肾发育不良 B.马蹄肾 C.重复肾 D.孤独肾 E.多囊肾 156、导致先天性肾盂输尿管连接部(UPJ)梗阻的常见原因是 A.结石 B.管腔内狭窄 C.腹膜后纤维化 D.迷走血管压迫
E.结核 157、异位输尿管口囊肿常来源于重复肾的 A.上位肾 B.下位肾 C.发育不良肾 D.对侧肾 E.以上都不是 158、电镜下UPJ梗阻病变部位平滑肌细胞间的改变是 A.胶原纤维减少 B.胶原纤维断裂 C.胶原纤维增多 D.胶原纤维缺乏 E.以上都不是 159、诊断小儿肾积水最常用的检查方法是 A.KUB B.彩色多普勒 C.B超 D.MRI E.IVP 160、切除积水肾的参考指标,大部分肾皮质厚度是 A.<3mm B.<5mm C.<2mm D.>3mm E.>2mm 161、UPJ梗阻治疗的目的是 A.切除病肾 B.解除梗阻,改善肾功能 C.肾盂整形 D.恢复肾脏形态 E.恢复肾功能 162、治疗UPJ梗阻的首选术式是 A.Y-V成形术 B.Anderson-Hynes肾盂成形术 C.输尿管镜下狭窄段内切开 D.输尿管狭窄球囊扩张术 E.双丁管留置术
多囊肾的遗传性——一谈控制遗传的重要性
多囊肾是一种常染色体显性遗传病,父母一方患病,子女的遗传机率都是二分之一。 和所有的遗传病一样,多囊肾也是由基因突变引起。导致多囊肾的基因突变在人群中的原发突变率在1/3000-1/5000左右,也就是说不考虑遗传因素,每3000到5000人中才会有一个多囊肾患者。但现在我国的多囊肾患者有200万以上,大致每600人中就有一个多囊肾患者。为什么有这么多的多囊肾患者呢?就是因为我们没有有效的控制遗传,多囊肾患者的基因突变位点不断遗传给下一代,导致多囊肾家族聚集。有的多囊肾大家族横跨4-5代人,整个40人左右的群体有20多个多囊肾患者,多囊肾的致病基因突变在整个家族里不断蔓延开来,成为挥之不去的痛苦。 关于多囊肾的遗传性,由于相关知识的匮乏,我们很多的多囊肾患者存在不少的认识误区。常见的有以下几种: 1.多囊肾的遗传概率很低,我见过一多囊肾患者,人家两个孩子都是健康的。 2.多囊肾是100%遗传的,我见过好几个多囊肾患者,他们的孩子都有多囊肾。 3.我多生几个孩子,总会有一个是健康的。 4.不用担心遗传问题,等我孩子多囊肾的时候,说不定医学进步多囊肾能根治了呢。 我们简单分析一下上面几种看法的误区在哪: 第一种,片面的看待多囊肾的遗传性,只看到一个多囊肾患者的两个孩子是健康的,没看到也许另外一个多囊肾患者的两个孩子都有多囊肾的情况,这两者的概率其实都不低,均为1/4。我们不能单凭自己知道的个例去判断多囊肾的遗传概率,以偏概全。 第二种,同样单凭自己的经验对多囊肾遗传下结论,很不科学。 第三种,这种想法非常可怕,一是对后代不负责任,二是很容易产生上述的多囊肾大家系,成为整个家族的痛苦之源。 第四种,这种想法看似很乐观,但没有认清遗传病的复杂性。包括多囊肾在内的所有遗
儿童多囊肾的两大分类
