相图计算理论相关
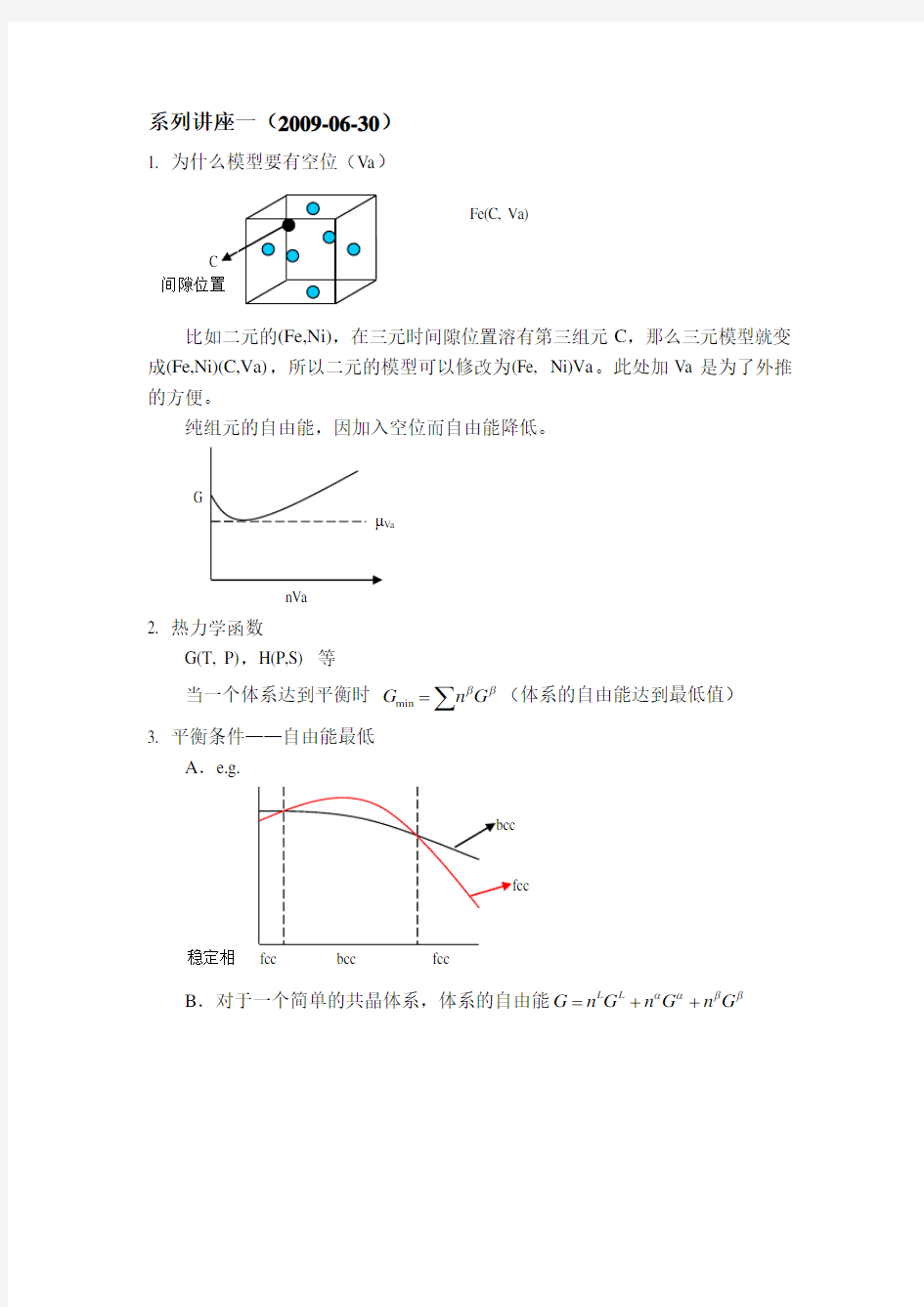

系列讲座一(2009-06-30)
1. 为什么模型要有空位(Va )
比如二元的(Fe,Ni),在三元时间隙位置溶有第三组元C ,那么三元模型就变成(Fe,Ni)(C,Va),所以二元的模型可以修改为(Fe, Ni)Va 。此处加Va 是为了外推的方便。
纯组元的自由能,因加入空位而自由能降低。
2. 热力学函数
G(T, P),H(P,S) 等
当一个体系达到平衡时 ∑=ββG n G min (体系的自由能达到最低值) 3. 平衡条件——自由能最低
A .e.g.
B .对于一个简单的共晶体系,体系的自由能ββααG n G n G n
G L L ++=
C .化学势的定义式 ααα
α
μB
n A A n G )(??=
系列讲座二(2009-07-02)
1. 相平衡时,混合自由能最低
证明一:化学势的定义式ααα
α
μB
n A A n G )(??=
∑=ααG n G
p
j k k j k k n j
k x k n j j
j j j n x x G n G n G n G n G n G n n n G )()()(,,????=????+=??+??=??≠≠∑∑∑ααα
αααααα n
n
n n x k j k k ==
∑ 其中,???
