甲基化引物设计完整攻略

1.获取启动子序列
第一种方法:UCSC
UCSC主页(https://www.360docs.net/doc/5c17113892.html,/)
点击菜单栏中Tools →Gene Sorter
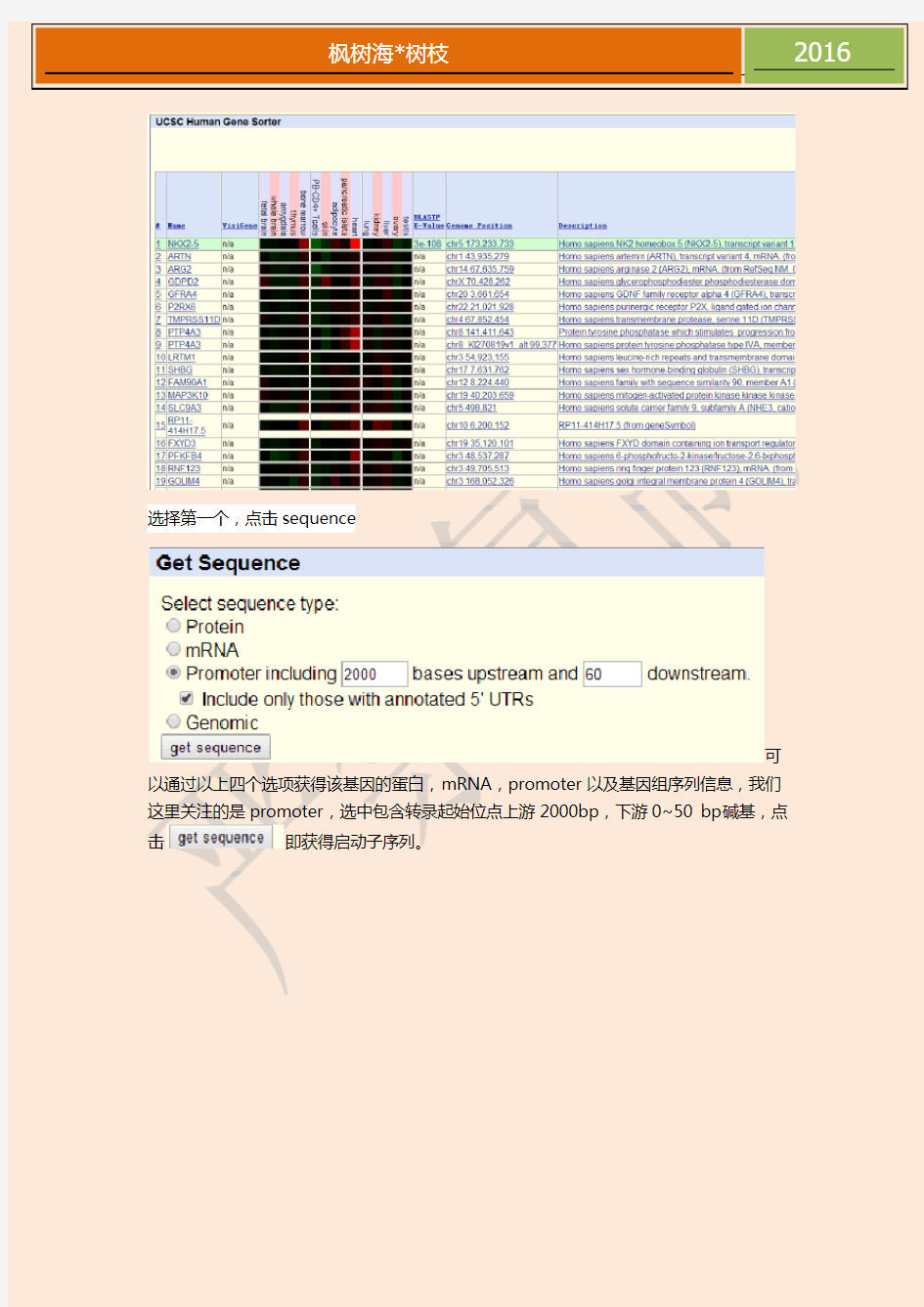
Genome 中选择物种选中Human,search框中键入基因名: NKX2-5,Go!出现以下页面,选择第一个,进入!
选择第一个,点击sequence
可以通过以上四个选项获得该基因的蛋白,mRNA,promoter以及基因组序列信息,我们这里关注的是promoter,选中包含转录起始位点上游2000bp,下游0~50 bp碱基,点击即获得启动子序列。
第二种方法 ensemble
Ensemble (https://www.360docs.net/doc/5c17113892.html,/index.html)
选择物种为human,键入基因名 NKX2-5 GO!