儿童多囊肾的两大分类 *导读:儿童多囊肾属于遗传性肾病,患有儿童多囊肾的患儿,在过去往往没有有效的治疗方法,所以患儿常活不到成年。…… 当前,儿童多囊肾属于遗传性肾病,患有儿童多囊肾的患儿,在过去往往没有有效的治疗方法,所以患儿常活不到成年;并且很多怀孕的女性在发现胎儿是婴儿多囊肾后都无奈的选择了终止妊娠;又由于多囊肾具有遗传性,所以怀孕女性在发现自己患有多囊肾以后,也有人停止妊娠。 通常情况下,根据年龄上的不同,儿童多囊肾可分为婴儿多囊肾和小儿多囊肾两大类。 1、婴儿型多囊肾 婴儿型多囊肾即常染色体隐性遗传型多囊肾。婴儿型多囊肾在超声下的表现大致有3类: 1)主要见于婴儿 此期婴儿多囊肾患儿超声波的特征为锥形强回声,图像类似髓质的肾钙质沉着; 2)主要见于绝大多数的新生儿和婴儿 该型的患儿具有典型的肾肥大和球形强回声影像,并偶尔位于皮质外部的2——3毫米处。 3)主要见于较大的小孩
此期婴儿多囊肾患儿被拍图像显示中央部有强回声,而皮质边缘的8——14毫米区域则无改变。其肝脏大小可正常或肥大,而回声则比肾脏弱。扩张的肝内胆管和由于纤维化导致的门脉末梢低,回声则很明显。随着年龄的增长,门脉纤维化不断发展,在较大的小孩,腹部超声可发现肝脾肿大和肝脏斑状强回声,门脉高压者可发现门脉倒流,肝内胆管扩张。 超声图像上婴儿型多囊肾在怀孕24周内即产生一定的征象,以 下声像图改变提示婴儿型多囊肾可能:1.双肾明显肿大;2.胎儿 膀胱缺如;3.羊水过少。 2、小儿型多囊肾 婴儿和小儿只是年龄上的界定,小儿型多囊肾和婴儿型多囊肾都是由于肾远曲小管和集合管连接错位,形成长形囊肿,儿童多囊肾患儿的年龄越小病情就会越重,预后也就越不好。婴儿型多囊肾多在出生后已有所表现,多数婴儿型多囊肾患儿在生后几个月便死于尿毒症;而小儿型多囊肾的发病率较婴儿型低,其存活年 龄也相对较长,但是小儿型多囊肾患儿也会在成年之前死亡。
多囊性肾发育不良
多囊性肾发育不良 多囊性肾发育不良种无功能的肾脏是由不相通的小囊以及相间于其中的纤维组织,初级小管,软骨灶组成,通常合并输尿管闭锁.其发生肿瘤,感染,高血压的可能性较低,有人建议手术切除患肾为好,也有人提出可观察随访. 基本内容 这种无功能的肾脏是由不相通的小囊以及相间于其中的纤维组织,初级小管,软骨灶组成,通常合并输尿管闭锁.其发生肿瘤,感染,高血压的可能性较低,有人建议手术切除患肾为好,也有人提出可观察随访. 疾病简介 肾发育不良是肾发生异常的临床后果,其典型病理组织学特征是出现原始肾小球和肾小管、软骨样化生等。 肾发育不良为肾脏未能进行正常生长发育的先天性疾患。大多呈散发性,少数有家族性倾向。病变程度取决于是单侧还是双侧肾累及以及影响哪一侧肾脏生长分化阶段。本病是新生儿最常见的腹部肿块原因之一,病变常呈单侧,与肾脏集合系统节段性狭窄有关,双侧者常有生命危险。多数患者产前超声即可确诊,临床表现为无症状性腹部肿块。患肾失去正常形态,被不规则的大小囊肿所代替,患侧肾脏无功能,并常伴有输尿管梗阻,对侧肾脏可有代偿性肥大,10%还有对侧输尿梗阻
肾血管,肾小管,集合管及尿路引流系统的发育异常可导致肾实质畸变,肾功能异常。肾发育不良renal hypoplasia的诊断可能需作肾脏活检。 疾病病因 发病因素不清,可能与肾脏在生长发育的某阶段受到外界各种理化及毒物因素的影响所致。病理上的重要特征是发现原始肾小管,并被不同分化阶段的肾实质组织所包绕,小管上皮细胞呈立方形或柱状,有时呈纤毛状,处于非成熟状态。可发现原始的或胎儿型肾小球、肾小管,呈软骨样化生。囊肿起源于集合管,也可起源于肾小球,大小及形态变异很大,有时可缺如。肾脏大小取决于所累及的生长发育阶段。部分肾发育不良病变呈局灶性。 取决于累及程度。