????
?
??-??=??≠≠j p p j
p p p n j
k
n j
k
n j k n n n n n n n n x ,,21)(
[]
k ij n n n
-=δ21
=
ij δ
k
j k
j ≠=01
G α
G β
[]
k ij x n
-=
δ1
∴∑∑∑===??-??+=??-??+=c
k k k
j c k k k ij c
k k j x G x x G G x G x x G G 11
1ααα
ααα
α
δμ 证明二:平衡时β
αμμA A =
ββααG f G f G +=
1010
021********=-+=-+=--=--ββα
αββ
ααβ
βααx x x x x f
x f x x f x f x 条件极值
令
)
1()1()()(212111221111-++-++--+--++=β
β
β
αααββααββααββααφφλλx x x x x f x f x x f x f x G f G f L
其中未知数有βαββααβαφφλλ,,,,,,,,,212121x x x x f f
)6(0)5(0)4(0)3(0)2(0)1(022
222211111122112211 =+-??=??=+-??=??=+-??=??=+-??=??=--=??=--=??β
βββααααααββββββα
αα
αβ
βββ
α
ααφλφλφλφλλλλλf x G f x L f x G f x L f x G f x L f x G f x L x x G f
L x x G f L
αα
αf x x ?-?+?)1()5()3(21: 02
211=-+??+??α
ααα
ααα
φG f x G f x x G f x 所以,??
????-??+??-=ααα
ααα
α
φG x G x x G x f 2211 (7)
根据(3)式又有:???
? ??-??-=12λφα
αααα
f x G f ,联立(3), (7)式有:
α
ααααααα
μλ2
2
22111=??+??-??-=x G x G x x G x G (8) 根据(5)式有:???
? ??-??-=22λφα
αααα
f x G f ,同理可得 则 12
22112λλαα
ααααα
=??+??-??-=x G x G x x G x G
用相同的方法,我们又可以得到:α
ααααααα
μλ11
22111=??+??-??-=x G x G x x G x G
所以,α
αμμ2
1= 系列讲座三(2009-07-03)
1. 吉布斯自由能模型
a. 单质元素
()()()
++++++++=-=--9
7132
,0,0ln hT
gT fT eT dT
T cT bT a T G
H T G T G i
SER i i i φ
φφ
b. 无序溶体模型
()()
n
x
x V V
L x x x x x L
x x G
G x x RT G
x G l
j i p p k k n
l j i l j i l
j i k k
k l
j i n
j i i i m
k k
j
i
k j i j i ex ex n i
i i n i
i
i ∑∑
∑∑
∑∑∑=≠≠==≠==-
+
=+
-=
++=,,)
(1,,,,)
(1,0)
,(,,,01ln φ
φφφ
c. 线性化合物模型
f n
i i i G G x G +=∑=1,0φ f G 为摩尔反应吉布斯自由能
d. 化合物能量模型
()()
()()()
∑∑∑∑-===++===v
v
j
q
i p v r q p r q p l
i m
p i p
i p
i id
s q p l s j q i p ref ex
id ref y y L L y
y f G G y y y G G G G G :,:,1
1:::ln
为组元
为点阵数s q p l ,,,
2. 零相分数线和相边界
a .什么是零相分数线(Zero Phase Fraction ZPF)
如图红色实线所示,在线上α相的相分数为0,就叫做α相的零相分数线。 由图也可以看出,相图中的相边界本质就是零相分区线,相图是由各个相的零相分区线构成的。
3. 三元相图中的两相杠杆定理
*x 处由杠杆定理ββααx f x f x +=*,取5.0=αf 的意思是α相始终占组成的
一半,在相图中这种情况的表示为:
三元相图中的两相杠杆定理
利用Pandat 软件可以将三元相图的两项杠杆线画出来:如图
x *
x
β
x
α
x B
T
β
α
γ
f α = 0.5
f α = 0
x B
T
β
α
γ
三元相图中,从绿线由杠杆定理可以判断相组成,也可以截取红线处的垂直截面图进行观察,如图:
x(Mg)
0.00.2
0.3
0.5
0.7
0.9
0.0
0.2
0.40.6
0.8
1.0
T [K ]
x(Al)
500
600
700
800
900
1000
1100
12000.0
0.1
0.2
0.3
0.4
0.5
0.6
T [K ]
x(Cu)
500
600
700
800
900
1000
1100
1200
0.0
0.1
0.2
0.30.4
0.5
0.6
0.7
系列讲座四(2009-07-07)
1. Chemical potentials Equilibrium Condition
φ
φφ
μμμT T T P P P j
j j ========= 212121 Chemical Potentials
()j
j
j j j j P RT P T ln ,+==*
μμμμ
Standard Reference Pressure —Every species has the same reference pressure.