选取基因 NKX2-5(Human Gene)点击左侧的sequence
后页面显示如下:
序列橙色区域的为外显子,第一个外显子前的序列是启动子,默认为600bp,,设置一下。点击左侧configure this page设置需要的启动子长度,一般我们需要2000 bp
(同上:),点
获得页面橙色序列前面部分即为启动子序列。
2.设计甲基化引物
利用Methyl primer1.0软件,将启动子序列复制进去
点击 Design Primers,选择select Target Sequence
一般将序列全部选中,后点击Next
则根据需要,选择BSP/MSP,点击Next即获得所需要引物信息。
点击显示所有引物信息
另外,在线设计
网址:https://www.360docs.net/doc/5c17113892.html,/cgi-bin/methprimer/methprimer.cgi 直接输入启动子序列,选择BSP/MSP,点击SUBMIT即可。
引物设计基本方法
Primer 5.0搜索引物: 1.Primer Length我常设置在18-30bp,短了特异性不好,长了没有必要。当然有特殊要求的除外,如加个酶切位点什么的。 2.PCR Product size最好是100-500bp之间,小于100bp的PCR产物琼脂糖凝胶电泳出来,条带很模糊,不好看。至于上限倒也不必要求苛刻。 3.Search parameters还是选Manual吧,Search stringency应选High,GC含量一般是40-60%。其它参数默认就可以了。 4.搜索出来的引物,按Rating排序,逐个送Oligo软件里评估。当然,搜索出的引物,其扩增产物很短,你可以不选择它,或是引物3端≥2个A或T,或引物内部连续的G或C太多,或引物3端≥2个G或C,这样的引物应作为次选,没得选了就选它。对于这样的引物,如果其它各项指标还可以,我喜欢在引物末端去掉一个不满意的或加上一个碱基,看看引物的评估参数有没有变好点。 Oligo 6.0评估引物: 1.在analyze里,Duplex Formation不管是上游引物、下游引物还是上下游引物之间,The most stable 3’-Dimer绝对值应小于4.5kcal/mol, The most stable Dimer overall绝对值一般应小于多少kcal/mol跟PCR退火温度有关,我几次实验感觉在PCR退火温度在65°的时候,The most stable Dimer ove rall 6.7kcal/mol没有问题。 2.Hairpin Formation根据黄金法则 3.False priming sites: Primer的priming efficiency应该是错配地方的4倍左右,更多当然更好。 4.在PCR栏,个人感觉其所显示的optimal annealing temperature数值值得参考。在PCR摸索条件的时候,退火温度为其数值加减2的范围就可以了。 5.Internal stability很重要:我们希望引物的内部稳定性是中间高、两边低的弧形,最起码保证3端不要过于稳定。下图1引物3端过于稳定,很容易导致不适当扩增。△G参照黄金法则,这其实很好理解:把一滴水放到大海里,这滴水就会不停的扩散分布,扩散的越厉害越稳定,所以△G绝对值越大结构越稳定。 最后说一句,敢于尝试就会成功。 第二贴 --科室工作很多,小医生了,没有办法,所以肯怕不能满足很多战友的要求(qq聊或帮助设计),在此表示抱歉。就楼上的问题我试着回答一下,不一定正确,供参考吧。 --1、两个评价系统不一样,个人感觉oligo评价引物好点,primer出来的引物,我一般按效率排序,再结合退火温度和引物长度,选择引物到oligo测试。这是初步的选择,其实引物到了oligo里,退火温度也不一样。 --2、3端的二聚体应该避免,这个要看你的退火温度决定,一个50°的退火温度肯定和65°对二聚体的影响不一样了,一般来讲尽量控制在-4.5kcal/mol以下(个人观点,很多东西真得还是需要自己摸索)。 --3、个人感觉3端有A无A影响不大,3端有T的没有经验。有T是不是一定不行,个人感觉不见得。软件是评估,法则也不是没有例外,不是1+1=2那么确定。 --4、错配和二聚体谁轻谁重,个人觉得“到致命的程度”谁都重要,我也说不好。我设计的时候,尽量两个都不得罪。 --5、GC含量并非不重要,它直接影响引物各端稳定性,3端来两个G或C,稳定性就上去了,粘在模板上很牢。所以我设计的时候,尽量避免这样的情况出现。 谈一下我学这个引物设计的过程吧:
引物探针设计简介
引物探针设计简介 已有2993 次阅读2009-1-1 20:48|个人分类:课堂集锦|系统分类:科研笔记 1.寡聚核苷酸引物的选择,通常是整个扩增反应成功的关键。所选的引物序列将决定PCR 产物的大小、位置、以及扩增区域的Tm值这个和扩增物产量有关的重要物理参数。好的引物设计可以避免背景和非特异产物的产生,甚至在RNA-PCR中也能识别cDNA或基因组模板。引物设计也极大的影响扩增产量:若使用设计粗糙的引物,产物将很少甚至没有;而使用正确设计的引物得到的产物量可接近于反应指数期的产量理论值。当然,即使有了好的引物,依然需要进行反应条件的优化,比如调整Mg2+浓度,使用特殊的共溶剂如二甲基亚砜、甲酰胺和甘油。计算机辅助引物设计比人工设计或随机选取更有效。一些影响PCR反应中引物作用的因素诸如溶解温度、引物间可能的同源性等,易于在计算机软件中被编码和限定。计算机的高速度可完成对引物位置、长度以及适应用户特殊条件的其他有关引物的变换可能性的大量计算。通过对成千种组合的检测,调整各项参数,可提出适合用户特殊实验的引物。因此通过计算机软件选择的引物的总体“质量”(由用户在程序参数中设定)保证优于通过人工导出的引物。需要指出的是,引物不必与模板完全同源,因此可包含启动子序列、限制酶识别位点或5'端的各种修饰,这种对引物的修饰不会妨碍PCR反应,而会在以后使用扩增子时发挥作用。 2.基本PCR引物设计参数引物设计的目的是在两个目标间取得平衡:扩增特异性和扩增效率。特异性是指发生错误引发的频率。特异性不好或劣等的引物会产生额外无关和不想要的PCR扩增子,在EB染色的琼脂糖凝胶上可见到;引物效率是指在每一PCR循环中一对引物扩增的产物与理论上成倍增长量的接近程度。①引物长度;特异性一般通过引物长度和退火温度控制。如果PCR的退火温度设置在近于引物Tm值(引物/模板双链体的解链温度)几度的范围内,18到24个碱基的寡核苷酸链是有很好的序列特异性的。引物越长,扩增退火时被引发的模板越少。为优化PCR反应,使用确保溶解温度不低于54℃的最短的引物,可获得最好的效率和特异性。总的来说,最好在特异性允许的范围内寻求安全性。每增加一个核苷酸,引物特异性提高4倍;这样,大多数应用的最短引物长度为18个核苷酸。引物设计时使合成的寡核苷酸链(18~24聚物)适用于多种实验条件仍不失为明智之举。②引物的二级结构包括引物自身二聚体、发卡结构、引物间二聚体等。这些因素会影响引物和模板的结合从而影响引物效率。对于引物的3'末端形成的二聚体,应控制其ΔG大于