完全性肾发育不良,双侧肾脏累及常在新生儿期死亡。单侧累及可表现为无症状,仅在以后的岁月中偶然被查出。病肾常有肾脏异位表现,如肾脏位于盆腔等。对侧健肾易发生肾盂积水,肾结石及尿路感染。部分肾发育不良者可无症状,偶可有巨大输尿管、巨大囊肿的表现。该病常伴其它血管发育异常。 临床表现 取决于累及程度。完全性肾发育不良,双侧肾脏累及常在新生儿期死亡。单侧累及可表现为无症状,仅在以后的岁月中偶然被查出。病肾常有肾脏异位表现,如肾脏位于盆腔等。对侧健肾易发生肾盂积水,肾结石及
各关节活动度
一、脊柱 (一)颈椎ROM 1、颈前屈0 ° -- 45° 2、颈后伸0 ° -- 45° 体位:端坐或直立轴心:下颌角固定臂:肩上 3、颈侧屈0 ° -- 45° 体位:端坐或直立轴心:第七颈椎棘突固定臂:双肩上 4、颈旋转0 ° -- 45° 体位:仰卧轴心:头顶固定臂:平床面 (二)胸和腰椎ROM 1、脊柱前屈0°-80° 体位:直立位轴心:L5棘突固定臂:体侧中线 运动测量:角度、弯腰手指离地距离、弯腰时第七颈椎棘突之第一骶椎长度1.6CM 2、脊柱侧屈0°-40° 体位:直立位轴心:S1 固定臂:背中线 运动测量:角度、侧屈时指尖与膝关节的距离 3、脊柱后伸0°-30° 体位:直立位轴心:S1 固定臂:体侧中线 4、脊柱旋转0°-45° 体位:仰卧或直立位轴心:头顶固定臂:平床面 二、上肢ROM (一)肩部关节 解剖及运动学概要 肩部的骨骼包括肱骨、锁骨、肩胛骨、胸骨、胸壁,组成肩部六个关节:盂肱、肩锁、胸锁、喙锁、肩肱、肩胛胸壁关节。盂肱关节是全身活动范围最大的一个关节,分别可在三个面围绕三个运动轴进行屈伸、内收、外展、内旋、外旋运动。 肩部的运动主要是盂肱、胸锁、肩锁、肩胛胸壁四关节配合协调共同完成。前屈 60º、外展30º、由盂肱关节完成,以后的运动由肩胛胸壁关节参与,运动比例2:1。 1、肩关节屈0°-180° 体位:坐或仰卧位轴心:肩峰固定臂:体侧中线 2、肩关节后伸0°-60° 轴心:肩峰固定臂:体侧中线 3、肩关节外展0°-180° 体位:坐或俯卧位轴心:肩峰固定臂:后上臂中线 4、肩关节水平外展0°-40° 体位:坐位轴心:肩峰固定臂:肩峰至颈后 5、肩关节水平内收0°-130°
辨析:多囊肾与肾囊肿
---------------------------------------------------------------最新资料推荐------------------------------------------------------ 辨析:多囊肾与肾囊肿 辨析: 多囊肾与肾囊肿多囊肾和肾囊肿是囊肿性肾脏病中最常见的两种,此外还有肾髓质囊肿,获得性肾囊肿等。 多囊肾是肾脏皮质和髓质出现的无数囊肿的一种遗传性肾病,常见的是常染色体显性遗传型(又叫成人型多囊肾),常见发病年龄为 40 岁以后,但从我们临床观察,由逐步前提之势,常表现为肾肿大,两侧受累程度可有明显差别,皮髓质满布大小不等的囊肿,直径 0、 1-数厘米。 约 20~50%的人伴多囊肝, 10~20%伴脑动脉病, 10%伴脾囊肿,由此可见,多囊肝也是多囊肾的一个并发症;而肾囊肿又叫单纯性肾囊肿,主要见于成年人, 50 岁以上更多见,常表现为一侧或两侧肾脏有 1 个或少数几个囊肿,直径一般 0.5 厘米,成孤立球形,多在皮质,并能改变肾外形。 单纯性肾囊肿与多囊肾有什么不同从发病机理上讲,与多囊肾不同,单纯性肾囊肿不是由先天遗传而是后天形成的。 过去曾认为它是由局部缺血造成,近年来的研究认为可能是由肾小管憩室发展而来。 随年龄增长,远端小管和集合管憩室增加,单纯性肾囊肿的发生率亦随之增加。 单纯性肾囊肿自然变化缓慢,有人曾用 B 超进行数年追踪 1 / 18