0P —reference pressure, 1atm
(
)00,P P T j j j ==μμ So, Chemical Potentials: 0
0ln P P RT j j j +=μμ
Total and External Pressures
total P —total pressure of gas external P —external pressure
At equilibrium external
total P P =
Molar Fraction of Species
tot
j j P P y =
∑==s
j j t o t
P P
1
11
=∑=s
j j
y
Now the Chemical Potentials can be expressed as:
external
()()j
j
j j tot j j tot j j
j j j y RT pas P RT y RT atm P RT P P RT P P RT P P P P RT P P RT ln 101325
ln ln ln ln ln ln ln
tot 0tot
00tot 0
0tot 0
0++=++=++=?
??
?
??+=+=μμμμμμ
Example
atm P O 2.02= a t m
P N 7.02= atm P P P N O tot 9.022=+=
9.02.02=
O y 9
.07
.02=N y 2.0ln 9
.02
.0ln 9.0ln ln ln 000
2
22
2
2
RT RT RT y RT P RT O O O tot O O +=++=++=μμμμ 7.0ln 9
.07
.0ln 9.0ln ln ln 00
2
2
2
2
2
RT RT RT y RT P RT N N N
tot N N +=++=++=μμμμ
Inert Gas Species
Why do we need inert gas species? a .Difficulty to maintain low pressure
b .Species partial pressures may be determined by the condensed phases Example
atm P O 2.02= a t m
P N 7.02= atm P P P P Ar N O tot 0.122=++= 2.02=O y 7.02=N y 1.0=Ar y
2.0ln 2.0ln 0.1ln ln ln 00
2
22
2
2
RT RT RT y RT P RT O O O
tot O O +=++=++=μμμμ 7.0ln 7.0ln 0.1ln ln ln 00
2
2
2
2
2
RT RT RT y RT P RT O O O
tot O O +=++=++=μμμμ
(Unchanged the chemical potentials)
Gibbs Energy
Example: Al-O
AlO Al O tot P P P P ++=2
2
2
2
ln ln 0O tot O O y RT P RT ++=μμ
Al tot Al Al y RT P RT ln ln 0++=μμ
AlO tot AlO AlO y RT P RT ln ln 0++=μμ
AlO AlO Al Al O O gas y y y G μμμ++=22
When atm P P P P AlO
Al O tot 5102-=++=,
When atm P P P P AlO Al O tot 0.12=++=,
When atm P tot 0.1=, but keep atm P P P AlO Al O 5
102-=++
222
22
ln ln1 O O O O O
RT P RT μμμμ=+=+=
2
12
O
μ
222
2
5
ln ln10O O O O RT P RT μμμ
-=+=+
2
12O μ212O μ
2. Binary system
Element A fcc 1 0 0 ! Element B bcc 1 0 0 !
FUNCTION G_A_Liq 298.15 0; 6000 N ! FUNCTION G_B_Liq 298.15 0; 6000 N !
FUNCTION G_A_fcc 298.15 -10000+10*T; 6000 N ! FUNCTION G_B_fcc 298.15 -1100+10*T; 6000 N !
FUNCTION G_A_bcc 298.15 -5000+10*T; 6000 N ! FUNCTION G_B_bcc 298.15 -9000+10*T; 6000 N !
FUNCTION G_A_hcp 298.15 -5000+10*T; 6000 N ! FUNCTION G_B_hcp 298.15 -6000+10*T; 6000 N !
Phase liquid % 1 1 ! Constituent liquid :A,B:!
Parameter G(liquid,A;0) 298 G_A_Liq; 6000 N ! 2
12O μ 212
O μG i b b s E n e r g y (J /m o l )
x(A)
-583.3-1166.7-1750.0-2333.3-2916.7-3500.0-4083.3-4666.7-5250.0
0.0583.31166.71750.02333.32916.73500.04083.34666.75250.05833.36416.77000.00.0
0.10.20.30.40.50.60.70.80.9 1.0
T [K ]
x(B)
300
400
500600700800
9001000
11000.00.10.20.30.40.50.60.70.80.91.0
Parameter G(liquid,B;0) 298 G_B_Liq; 6000 N !
Phase fcc % 1 1 ! Constituent fcc :A,B:!
Parameter G(fcc,A;0) 298 G_A_fcc; 6000 N ! Parameter G(fcc,B;0) 298 G_B_fcc; 6000 N !
Phase bcc % 1 1 ! Constituent bcc :A,B:!
Parameter G(bcc,A;0) 298 G_A_bcc; 6000 N ! Parameter G(bcc,B;0) 298 G_B_bcc; 6000 N !
②当添加一个定化学计量比化合物A 3B 7时, At T=800K, G(A3B7) = -6100 J/mole atoms Data base 文件表述:
Phase A3B7 % 2 3 7 ! Constituent A3B7 :A:B:!
③当将化合物模型改成两个亚点阵时,(A,B)3(A,B)7
Phase A3B7 % 2 3 7 ! Constituent A3B7 :A,B:A,B:!
Parameter G(A3B7 ,A:A;0) 298 10*G_A_hcp; 6000 N !
Parameter G(A3B7 ,A:B;0) 298 3*G_A_hcp + 7*G_B_hcp -100000+20*T ; 6000 N ! Parameter G(A3B7 ,B:A;0) 298 3*G_B_hcp + 7*G_A_hcp + 0; 6000 N ! Parameter G(A3B7 ,B:B;0) 298 10*G_B_hcp; 6000 N !