引物设计大全
引物设计和Primer-BLAST的应用 Lv Peng 2015.11.18
CONTENT 1.PCR-引物设计目的 2.引物设计原则 3.设计引物软件 4.在线设计工具 5.probeBase 简介
1.1PCR(Polymerase Chain Reaction) 聚合酶链式反应 1971 Khorana 提出设想 1985 Kary Mullis 发明了PCR 1986年5月 Mullis在冷 泉港实验室 做专题报告 冷泉港实验室(The Cold Spring Harbor Laboratory,缩写CSHL),又译为科尔德斯普林实验室。
几不同的PCR技术 1.扩增已知序列两侧DNA的PCR:反向PCR(Inverse PCR,IPCR)、锚定PCR(anchored PCR)、RACE(Rapid Amplification of cDNA Ends)、连接介导的PCR(ligation-mediated PCR,LM-PCR); 2.检测有限量稀有靶序列,即一对引物扩增产物不足以以通过凝胶电泳观察到的时:巢式PCR(nested PCR); 3.快速、灵敏、特异而准确定量的PCR:实时荧光定量PCR (real-time quantitative PCR,RQ-PCR)。
特性 优化 碱基组成 (G+C )含量应在40%-60%,4种碱基要分布均匀;长度 一般为18-27个核苷酸长度。上下游引物长度差别不能大于3bp ;重复和自身互补序列 不能有大于3bp 的反向重复序列或自身互补序列存在;上下游引物互补性一个引物的3’末端序列不能结合到另一个引物的任何位点上; 解链温度(Tm ) 两个引物的Tm 值相差不能大于5℃,扩增产物与引物的Tm 值相差不能大于10℃3’末端 引物3’末端碱基尽量为G 或C ,不能使3’末端有NNGC 或NNCG 序列引物序列不要有局部的GC rich 或AT rich (特别是3’端),避开T/C 或A/G 的连续结构 1.2引物设计原则 引物特性及优化设计
利用INTERNET设计PCR引物举例
利用IN T ERN ET设计PCR引物举例 周咏东 华西医科大学附属第一医院眼科(610041) 国际互联网上信息资源十分丰富,给人们的生活、学习和工作带来了极大的方便。过去,需要设计PCR引物时,研究人员需要查阅大量文献,有时因无法查到原文,或无相关报道,会使研究工作一开始就不能顺利开展。笔者在科研中发现,有许多科研人员未能掌握I NT ERN ET上有关引物设计的共享资源,故结合实际应用经验予以介绍。使你的科研工作如虎添翼。 IN T ER NET上设计引物,分为两个步骤: 1 检索待扩增基因的DN A序列 首先接入IN T ERN ET,然后键入网址:w w w.ncbi.nlm. nih.g ov便进入了美国国家医学图书馆的生物技术信息中心的主页。在“Sear ch”右边的检索框内选择“G enBank”,然后在“fo r”右边的框内键入你检索的基因序列名,如“human Bcl-2cDN A sequence”,点击“Go”,检索就开始了。 出现的下一网页是“Cur rent Q uery”即告诉你检索出相关文献的数目。如你对结果不满意,该网页下半部分有“A dd T er m(s)to Q uer y”和“M o dify Cur rent Q uer y”两栏供你重新检索;如你对检检索结果满意,即可点击“Retr iev e XX”(X X 为查出的文献数)。接着即显示了刚才调出的文献名。你可选择一篇最符合的,然后将“Display”键右边的框内选择为FA ST A r epor t”(这是下一步设计引物所规定的),点击“D is-play”健,你要的序列就显示出来了。 最后,将这段序列全选,在“编辑”栏中点击“复制”,将此窗口最小化,重开窗口,进入下一步骤。 2 引物的设计 在新开的窗口中,健入网址w ww.g eno me.w https://www.360docs.net/doc/5c17113892.html, 你即进入了“Whitehead Instit ut e for Bio medical R eser ch/ M IT Cent er for Genome Resear ch”的主页。在主页中先找到“G enome Center Softw ar e”标题,在其中的标题为“Ex peri-mental W eb-based Softw ar e”中,点击“W WW.P rimer P ick-ing(Pr imer3)”,此项,我们将用此网上软件设计引物。 网页上显示为“P r imer3o ld V ersio n”及“Click Her e T o T r y N ex t Ver sio n”,这两个版本大同小异,随便用哪一个。关键是在“Paste sour ce sequence belo w”文字下方的大空框内,粘贴上第一步查出的那段序列。然后根据自己的要求,对列出的各项引物设计指标作相应变动,否则为默认。确认指标设定完毕后,点击粘贴序列框下方的“Pick Pr imer s”键,你即得到了所需的引物,显示在“P rimer3O utput”网页上,共有5对,你可按需任选其一。 通过上述两个步骤,你如愿以偿,是不是很简便、快捷?快动手试一试吧!。 编辑 陈小娜 2.1.2非标准数字影像设备上网直接接入模块 该模块用于解决一部分带有数字网络接口,但又仅符合生产厂家内部标准的设备上网问题。以往,生产数字影像设备的众多厂家由于没有统一的上网标准,使医院相当一部分数字影像设备难以上网,因此,该模块须克服许多困难和问题,在实践中逐渐地、局部性地予以实现和完善。 2.1.3 非标准数字影像设备上网间接接入模块 此模块专用于对医院过去引进的,生产厂商根本就没提供上网接口的一部分早期非标准数字影像设备,通过对影像重新A/D的方法让它们间接上网。 