观察,发现其中只有少部分发生变化,主要是数目的增加,其次是大小的轻度增加,少数轻度缩小。 单纯性肾囊肿一般不会出现症状,常因其他目的作尿路 X 线、腹部 B 超或 CT 检查时无意被发现。 可以出现血尿和局部疼痛,也可出现肾盏梗阻和继发感染,但不会导致肾功能衰竭。 一、病因病机方面: 多囊肾是常染色体遗传病,多发肾囊肿可以是先天的(胚胎时期形成),可是创伤、炎性、肿瘤等引起。 二、家族史: 多囊肾家庭成员中有类似患者,多发肾囊肿患者家族中往往无类似患者。 三、囊内容物: 多囊肾内为尿液,多发肾囊肿内是体液(似血浆)其中含红细胞等。 四、并发症及危害: 多囊肾可出现血尿、高血压、浮肿、肾功能不全、尿毒症,多发肾囊肿一般不会出现高血压、肾功能不全等。 五、多囊肾可以控制囊体发展消除症状,多发肾囊肿控制或缩小囊体,较小者可消除。 1、尿的检查尿常规正常,若囊中压迫肾实质或合并有囊内感染,尿中可出现小量红细胞和白细胞。
肾囊肿+干预措施
肾囊肿+干预措施 肾囊肿是肾脏内出现的、大小不等的、与外界不相通的囊性肿块的总称。 临床上,肾囊肿可分为单纯性肾囊肿、获得性肾囊肿、成人型多囊肾三类。 1、单纯性肾囊肿 单纯性肾囊肿可能是一种先天性肾脏异常,由于多数出现在单侧,故称单纯性肾囊肿。单纯性肾囊肿的发病率可随年龄增长而增高,统计数据显示,50岁以上的人进行B超检查,可有50%存在这种囊肿类型。 2、获得性肾囊肿 获得性肾囊肿主要是在尿毒症或透析治疗后才发生的,所以与年龄无关,而与血液透析的时间有关。调查显示,透析时间超过3年的,大多数患者会出现获得性肾囊肿。获得性肾囊肿在一个肾内至少可以并存4个,直径多为2—3公分,有些还可以发生感染甚至癌变。 3、成人型多囊肾 成人型多囊肾是一种先天性、遗传性疾病。患有成人型多囊肾的患者,其肾脏实质内充满数不清的圆形囊肿,囊内含有液体,小的肉眼看不到,大的可有数厘米。患者临床上多有夜尿增多、腰痛、高血压等症状表现,尿检可见血尿、少量蛋白尿。成人型多囊肾可缓慢地发展为慢性肾衰,有10%的患者伴有肾结石,30%的患者伴有多囊肝。 肾囊肿的引发原因,不外乎以下几种因素。
1、先天发育不良或者基因突变 先天性发育不良,容易造成造成髓质海绵肾、发育不良性多囊肾病等。不过患者的基因一般无异常。胚胎形成时的基因突变,也可导致肾囊肿的出现。 2、感染 感染容易导致患者的内分泌出现紊乱,从而产生有利于囊肿的环境条件,使囊肿的内部因素活性增强,促进肾囊肿的生成、长大。且身体任何部位的任何感染,都会通过血液进入肾脏,从而影响肾囊肿。 3、情绪 情绪也容易引起人体的内分泌失调,从而通过神经体液作用改变人体的内环境,对肾囊肿造成影响。并且,不良的情绪还可使人的免疫力降低,利于病菌和病毒的侵袭,从而使囊肿受到影响。 4、饮食 过饥、过饱、过食肥甘厚味等饮食不节行为,饮食不洁行为,以及饮食偏嗜行为,都容易使患者体内的分泌出现紊乱,直接或间接地影响着肾囊肿的变化发展。 5、毒素 诸如农药、某些化学药剂、放射线、污染等毒素,都会对人体的各个器官产生伤害。尤其是一些具有肾毒性的药物,使用不当很容易造成肾损害而诱发肾囊肿。 6、劳逸失宜 经常劳累过度,会使自身的免疫能力下降,从而导致肾囊肿的发生。