G [J ]
x(B)-500-1000-1500-2000-2500-3000-3500-4000-4500-5000-5500-6000-6500-7000
05001000150020002500300035004000450050005500600065007000
0.0
0.1
0.2
0.3
0.40.50.60.70.80.9 1.0
T [K ]
x(B)
300
400
500600700800
900100011000.00.10.20.30.40.50.60.70.80.91.0
G(A3B7 ,A:A;0) = 3000 J/mole atoms y(A#1) = 0.917536 y(B#1) = 0.082464 G(A3B7 ,B:A;0) = 2700 J/mole atoms y(A#2) = 0.035342 y(B#2) = 0.964658
G(A3B7 ,A:B;0) = -6100 J/mole atoms G(A3B7 ,B:B;0) = 2000 J/mole atoms
④当用四个亚点阵模型时,(A)1(A,B)2(A,B)3(B)4
Phase A3B7 % 4 1 2 3 4 ! Constituent A3B7 :A:A,B:A,B:B:!
Parameter G(A3B7 ,A:A:A:B;0) 298 6*G_A_hcp + 4*G_B_hcp; 6000 N !
Parameter G(A3B7 ,A:A:B:B;0) 298 3*G_A_hcp + 7*G_B_hcp -100000+20*T ; 6000 N ! Parameter G(A3B7 ,A:B:A:B;0) 298 6*G_B_hcp + 4*G_A_hcp + 0; 6000 N ! Parameter G(A3B7 ,A:B:B:B;0) 298 1*G_A_hcp + 9*G_B_hcp; 6000 N !
y
x(B)
0.0
0.10.20.30.40.50.60.70.80.91.00.00.10.20.30.40.50.60.70.80.91.0
G [J ]
x(B)
-500-1000-1500-2000-2500-3000-3500-4000-4500-5000-5500-6000-6500-7000
05001000150020002500300035004000450050005500600065007000
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
G [J ]
x(B)
-500-1000-1500-2000-2500-3000-3500-4000-4500-5000-5500-6000-6500-7000
05001000150020002500300035004000450050005500600065000.0
0.1
0.2
0.3
0.40.50.60.70.80.9 1.0
T [K ]
x(B)
300
400
500600700800
90010000.00.10.20.30.40.50.60.70.80.91.0
G(A3B7 ,A:A:A:B;0) = 2600 J/mole atoms y(A#2) = 0.993508 y(B#2) = 0.006492 G(A3B7 ,A:B:A:B;0) = 2400 J/mole atoms y(A#3) = 0.004328 y(B#3) = 0.995672
G(A3B7 ,A:A:B:B;0) = -6100 J/mole atoms G(A3B7 ,A:B:B:B;0) = 2100 J/mole atoms
y
x(B)0.0
0.10.20.30.40.50.60.70.80.91.00.00.10.20.30.40.50.60.70.80.91.0
G [J ]
x(B)
-500-1000-1500-2000-2500-3000-3500-4000-4500-5000-5500-6000-6500-7000
05001000150020002500300035004000450050005500600065007000
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
G [J ]
x(B)-500-1000-1500-2000-2500-3000-3500-4000-4500-5000-5500-6000-6500-7000
05001000150020002500300035004000450050005500600065000.0
0.1
0.2
0.3
0.40.50.60.70.80.9 1.0
T [K ]
x(B)
300
400
500600700800
90010000.00.10.20.30.40.50.60.70.80.91.0
物理化学相图小知识