2.2 数字影像会诊中心模块 建立医院数字影像局域网与PA CS系统,其核心意义在于能够方便地汇集各种各类检查的数字影像,提供给专家进行综合会诊。因此,在PA CS局域网基础上,建立硬件设施过硬、图像显示清晰、显示技术优良,能同时方便地显示各种数字诊断影像的数字化影像会诊中心,在很大程度上将代表整个项目的临床诊断价值与水平。2.3 数字影像局域网网络工程模块 网络工程模块包括:网络服务器、数据存储与管理软件系统模式、网络布局与工作站分布、网络构架与布线等等。此部分模块归属于计算机Intr anet信息网络工程范畴,对当前医院影像局域网In-tr anet的软硬件性能指标及设备系统的可扩展性具有决定性的作用。 2.4 各工程模块的信息流与局域网系统P ACS软件模块 如果说硬件是骨架,软件就是血液。建立了良好的硬件平台以后,必须要建立合理的网络信息流向和配以优良的PA CS 软件系统,才能保证整个系统能够正常运行。因此,这是建立数字影像会诊中心时,必须很好建立的又一个重要模块。 总之,医院建立数字影像局域网,并逐步过度到最终院际广域网连接已是二十一世纪医院计算机网络发展的必然趋势。我们必须紧跟这一时代发展潮流,扎扎实实从手上工作做起,理清各个系统模块关系,做好各方面技术储备,为医院建立数字影像局域网及P A CS系统作好准备。 编辑 杨立新 142?医学信息2001年3月第14卷第3期 Internet应用●
引物设计的11条黄金法则
引物设计的11条黄金法则
PCR引物设计的11条黄金法则 1.引物最好在模板cDNA的保守区内设计。DNA序列的保守区是通过物种间相似序列的比较确定的。在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该基因的保守区。 2.引物长度一般在15~30碱基之间。 引物长度(primerlength)常用的是18-27bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于TaqDNA聚合酶进行反应。 3.引物GC含量在40%~60%之间,Tm值最好接近72℃。 GC含量(composition)过高或过低都不利于引发反应。上下游引物的GC含量不能相差太大。另外,上下游引物的Tm值(meltingtemperature)是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度。有效启动温度,一般高于Tm值
5~10℃。若按公式Tm=4(G+C)+2(A+T)估计引物的Tm值,则有效引物的Tm为55~80℃,其Tm值最好接近72℃以使复性条件最佳。 4.引物3′端要避开密码子的第3位。 如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。 5.引物3′端不能选择A,最好选择T。 引物3′端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T 时,错配的引发效率大大降低,G、C错配的引发效率介于A、T之间,所以3′端最好选择T。 6.碱基要随机分布。 引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发(Falsepriming)。降低引物与模板相似性的一种方法是,引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。尤其3′端
引物设计原则(含Realtime引物)
1.引物最好在模板cDNA的保守区内设计。 DNA序列的保守区是通过物种间相似序列的比较确定的。在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该基因的保守区。 2.引物长度一般在15~30碱基之间。 引物长度(primer length)常用的是18-27 bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于Taq DNA 聚合酶进行反应。 3.引物GC含量在40%~60%之间,Tm值最好接近72℃。 GC含量(composition)过高或过低都不利于引发反应。上下游引物的GC含量不能相差太大。另外,上下游引物的Tm值(melting temperature)是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度。有效启动温度,一般高于Tm值5~10℃。若按公式Tm= 4(G+C)+2(A+T)估计引物的Tm值,则有效引物的Tm为55~80℃,其Tm 值最好接近72℃以使复性条件最佳。 4.引物3′端要避开密码子的第3位。 如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。 5.引物3′端不能选择A,最好选择T。 引物3′端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T时,错配的引发效率大大降低,G、C 错配的引发效率介于A、T之间,所以3′端最好选择T。 6. 碱基要随机分布。 引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发(False priming)。降低引物与模板相似性的一种方法是,引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。尤其3′端不应超过3个连续的G或C,因这样会使引物在GC富集序列区错误引发。 7. 引物自身及引物之间不应存在互补序列。 引物自身不应存在互补序列,否则引物自身会折叠成发夹结构(Hairpin)使引物本身复性。这种二级结构会因空间位阻而影响引物与模板的复性结合。引物自身不能有连续4个碱基的互补。 两引物之间也不应具有互补性,尤其应避免3′ 端的互补重叠以防止引物二聚体(Dimer与Cross dimer)的形成。引物之间不能有连续4个碱基的互补。 引物二聚体及发夹结构如果不可避免的话,应尽量使其△G值不要过高(应小于4.5kcal/mol)。否则易导致产生引物二聚体带,并且降低引物有效浓度而使PCR 反应不能正常进行。 8. 引物5′ 端和中间△G值应该相对较高,而3′ 端△G值较低。 △G值是指DNA 双链形成所需的自由能,它反映了双链结构内部碱基对的相对稳定性,△G 值越大,则双链越稳定。应当选用5′ 端和中间△G值相对较高,而3′ 端△G值较低(绝对值不超过9)的引物。引物3′ 端的△G 值过高,容易在错配位点形成双链结构并引发DNA 聚合反应。(不同位置的△G值可以用Oligo 6软件进行分析) 9.引物的5′端可以修饰,而3′端不可修饰。 引物的5′ 端决定着PCR产物的长度,它对扩增特异性影响不大。因此,可以被修饰而不影响扩增的特异性。引物5′ 端修饰包括:加酶切位点;标记生物素、荧光、地高辛、Eu3+等;引入蛋白质结合DNA序列;引入点突变、插入突变、缺失突变序列;引入启动子序列等。引物的延伸是从3′ 端开始的,不能进行任何修饰。3′ 端也不能有形成任何二级结构可能。 10. 扩增产物的单链不能形成二级结构。