多囊肾
多囊肾 多囊肾病(polycystic kidney disease,PKD)是一种常见的遗传性肾脏病,主要表现为双侧肾脏出现多个大小不一的囊肿,囊肿进行性增大,最终破坏肾脏结构和功能,导致终末期肾功能衰竭。 根据遗传方式不同,可分为常染色体显性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)和常染色隐性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD)。常染色体隐性遗传型多囊肾,发病于婴儿期,临床较罕见;常染色体显性遗传型多囊肾,常于青中年时期被发现,也可在任何年龄发病。 ADPKD是一种最常见的单基因遗传性肾病,发病率1/1000~1/4000,发病年龄多在30~50岁,故既往又称之为“成人型多囊肾病”,实际上该病可发生于任何年龄,甚至胎儿,故“成人型”这一术语并不准确,现已废用。ADPKD除累及肾脏外,还可伴有肝囊肿、胰腺囊肿、颅内动脉瘤、心脏瓣膜异常等,因此,它也是一种系统性疾病。目前已经明确引起多囊肾病的突变基因主要有PKD1HE PKD2两种。60岁以上患者将有50%将发展至终末期肾衰竭,占终末期肾衰竭病因的5~10%。 ARPKD是一种隐性遗传性肾病,一般在婴儿期即有明显表现,因此过去称为“婴儿型多囊肾病”,少部分发生于儿童或青少年。发病率约1/1万~1/4万,常伴有肝脏受累,表现为肝囊肿。目前已发现其发病与PKHD1基因有关。ARPKD患儿中,50%在出生后数小时至数天内死于呼吸衰竭或肾衰竭,存活至成人者主要特征是肾集合管纺锤形扩张,进展至肾衰竭,同时伴有肝内胆管扩张、先天性肝纤维化,临床表现为门脉高压症。由于ARPKD是一种少见病,多发生于儿童,故本文仅介绍ADPKD。 发病原因 本病为常染色体显性遗传,按其遗传规律,代代发病,男女患病几率均等。父母一方患病,子女发病几率50%。但约有40%的患者无家族一传十,可能为患者自身基因突变所致。 目前已知ADPKD突变基因有两个,按照发现前后分别命名为PKD1和PKD2。PKD1位于第16染色体短壁(16p13.3),基因长度52kb,有46个外显子,mRNA为14kb。PKD2位于第4染色体长臂(4q22~23),基因长度68kb,有15个外显子,mRNA约2.9kb。第3个基因(PKD3)可能存在,但尚未在染色体上定位和克隆。PKD1和PKD2的蛋白表达产物分别成为多囊蛋白1和多囊蛋白2。迄今报道的PKD1和PKD2基因突变形式分别为81中和41种,包括错义突变、无义突变、剪切错误、缺失、插入和重复等。
出生缺陷十种
发育的战争——健康PK 出生缺陷十种出生缺陷 基础医学院 2012 级影像二大班