1.相律的有关概念与相律表达式 (1)独立组份数C=S-R-R′。S为物种数,R为独立化学反应计量式数目。R′ 为同一相中独立的浓度限制条件数(包括不同物种依反应计量式比例关系及离子物种电中性条件) (2)自由度数f,系指相平衡体系中相数保持不变时,所具有独立可变的强度变量数。 (3)相律内容及其数学表达式。相律就是揭示pVT平衡系统中自由度数、独立组份数和相数三者之间的制约关系。 表达式为:f=C-Φ+2;式中(式中 2 指T、p两强度变量) 当T、p中有任一固定,则表达式为:条件自由度数f*=C-Φ+1 当考虑除T、p、X B以外的其他变量或相间有某种限制时,则表达式为f=C-Φ+n;(式中n≥2)(4)相律的局限性与应用的关键性。相律是一个定性规律,它指明特定条件下该平衡系统至多存在的相数及其相应的独立变量数,但不能指明是哪些相共存?哪些性质可作为独立变量及其它们之间的定量关系?相律对单相与复相都适用,但应用相律时,首先要考察系统是否满足相律成立的条件,并确定系统的组份数。 2.单组份系统的相图与特征 (1)单组份系统相律与相图:因C=1 ,故相律表达式为f=3-Φ。显然f最小为零,Φ最多应为 3 ,因相数最少为 1 ,故自由度数最多为 2 。相图是用几何图形来描述多相平衡系统宏观状态与T、p、X B(组成)的关系。在单组份相图中有单相的面、两相平衡线和三相平衡的点,自由度分别为f=2、f=1、f=0。 (2)单组份相变的特征与类型。相变是一个连续的质的飞跃。相平衡时物质在各相中的化学势相等,相变时某些物理性质有突变。根据物性的不同变化有一级相变和连续相变(包括二级相变等高阶相变)之分;前者广为存在如气、液、固之间转变,其特点是物质在两相中的化学势一级导数不相等,且发生有限的突 变〔即〕,此 类相变平衡曲线斜率符合克拉贝龙方程。后者如氦He(Ⅰ)与He(Ⅱ)的转变。正常状态与超导状态的转变,其特点是化学势的一级导数在相变点连续〔即V1=V2,S1=S2〕,但化学势二级导数 在相变点附近则迅速变化,出现一个极大峰如; 或。二级相变平衡曲线斜率符 合爱伦菲斯(Ehrenfest)方程: 3.克拉贝龙—克劳修斯方程及其应用条件 (ⅰ)克拉贝龙方程:适用于单组份系统两相间平衡 (ⅱ)克拉贝龙—克劳修斯方程:适用与其中含气相的两相间平衡,且气相应服从理想气体状态方程。
二维相图和三维相图的计算
二维相图和三维相图的计算 描述二维相图和三维相图计算的算法。虽然零相分数的概念是用于计算二维相图,单相分数的概念被应用于计算三维相图。三维相图可以更好的观察等高线,例如在三维相边界的等温线。零相分数和单相分数的概念已经被推广到任何属性的等高线。 引言 材料是现代科技的基石。材料目前面临的挑战是设计新材料,改进现有的技术,以满足新技术的需要。为了提高材料的研究效率,概念集成计算材料工程(ICME)已被提出并应用在材料研究和工业应用中。在近十年在ICME领域有许多重大的成就。在ICME领域中的一个最重要的组成部分是相位特性变化的模拟,如热力学,动力学和力学性能的模拟。所有相得相关属性与相平衡密切相关,它可以图形化地呈现在相图中。 相图,通常被称为材料的设计图,在材料设计中起重要作用。在早期,大多数的相图通过实验测量并且局限于一元,二元和三元系统。计算相图可以追溯到Van Laar 和meijering 两人。他们计算了一些简单的二元和三元相图。1970年,计算机作为一个新的材料研究方法由考夫曼等人开创的相图计算标志着相图计算的开始。相图计算方法在ICME中已经成为一个重要的仿真方法。 相图被收集在一起便于查阅运用。随着互联网的效率越来越高,许多常用的在线相图是在网上可以找到。收集的相图和网上的资源大多数是二维(2D)静态图并且局限于低阶系统。然而,在实际应用中,多组分相图通常在手册或在线相图数据库中不可用。为了计算多组元相图的有用性,计算机软件和多组分热力学数据库是必要的。近年来,一些相图计算软件,如Pandat, Thermo-Calc, 和FACTSage已经被开发用来解决这个问题。
5.静力学:相图、物理平衡复习
静力学的基本方程大家都是熟知的,对于每个物体写合力为零+合力矩为零,但是这样的后果是经常造出一个n 元一次方程组(n 元不等式的处理办法我们在上一讲已经部分解决)。如何省去不必要的麻烦是这一讲的目标。正是“去掉所有不必要的东西”,“用同一个方程表述尽量多的情况”这样的想法诱发了拉格朗日等人建立了理论力学。如今理论力学已经几乎现代物理的标准描述方式。 静力学化简的基本原理在于,约束力是“要多大,有多大”。约束力和约束总是成对出现,增加一个几何条件,增加一个未知的力。如果能让一个约束力不出现在方程中,方程组就会从一个n 元一次 方程组变成1n -元一次方程组。要知道在n 很大的时候,解方程计算量大约是3 3 n ,从而导致你的运算 时间随着3n 增长,计算正确率随着3 3 (1)n q -下降,其中q 是你一次计算犯错的概率。所以要干事情就是不让约束力出现。 第一部分:矢量力学的基本方法 1、 在垂直于约束力方向写方程,也就是高中天天念叨的“在XX 方向的分力” 2、 通过合理选取支点,让一些力不出现。 