甲基化引物探针设计方法
本文叙述了一种用于甲基化分析的探针法定量PCR的引物和探针设计方法,目前用于甲基化检测的引物探针设计工具非常多,都有使用成功的案例,经过初步多方尝试,本文中叙述的为本人认为较为靠谱的方法。Oligo7的优势在于专业,参数详尽且可自由设置,模块化设计,学会后使用便利。专业的活就是要专业的用专业的工具干。
首先是进行序列转换,有较多的在线工具和联机软件都可实现,这里使用https://www.360docs.net/doc/5c17113892.html,/methprimer/,较为简单直观。
直接将目标序列放入如上图的编辑框中,此也可直接用于相关引物的设计,不过本人没使用过,因为不能设计探针。submit后就有转化后的序列信息,如下图: 以上详细标记了CpG位置和非CpG位置的C,可直接复制到Word标注使用,下面就可以使用Oligo7利用上边的序列设计引物和探针了,如果是设计非甲基化引物探针,则使用原始序列。
关于引物和探针的一些主要参数,主要参考invtrogen的建议: Primer设计的基本原则: a)引物长度一般在18-35mer。 b)G-C含量控制在40-60%左右。 c)避免近3’端有酶切位点或发夹结构。 d)如果可能避免在3’端最后5个碱基有2个以上的G或C。 e)如果可能避免在3’端最后1个碱基为A。 f)避免连续相同碱基的出现,特别是要避免GGGG或更多G出现。 g)退火温度Tm控制在58-60C左右。 h)如果是设计点突变引物,突变点应尽可能在引物的中间。 T aqMan 探针设计的基本原则: a)T aqMan 探针位置尽可能靠近扩增引物(扩增产物50-150bp),但不能与引物重叠。 b)长度一般为18-40mer 。 c)G-C含量控制在40-80%左右。 d)避免连续相同碱基的出现,特别是要避免GGGG或更多G出现。 e)在引物的5’端避免使用G。 f)选用比较多的碱基C。 g)退火温度Tm控制在68-70℃左右。 另:目标变异碱基最好在3’末端或3’末端-1位置,保证扩增特异性,对于甲基化,则最好是C。
简并引物设计原则
The central role of UDPGDH played in capsule and other polysaccharides synthesis. KPS, capsule polysaccharide; LPS,lipopolysaccharide 简并引物设计方法 (1)利用NCBI搜索不同物种中同一目的基因的蛋白质或cDNA编码的氨基酸序列因为密码子的关系,不同的核苷酸序列可能表达的氨基酸序列是相同的,所以氨基酸序列才是真正保守的。首先利用NCBI的Entrez检索系统,查找到一条相关序列即可。随后利用这一序列使用BLASTP(通过蛋白查蛋白),在整个NR数据库中查找与之相似的氨基酸序列。 (2)对所有的序列进行多序列比对将搜索到的同一基因的不同氨基酸序列进行多序列比对,可选工具有Clustal W/X,也可在线分析。所有序列的共有部分将会显示出来。“*”表示保守,“:”表示次保守。 (3)确定合适的保守区域设计简并引物至少需要上下游各有一个保守区域,且两个保守区域相距50~400个氨基酸残基为宜,使得PCR产物在150~1200bp 之间,最重要的是每一个保守区域至少有6个氨基酸的保守区,因为每条引物至少18bp左右。 若比对结果保守性不是很强很可能找不到6个氨基酸序列的保守区,这时可以根据物种的亲缘关系,选择亲缘关系近的物种进行二次比对,若保守性仍达不到要求,则需进行三次比对,总之,究竟要选多少序列来比对,要根据前一次的结果反复调整。最终目的就是有两个6个氨基酸且两者间距离合适的保守区域。 (4)利用软件设计引物当得到保守区域后,就可以利用专业的软件来设计引物了,其中Primer 5.0 支持简并引物的设计,将参与多序列比对的序列中的任一条导入Primer 5.0 中,将其翻译成核苷酸序列,该序列群可用一条有简并性的核苷酸链来表示(其中R=A/G,Y=C/T,M=A/C,K=G/T,S=C/G,W=A/C/T,B=C/G/T,V=A/C/G,D=A/G/T,N=A/C/G/T,该具有简并性的核苷酸链必然包含上一步中找到的氨基酸保守区域的对应部分,在Primer 5.0 中修改参数,令其在两个距离合适的保守的nt区域内寻找引物对,总之要保证上下游引物都落在该简并链的保守区域内,结果会有数对,分数越高越好。 (5)对引物的修饰若得到的引物为: 5-NAGSGNGCDTTANCABK-3 则简并度=4×2×4×3×4×3×2=2304,很明显该条引物的简并度很高不利于PCR,可以通过次黄嘌呤代替N(因为次黄嘌呤可以很好的和4种碱基配对)和根据物种密码子偏好这两种方法来降低简并度。 这样设计出来的简并引物对,适用于比对的氨基酸序列所属物种及与这些物种分类地位相同的其他物种。 简并引物设计原则
引物设计1