常见十种出生缺陷 出生缺陷是指婴儿出生前发生的身体结构、 功能或代异常。通常包括先天畸形、 染色体 异常、遗传代性疾病、功能异常如盲、聋和智力障碍等。 为了更好地预防出生缺陷,我们需 要进一步了解它们。以下是常见的十种出生缺陷。 先天性心脏病 临床特征:先天性心脏病的症状轻重,主要决定于心脏畸形的位置和程度。 轻者无症状, 查体时发现,重者可有活动后呼吸困难、紫绀、晕厥等,年长 儿可有生长发育迟缓。症状有 根据血液动力学结合 病理生 理变化,可发为三类: 一、无分流类。左、右两侧 无分流, 无紫绀,如肺动脉 口狭窄,主动脉 狭窄,主动 脉缩窄,原发性肺动脉 扩, 原发性肺动脉高压或右位心 等。二、左至右分流类。在 左、右 心腔或主、肺动脉间 有异常通道, 左侧压力高于 隔缺损,动脉导管未闭,主肺动脉隔缺损;三、右至左分流类。右心腔或肺动脉压力异常增 高,血流通过异 常通道流入左心腔或主动脉。 图1-1法洛四联症 发病原因: 由于胎儿心脏在发育过程中受到干扰,使部分发育停顿或缺陷,以及部 此外还有心衰、紫绀、蹲踞、发育障碍等症状。 右侧,左侧动脉血通过异常通道进入右侧静脉血中 ---左向右分流,如心房间隔缺损,心室间 无与表现还与疾病类型和有无并发症有关。
分该退化者未能完全退化所致。 一、胎儿周围环境因素。妊娠早期子宫病毒感染,以风疹病毒感染后多见,常引起动脉 导管未闭及肺动脉口狭窄,其次为柯萨奇病毒感染(Coxsakie )可引起心膜弹力纤维增生症, 此外羊膜病变,胎儿周围机械压迫,母体营养障碍,维生素缺乏及代病,母体用细胞毒类药物或较长时间放射线照射,均可能与本病发生有关。 二、遗传因素。5%先心病患者发生于同一家族,其病种相同或近似,可能由于基因异常或染色体畸变所致。 图1-2室缺 三、其他。高原地区动脉导管未闭及房间隔缺损发病率较高,发生可能与缺氧有关。有 些先心病有性别倾向性。 预防与治疗原则:1?在怀孕早期(3个月之前)尽量别在电脑前、微波炉等磁场强的地方坐太长时间,因这时的胎儿还不稳定,各个器官还正在成形阶段,很可能造成孩子先天性心脏病。 2. 、早期检查诊断先心病后,尽量让孩子保持安静,避免过分哭闹,保证充足的睡眠。 大些的孩子生活要有规律,动静结合,既不能在外边到处乱跑(严格禁止跑跳和剧烈运动),也不必整天躺在床上,晚上睡眠一定要保证,以减轻心脏负担。 心功能不全的孩子往往出汗较多,需保持皮肤清洁,夏天勤洗澡,冬天用热毛巾擦身(注 意保暖),勤换衣裤。多喂水,以保证足够的水份。 保持大便能畅,若大便干燥、排便困难时,过分用力会增加腹压,加重 心脏的负担,甚至会产生严重后果。 居室保持空气流通,患儿尽量避免到人多拥挤的公共场所逗留,以减少 呼吸道感染的机会。应随天气冷暖及时增减衣服,密切注意预防感冒。 定期去医院心脏心科门诊随访,严格遵照医嘱服药,尤其是强心、利尿 药,由于其药理特性,必须绝对控制剂量,按时、按疗程服用,以确保疗 效。每次服用强心药前,须测量脉搏数,若心率过慢,应立即停服,以防药 物毒性作用发生,危及孩子生命。 手术一般取决于畸形的类型和严重程度,适合手术矫正者的手术时机及术前心功能状 况,有无合并症而定。无分流类或者左到右分流类,轻者无症状、心电图和X-线无异常者, 以及中,重度均可通过手术矫正,预后较佳,若已产生严重肺动脉高压双向分流则预后较差, 右至左分流或复合畸形者,病情较重者,应争取早日手术。轻者可选择手术时机,以10岁左右为佳。 、多指 多指症是指正常手指以外的手指赘生,或者足手指的指骨赘生,或足单纯软组织成分赘 生,或足掌骨赘生等。多指畸形是一种染色体的疾病这是手及上肢先天性畸形中最多见的