注意 I 只有合外力等于0的体系才能导致体系不同点为支点计算力矩相同 II 同一个物体选择两个支点写方程,相当于一个力矩方程+一个不平行于支点连线方向的受力方程。 3、 通过以整体为对象,将内部相互作用消去(相当于用目测的办法把两个方程加了一下) 4、 即将滑动的时候将摩擦力和支持力合成一个确定方向,不定大小的力。然后配合矢量图或者三 力汇交解决问题。(相当于用目测的办法把f N μ=这个式子带入了) 5、 计算的主要复杂度来源于将方程相加时候,要把同类项都乘一遍,再加一遍,如果能每次都能“用一个方程消灭一个未知数”,那么你解的就是n 个一元一次方程,而不是n 元一次方程组, 正确概率会变成(1')n q -,时间变成n ,显然好了很多。以5为最后目标,以1-4为手段,绝大部分静力学暴力计算题都可以合理时间内解决。 附加说明两点 1 明白这些原理的老师命的题可以使得以上做法全部失效,参见学而思出的《第29届复赛模拟试题汇编》(六套),在https://www.360docs.net/doc/6d1886254.html, 下载。 2 如何在压根没时间解方程时候,只通过解方程混分,参见暑期最后一节课的骗分学导论。 本讲导学 第4讲 静力学化简 知识模块
二元相图计算
《二元相图计算》创新课程作业 学生:于永龙班级:焊接2班学号:10850212 一名词解释 1. 体系 体系就是我们研究的对象的总和。 2. 环境 系统以外又与系统密切相关的部分称为环境,环境必须是与系统有相互影响的有限部分。 3. 组元 组成合金的独立的、最基本的单元称为组元,组元可以是组成合金的元素或稳定的化合物。 4. 相 系统中物理性质和化学性质完全相同的均匀部分称为相。 5. 相律 表示平衡物系中的自由度数,相数及独立组分数之间的关系。数学表达式:?=C-Ф+2 6. 杠杆定律 在结晶过程中,液、固二相的成分分别沿液相线和固相线变化。 7. Gibbus自由能 G=H-TS, G叫做吉布斯自由能。 8. 化学势 等温等压下,在一定浓度的溶液中,加入微量组分B,而引起系统吉布斯函数对组分B物质的量的变化率。 9. 理想溶液 宏观定义:溶液中的任一组分在全部浓度范围内都符合拉乌尔定律的溶液称为理想溶液。 分子模型定义:各组分分子的大小及作用力彼此相似,当一种组分的分子被另一种组分的分子取代时,没有能量的变化或空间结构的变化,即就是当各组分混合成溶液时,没有热效应和体积的变化。 10. 拉乌尔定律 如果溶质是不挥发性的,即它的蒸气压极小,与溶剂相比可以忽略不计,则在一定的温度下,稀溶液的蒸气压等于纯溶剂的蒸气压与其克分子分数的乘积。 二读书报告 关于《相图分析及应用》的读书报告 相图在冶金,化工等工业生产部门及矿物、化学等科学研究领域有着广泛应用和重要指导意义,是解决一些实际问题不可缺少的工具。在生产及新产品开发过程中,人们经常要遇到相图基础知识和应用相图解决一些实际问题,而《相图
相图计算理论相关
系列讲座一(2009-06-30) 1. 为什么模型要有空位(Va ) 比如二元的(Fe,Ni),在三元时间隙位置溶有第三组元C ,那么三元模型就变成(Fe,Ni)(C,Va),所以二元的模型可以修改为(Fe, Ni)Va 。此处加Va 是为了外推的方便。 纯组元的自由能,因加入空位而自由能降低。 2. 热力学函数 G(T, P),H(P,S) 等 当一个体系达到平衡时 ∑=ββG n G min (体系的自由能达到最低值) 3. 平衡条件——自由能最低 A .e.g. B .对于一个简单的共晶体系,体系的自由能ββααG n G n G n G L L ++=
C .化学势的定义式 ααα α μB n A A n G )(??= 系列讲座二(2009-07-02) 1. 相平衡时,混合自由能最低 证明一:化学势的定义式ααα α μB n A A n G )(??= ∑=ααG n G p j k k j k k n j k x k n j j j j j n x x G n G n G n G n G n G n n n G )()()(,,????=????+=??+??=??≠≠∑∑∑ααα αααααα n n n n x k j k k == ∑ 其中,??? ???? ? ??-??=??≠≠j p p j p p p n j k n j k n j k n n n n n n n n x ,,21)( [] k ij n n n -=δ21 = ij δ k j k j ≠=01 G α G β