1-2890(引物1) #1: Product of length 640 (rating: 171) Contains region of the molecule from 1 to 640 Tm: 72.1 C TaOpt: 48.8 C GC: 32.3 Sense Primer: CCTGGTTAATCCAAATCAC Similarity: 100.0% Length: 19 Tm: 44.1 C GC: 42.1 dH: -142.0 kcal/mol dS: -372.4 cal/mol dG: -29.2 kcal/mol Antisense Primer: GACAGGCCCTAATTAAGTT Similarity: 100.0% Length: 20 Tm: 45.0 C GC: 42.1 dH: -158.0 kcal/mol dS: -418.4 cal/mol dG: -31.5 kcal/mol Tm Difference: 0.9 GC Difference: 0 #1: Product of length 540 (rating: 171) Contains region of the molecule from 1 to 540 Tm: 72.2 C TaOpt: 49.4 C GC: 33.1 Sense Primer: CCTGGTTAATCCAAATCACT Similarity: 100.0% Length: 20 Tm: 45.8 C GC: 40.0 dH: -149.8 kcal/mol dS: -393.2 cal/mol dG: -30.8 kcal/mol Antisense Primer: ATAAGATTTGAGGTCAGCCA Similarity: 100.0% Length: 20 Tm: 46.4 C GC: 40.0 dH: -147.7 kcal/mol dS: -386.3 cal/mol dG: -30.7 kcal/mol Tm Difference: 0.6 GC Difference: 0.0 1-2890(引物2) #1: Product of length 603 (rating: 171) Contains region of the molecule from 514 to 1116 Tm: 73.9 C TaOpt: 50.3 C GC: 37.0 Sense Primer: TTGAAGATGGCTGACCT Similarity: 100.0% Length: 18 Tm: 42 C GC: 47.1 dH: -129.6 kcal/mol dS: -335.0 cal/mol dG: -27.9 kcal/mol Antisense Primer: GGAGGCCCTTTAACTTAA
引物设计步骤与要点
引物设计step by step 1、在NCBI上搜索到目的基因,找到该基因的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy该编码序列作为软件查询序列的候选对象。 2、用Primer Premier5搜索引物 ①打开Primer Premier5,点击File-New-DNA sequence, 出现输入序列窗口,Copy目的序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。点击Primer,进入引物窗口。 ②此窗口可以链接到“引物搜索”、“引物编辑”以及“搜索结果”选项,点击Search按钮,进入引物搜索框,选择“PCR primers”,“Pairs”,设定搜索区域和引物长度和产物长度。在Search Parameters里面,可以设定相应参数。一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200bp的产物电泳跑得较散,所以可以选择300~500bp. ③点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口”中,显示出该引物的综合情况,包括上游引物和下游引物的序列和位置,引物的各种信息等。 ④对于引物的序列,可以简单查看一下,避免出现下列情况:3’不要出现连续的3个碱基相连的情况,比如GGG或CCC,否则容易引起错配。此窗口中需要着重查看的包括:Tm 应该在55~70度之间,GC%应该在45%~55%间,上游引物和下游引物的Tm值最好不要相差太多,大概在2度以下较好。该窗口的最下面列出了两条引物的二级结构信息,包括,发卡,二聚体,引物间交叉二聚体和错误引发位置。若按钮显示为红色,表示存在该二级结构,点击该红色按钮,即可看到相应二级结构位置图示。最理想的引物,应该都不存在这些二级结构,即这几个按钮都显示为“None”为好。但有时很难找到各个条件都满足的引物,所以要求可以适当放宽,比如引物存在错配的话,可以就具体情况考察该错配的效率如何,是否会明显影响产物。对于引物具体详细的评价需要借助于Oligo来完成,Oligo自身虽然带有引物搜索功能,但其搜索出的引物质量感觉不如Primer5. ⑤在Primer5窗口中,若觉得某一对引物合适,可以在搜索结果窗口中,点击该引物,然后在菜单栏,选择File-Print-Current pair,使用PDF虚拟打印机,即可转换为Pdf文档,里面有该引物的详细信息。 3、用Oligo验证评估引物 ①在Oligo软件界面,File菜单下,选择Open,定位到目的cDNA序列(在primer中,该序列已经被保存为Seq文件),会跳出来两个窗口,分别为Internal Stability(Delta G)窗口和Tm窗口。在Tm窗口中,点击最左下角的按钮,会出来引物定位对话框,输入候选的上游引物序列位置(Primer5已经给出)即可,而引物长度可以通过点击Change-Current oligo length来改变。定位后,点击Tm窗口的Upper按钮,确定上游引物,同样方法定位下游引物位置,点击Lower按钮,确定下游引物。引物确定后,即可以充分利用Analyze 菜单中各种强大的引物分析功能了。
分子生物学--引物设计
PCR引物设计与分析 摘要:本文简单的介绍了PCR技术以及PCR引物设计原则和技巧,并以一段序列为例,介绍两种引物设计软件的使用方法。一般性引物自动搜索可采用“Premier Primer 5”软件,而引物的评价则可采用“Oligo 6”软件。 关键词:PCR;引物设计;软件; 1PCR 聚合酶链式反应(英文全称:Polymerase Chain Reaction),简称PCR。聚合酶链式反应(PCR)是体外酶促合成特异DNA片段的一种方法,由高温变性、低温退火(复性)及适温延伸等几步反应组成一个周期,循环进行,使目的DNA 得以迅速扩增,具有特异性强、灵敏度高、操作简便、省时等特点。它不仅可用于基因分离、克隆和核酸序列分析等基础研究,还可用于疾病的诊断或任何有DNA,RNA的地方。 PCR又称无细胞分子克隆或特异性DNA序列体外引物定向酶促扩增技术。 