[] k ij x n -= δ1 ∴∑∑∑===??-??+=??-??+=c k k k j c k k k ij c k k j x G x x G G x G x x G G 11 1ααα ααα α δμ 证明二:平衡时β αμμA A = ββααG f G f G += 1010 021********=-+=-+=--=--ββα αββ ααβ βααx x x x x f x f x x f x f x 条件极值 令 ) 1()1()()(212111221111-++-++--+--++=β β β αααββααββααββααφφλλx x x x x f x f x x f x f x G f G f L 其中未知数有βαββααβαφφλλ,,,,,,,,,212121x x x x f f )6(0)5(0)4(0)3(0)2(0)1(022 222211111122112211 =+-??=??=+-??=??=+-??=??=+-??=??=--=??=--=??β βββααααααββββββα αα αβ βββ α ααφλφλφλφλλλλλf x G f x L f x G f x L f x G f x L f x G f x L x x G f L x x G f L αα αf x x ?-?+?)1()5()3(21: 02 211=-+??+??α ααα ααα φG f x G f x x G f x 所以,?? ????-??+??-=ααα ααα α φG x G x x G x f 2211 (7)
材料设计与热力学相图计算
哈尔滨工业大学材料热力学论文——相图计算及其在材料设计中的应用 指导老师:郑明毅 学生:孙永根 学号:11S109048
相图计算及其在材料设计中的应用 摘要 本文首先介绍了材料设计所遇到的困难以及CALPHAD技术的出现及应用。CALPHAD 技术综合利用计算热力学、动力学模拟及实验数据规范评估来优化材料的成分、相(含亚稳相)组成、组织结构及加工处理过程,进而改善材料性能,是二十世纪八十年代出现了计算材料学这一新学科的重要组成部分。 本文分别简要介绍了计算相图(CALPHAD技术)在ZA52-xY镁合金的合金设计及建立Mg-Ca-Ce三元体系热力学系统中的应用,凸显了CALPHAD技术在计算多元体系相图中的优势。 1 材料设计与热力学相图计算 1.1 材料设计的途径及CALPHAD技术 在以往的材料开发上,通常采用“试错法”来实现,即材料开发人员通过大量的实验和经验来选择材料的成分、稳定工艺参数。这样即消耗了大量的人力和物力,又不利于系统地探讨材料改性的机理。 材料科学研究面临的突出问题可以归结到两个方面:(1)由于研究对象的复杂性,现有理论模型无法突破局限性,对一些错综复杂问题的处理难以令人满意;(2)虽然新的实验技术、仪器和设备不断涌现,在一定范围内为实验研究提供了新的途径,但大都极为昂贵。材料制备中一个不容忽视的问题是:我们对具有一定组织和性能的多组元或多相材料的成分缺乏可预见性。相图常常作为确定材料制各工艺路线(包括成分配比、合成和处理)的唯一依据。但是,对于多元、多相新兴材料,绝大多数情况下只能找到其构成元素间的二元相图,而三元和三元以上的多元相图非常有限。因此,对多组元合金制备时成分的确定相当缺乏理论指导,而试验尝试的方法盲目性较大,又非常耗时耗力。 由上述可见,传统的材料研究方法存在不少局限性。对于新材料研制,单纯依靠理论研究和实验尝试都不能保证科学性和高效性。 随着近一个世纪合金理论的积累和几十年来计算机技术的迅速发展,20世纪60年代相计算(PHACOMP)技术在Ni基高温合金成分设计上的成功应用揭开了合金设计的序幕。虽然那仍是一种依赖于经验的相平衡成分计算,至少让材料学家体会到相平衡信息对于合金设计是多么的重要;70年代出现的CALPHAD技术已经是在追求利用普遍适应性的热力学模型获得多元体系中所有物相(包括亚稳相)的特征函数,再通过严格的热力学理论,得到多元体系的所有物相的热力学性质,使材料设计由经验设计向科学设计转变。 CALPHAD技术综合利用计算热力学、动力学模拟及实验数据规范评估来优化材料的成分、相(含亚稳相)组成、组织结构及加工处理过程,进而改善材料性能,是二十世纪八十年代出现了计算材料学这一新学科的重要组成部分。CALPHAD技术利用实验测定的相平衡信息和热化学数据,对相关研究体系进行严格的热力学优化,获得体系中包括亚稳相在内所有物相的热力学特征函数(通常为Gibbs自由焓),虽然它仍依赖于由实验获得低元体系的数据参数,但可以说,多元体系的所有热化学性质尤其是相转变驱动力、相转变所需克服的势垒及亚稳相关系的获得过程已经达到了真正意义上的理性阶段。人们对实验测定相关系在新材料研发特别是材料设计上的重要性是有足够认识的,但只有在通过CALPHAD技术来获得所有热化学性质之后,相图测定和相平衡研究才真正成为了材料设计的一部分。 目前,材料设计领域富有挑战性的课题就是如何在不同层次一材料的成分设计、显微结构、性能和制备工艺之间搭桥,从而达到从材料微观结构到宏观性能的预测和设计。
铁碳合金相图相关计算
铁碳合金相图相关图像算式问题整理 Gary 问答题: 图像总结:
工业纯铁 亚共析钢 共析钢 过共析钢 亚共晶白口铸铁 共晶白口铸铁 过共晶白口铸铁 占比计算: 1.工业纯铁(<=0.0218%C ) 2.亚共析钢(0.0218%~0.77%C ) 3 III Fe C 3III ++Fe C αγαγα γγααα→→→???→???→????→3III Fe C 100%6.69x W = ?3III Fe C 6.69-=1-100%6.69x W W α= ?