类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性--退火(复性)--延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加热至90~95℃一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55~60℃,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板--引物结合物在DNA聚合酶的作用下,于70~75℃,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链重复循环变性--退火--延伸三过程,就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。 参加PCR反应的物质主要有五种即引物、酶、dNTP、模板和Mg2+。引物是PCR特异性反应的关键,PCR 产物的特异性取决于引物与模板DNA互补的程度。理论上,只要知道任何一段模板DNA序列,就能按其设计互补的寡核苷酸链做引物,利用PCR就可将模板DNA在体外大量扩增。 2引物设计原则及注意
探针的设计原则
实时荧光Taqman 探针设计的几个要点 实验室很多同学都要做Real time PCR实验,实验室的师兄师姐都会有很多宝贵意见,不过也有实验室前没有做过的,查找了下资料和大家分享下关于实时荧光Taqman探针设计、实时荧光PCR探针的选择、 引物的设计及评价。 荧光探针法是用序列特异的荧光标记探针来检测产物,探针法的出现使得定量PCR技术的特异性比常规PCR技术大大提高。目前较常提及的有TaqMan探针、FRET杂交探针(荧光共振能量传递探针)和分子信 标Molecular Beacon。 广泛使用的TaqMan探针法是指PCR扩增时在加入一对引物的同时另外加入一个特异性的荧光探针,该探针只与模板特异性地结合,其结合位点在两条引物之间。探针的5′端标记有荧光报告基团(Reporter, R),如FAM、VIC等,3′端标记有荧光淬灭基团(Quencher, Q),如TAMRA等。当探针完整的时候,5′端报告基团经仪器光源激发的荧光正好被近距离的3′端荧光基团淬灭,仪器检测不到5′端报告基团所激发的荧光信号(就是说5’荧光基团的发射波长正好是3’ 荧光基团的吸收波长,因而能量被吸收传递到3’荧光基团而发出其它荧光)。随着PCR的进行,Taq酶在链延伸过程中遇到与模板结合的探针,其5′-3′外切酶活性(此活性是双链特异性的,游离的单链探针不受影响)就会将切割探针,释放5′端报告基团游离于反应体系中,远离3′端荧光淬灭基团的屏蔽,5′端报告基团受激发所发射的荧光信号就可以被探头检测到。也就是说每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。报告信号 的强度就代表了模板DNA的拷贝数。 (请注意,该图显示的不是普通的Taqman探针法,而是Taqman MGB探针法)Taqman探针检测的是积累荧光。常用的荧光基团有FAM,TET,VIC,HEX等等。当探针完整的时候,由于3′端的荧光淬灭基团在吸收5′端报告基团所发射的荧光能量,本身会发射波长不同的荧光而导致本底高,因此TaqMan探针近来又有新的发展——TaqMan MGB探针。MGB探针的淬灭基团采用非荧光淬灭基团(Non-Fluorescent Quencher),本身不产生荧光,可以大大降低本底信号的强度。同时探针上还连接有MGB (Minor Groove Binder)修饰基团,可以将探针的Tm值提高10°C左右。因此为了获得同样的Tm值,MGB探针可以比普通TaqMan探针设计得更短,既降低了合成成本,也使得探针设计的成功率大为提高——因为在模板的DNA碱基组成不理想的情况下,短的探针比长的更容易设计。实验证明,TaqMan MGB探针对于富含A/T 的模板可以区分得更为理想。 Taqman探针法已经得到广泛使用,不过有人认为这种技术利用了Taq酶5`—3`外切酶活性,一般试剂厂家只给Taq酶的聚合酶活性定标,没有同时给Taq酶5`—3`外切酶活性定标,不同批号试剂之间会给定量带来差异。另外对探针的熔点温度(Tm)仅要求其高于60°C,这就使不同试剂盒之间的特异性参差不齐,难 于做质控检测。 Real time PCR Taqman探针设计、实时多重PCR探针的选择和引物的设计及评价 一、实时荧光Taqman探针设计 总原则:探针选择要保守,引物选择要保守,因此必须找一段100-200bp相对要保守的片段来设计引物与探针。即real-time PCR的扩增片段是50bp----150bp。当找不到150bp的保守片段时,必须确保探针的 片段是保守的。
LAMP技术原理和引物设计
LAMP原理及引物设计与实例 .LAMP引物的设计 LAMP引物的设计主要是针对靶基因的六个不同的区域,基于靶基因3' 端的F3c、F2c和Flc区以及5' 端的Bl、B2和B3区等6个不同的位点设计4种引物。 FIP(Forward Inner Primer):上游内部引物,由F2区和F1C区域组成,F2区与靶基因3’端的F2c区域互补,F1C区与靶基因5' 端的Flc区域序列相同。 F3引物:上游外部引物(Forward Outer Primer),由F3区组成,并与靶基因的F3c区域互补。 BIP引物:下游内部引物(Backward Inner Primer ),由B1C和B2区域组成,B2区与靶基因3' 端的B2c区域互补,B1C域与靶基因5' 端的Blc区域序列相同. B3引物:下游外部引物(Backward Outer Primer ),由B3区域组成,和靶基因的B3c区域互补。 2.扩增原理 60-65℃是双链DNA复性及延伸的中间温度,DNA在65℃左右处于动态平衡状态。因此,DNA在此温度下合成是可能的。利用4种特异引物依靠一种高活性链置换DNA聚合酶。使得链置换DNA合成在不停地自我循环。扩增分两个阶段。 第1阶段为起始阶段,任何一个引物向双链DNA的互补部位进行碱基配对延伸时,另一条链就会解离,变成单链。上游内部引物FIP的F2序列首先与模板F2c结合(如图B所示),在链置换型DNA聚合酶的作用下向前延伸启动链置换合成。外部引物F3与模板F3c结合并延伸,置换出完整的FIP连接的互补单链(如图C所示)。FIP上的F1c与此单链上的Fl 为互补结构。自我碱基配对形成环状结构(如图C所示)。以此链为模板。下游引物BIP与B3先后启动类似于FIP和F3的合成,形成哑铃状结构的单链。迅速以3' 末端的Fl区段为起点。以自身为模板,进行DNA合成延伸形成茎环状结构。该结构是LAMP基因扩增循环的起始结构。 第2阶段是扩增循环阶段。以茎环状结构为模板,FIP与茎环的F2c区结合。开始链置换合成,解离出的单链核酸上也会形成环状结构。迅速以3’末端的B1区段为起点,以自身为模板。进行DNA合成延伸及链置换.形成长短不一的2条新茎环状结构的DNA,BIP引物上的B2与其杂交。启动新一轮扩增。且产物DNA长度增加一倍。在反应体系中添加2条环状引物LF和LB,它们也分别与茎环状结构结合启动链置换合成,周而复始。扩增的最后产物是具有不同个数茎环结构、不同长度DNA的混合物。且产物DNA为扩增靶序列的交替反向重复序列。 https://www.360docs.net/doc/5c17113892.html,MP的特点 LAMP与以往的核酸扩增方法相比具有如下优点: (1)操作简单,LAMP核酸扩增是在等温条件下进行,对于中小医院只需要水浴锅即可,产
引物设计的原理与方法
引物设计的原理与方法 This model paper was revised by the Standardization Office on December 10, 2020
PCR引物设计的原理及方法 阎振鑫S111666(四川大学生命科学学院细胞生物学成都 610014) 摘要:自20世纪后期发展了PCR技术以来,PCR已经改变了整个生物学研究的进程。而PCR反应的第一步就是设计引物,引物设计的好坏直接关系到PCR的成败。PCR引物设计有许多的原则必须要遵循:引物与引物之间避免形成稳定的二聚体或发夹结构,引物与模板的序列要紧密互补。引物不能在模板的非目的位点引发DNA聚合反应等。另外,引物的设计方法也越来越多,出现了许多专门的设计软件和网站,如:PrimerPremier5.0等。 关键词:PCR 引物原理方法 NCBI PrimerPremier5.0 PCR primer design principle and method YanZhenxin (sichuan Univercity, Life science college cell biology chengdu 610014 ) Abstract: When PCR technology was find, PCR has changed all of the program in research of biology. The design of primer is the frist step of PCR. It is relation to the fate of PCR. There are some principals must be obey: dipolymer and hairpin structure must be avoid between different primers. The DNA polymerization reaction should not be triggered at the wrong site. Therefore, there are more and more methods of design primer, include the professional softwares and professional web site. Key word: PCR primer principle NCBI PrimerPremier5.0 聚合酶链式反应(Polymerase chain reaction。PCR)是20世纪后期发展起来的 一种体外扩增特异DNA片断的技术。具有快速、简便及高度敏感等优点,能极大地缩短目的基因扩增时间[1]。因此,其一直是生物学者们致力于构建cDNA文库、基因克隆以及表达调控研究的必要前提和基础[2]。PCR的第一步就是引物设计。引物设计的好坏,直接影响了PCR的结果,因此这一步很关键。成功的PCR反应既要高效,又要特异性扩增产物,因此对引物设计提出了较高的要求。引物设计需要注意的地方很多,在大多数情况下,我们都是在知道已知模板序列时进行PCR扩增的。在某些情况比如构建文库的时候也会在不知道模板序列的情况下进行设计。这个时候随机核苷酸序列
PCR引物设计过程
PCR引物设计过程 (一)设计引物前应的准备工作: 1.准备载体图谱,大致准备把片断插在那个部分 2.对片断进行酶切分析,确定一下那些酶切位点不能用3.准备一本所买公司的酶的商品目录,便于查酶的各种数 据及两种酶是否可以配用 (二)引物的结构:5’—保护碱基+酶切位点+引物配对区—3’1.两个酶切位点 2.酶切位点的保护碱基 3.5’端保护碱基 4.3’端保护碱基 5.引物配对区 (三)设计引物所要考虑的问题 1.酶切位点 两个酶切位点应是载体上的,所连接片断上没有这两个位点,且距离不能太近,否则往往导致两个酶都切不好。因此,两个酶切位点要紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点,最好隔四个核苷酸。且不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。2.酶的选择
最好使用双酶切效率高的,但两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切,最好使用具有共同buffer,且较常用的酶(如hind3,bamh1,ecor1等),这样可以省钱。 3.Tm的计算。 Tm是由互补的DNA区域决定的,而不互补的区域对DNA 的溶解是没有作用的。因此,对于引物的Tm,只有和模板互补的区域对Tm才有贡献。计算Tm时,只计算互补的区域(除非你的酶切位点也与模板互补)。设计引物的时候,先不管5'端的修饰序列,把互补区的Tm控制在55度以上(我喜欢控制在58以上,具体根据PCR的具体情况,对于困难的PCR,需要适当提高Tm),再加上酶切位点和保护碱基,这样的引物通常都是可用的,即使有小的问题,也可以挽回。 Tm温度高的引物就比较容易克服3’发卡、二聚体及3'非特异结合等问题。简单的计算公式可以用2+4的公式。若你计算的Tm值达到了快90 ,不包括酶切位点。引物公司给你发的单子是包括酶切位点的。自己可以再估计一下。如你设计了带酶切位点的引物,总长分别为29、33个碱基,去掉酶切位点和保护碱基,分别为17、21个碱基。引物公司给的单子是70多度,实际用的只有50度,用55度扩的结果也差不多。