3.共析钢(0.77%C ) 4.过共析钢(0.77%~2.11%C ) 5.亚共晶白口铸铁(2.11%~4.3%C ) 6.共晶白口铸铁(4.3%C ) 7.过共晶白口铸铁(4.3%~6.69%C ) 3 III Fe C P 3III ++P +P+Fe C αγαγγγααα→→→???→???→????→P -0.0218 100% 0.77-0.0218x W = ?3III Fe C P -100%6.69x W W = ??(1)3III P Fe C =1--W W W α?()100% P 3P F+Fe C γγ→???→析() P 100% W =F 6.69-0.77 100% 6.69W = ?3Fe C 0.77 100%6.69W = ?析3 II Fe C P 3II 3II +Fe C P+Fe C γγγγ→→????→???→3Fe C 0.77 100% 6.690.77x W -= ?-II 3II P Fe C 6.69(1)100%= 100% 6.690.77x W W -=-??-d 3II L L Fe C L γ d L L++L γγγ→→→???→???→????→P '3II d 3II d F e C L P+Fe C +L γγ→++??? →'d d L L 2.11= 100%4.30 2.11x W W -=?-3Fe C 2.110.77 4.30100% 6.690.77 4.30 2.11x W --=??--II '3d P Fe C L 6.69-2.11 4.301--= 100% 6.69-0.77 4.30 2.11x W W W -=??-II d L L P 'd d 3L L L (P+Fe C ) γ→→???→???→共晶'd d L L =100% W W =3P Fe C 6.69-4.30 1-= 100% 6.69W W =?晶3I d L Fe C L L P '3I d 3I d 3I L L+Fe C L +Fe C L +Fe C γ→→→????→???→???→3Fe C 4.30 100% 6.69 4.30x W -= ?-I '3d Fe C L 6.691100% 6.69 4.30x W W -=-= ?-I
相图动力学计算步骤及方法CuNi
一:Cu-NI 互扩散计算步骤: 1. setup SYS: go data------进入数据库 THERMODYNAMIC DATABASE module running on PC/WINDOWS NT Current database: SGTE Alloy Solutions Database v4 V A /- DEFINED B2_BCC BCC_B2 L12_FCC L102_FCC REJECTED GAS:G REJECTED IONIC_LIQUID:Y OXIDE_LIQUID:Y REJECTED TDB_SSOL4:sw user data------转化至用户自定义数据库(打开Cu-Ni.TDB热力学数据库) TDB_USER: define-species------定义元素 SPECIES: CU NI CU NI DEFINED TDB_USER: rej ph *------屏蔽所有相 LIQUID:L FCC_A1 REJECTED TDB_USER: rest ph fcc------保留fcc相
FCC_A1 RESTORED TDB_USER: get------获取数据 TDB_USER: app user------添加用户数据库(打开CU-NI(自己定义).TDB动力学数据库) TDB_APP: def-sp SPECIES: CU NI CU NI DEFINED TDB_APP: rej ph * FCC_A1 REJECTED TDB_APP: res ph fcc FCC_A1 RESTORED TDB_APP: get TDB_APP: go par------进入PARROT模块 PARROT VERSION 5.3d RUNNING ON PC/WINDOWS NT PARROT: go d-m------进入dictra-monitor模块 NO TIME STEP DEFINED DIC>
相图的热力学基础
相图的热力学基础 合金相图尽管都是由实验测绘的,但其理论基础却是热力学。因此,了解一些相图热力学的基本原理,对正确测绘相图、正确理解和应用相图均有重要意义。现在,对于一些简单类型相图已能利用组元的热力学参数进行理论计算。理论算出的相图与实验测绘的基本符合。由于电子计算机的出现,促使理论计算相图有了显著进展。特别是对一些实验测绘有困难的领域,如超高温、高压和低温等方面的相图工作,理论计算更有其重要意义。 一、两相混合的自由能 在一定温度下,当某成分合金分解成两个混合相时,如果忽略它们的界面能,则在自由能一成分图上,此合金和两个混合相的自由能值必在一条直线上,如图3—72所示。设合金为x,其摩尔自由能为G(高度为bx),当它分解为x1和x2两相后,其摩尔数分别为n1和n2,靡尔自由能分别为G1(高度为ax1)和G2(高)。此时合金的成分x和摩尔自由能G可分别用下式表示: 度为cx 2 式(3-22)表明,ab线和bc线的斜率相等,所以a、b和c三点在一直线上,即是说,两个相混合后的自由能值(b)就在此两相的自由能值的连线上,而b点的位置可由两个相的摩尔数(n1和n2)按杠杆定律决定,即
二、溶体的自由能一成分曲线 溶体是指两种以上组元组成的均匀单相溶体,如溶液和固溶体。已知吉布斯自由能G(简称自由能)的一般表示式为 式中H为焓(热函),S为熵,T为绝对温度。 1、焓:在温度T时,溶体的焓是由构成它的原子之间的结合能及其热能之和组成的,即 式中Ho为OK时原子间的结合能,Cp为等压热容。 T CpdT/T和混合熵△Sm。 2、熵:也是由两项组成,即升高温度时的温熵∫ 根据热力学第三定律,在温度OK时,如果是纯组元或化合物,其结构处于理想完整状态,两项熵值皆为零。如果是由两种以上原子组成的溶体,由于两种原子存在不同的排列方式,使得混合熵不为零。故在温度T时,溶体的熵值S为 3、溶体自由能的表达式 将式(3-24、25)代入式(3-23)中,即得在温度T时溶体自由能的表达式: 溶体的Cp值难于理论计算,只能用实验测出。 下面介绍Hm和△Sm值的近似求法。(此处省略,详见本文最后(一)或书本p100-104)