土壤砷锑铋硒碲的测定乙醇增强氢化物发生ICPMS法
FHZDZTR0139 土壤砷锑铋硒碲的测定乙醇增强氢化物发生ICPMS法
F-HZ-DZ-TR-0139
土壤—砷锑铋硒碲的测定—乙醇增强氢化物发生ICP-MS法
1 范围
本方法适用于地质样品土壤及生物和植物样品中砷,锑,铋,硒,碲的测定。方法的检出限为(3σpg/mL):As 71,Sb 10,Bi 9,Se 6,Te 8。
2 原理
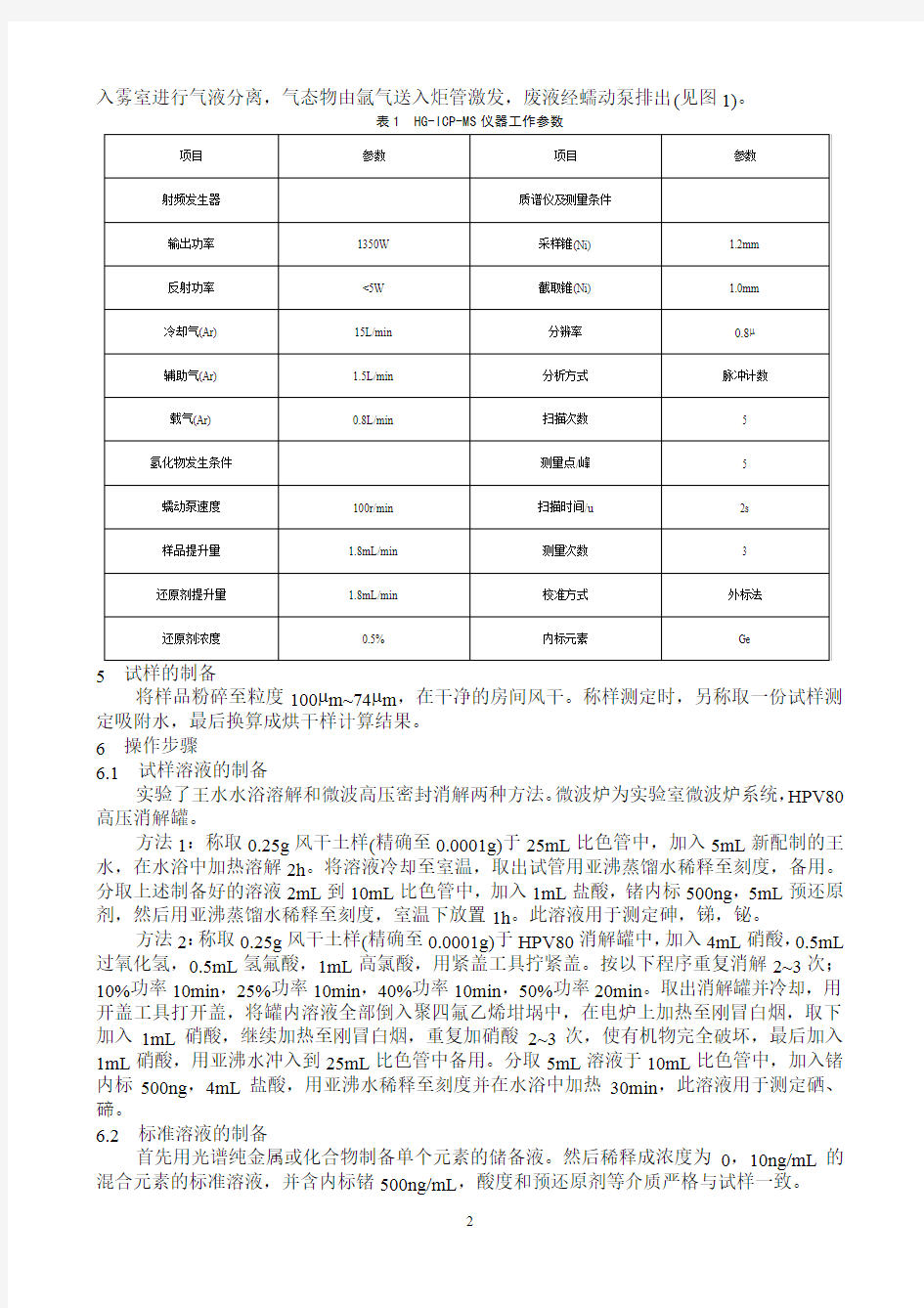
用电感耦合等离子体(ICP)作为离子源,借助电感耦合等离子体-质谱(ICP-MS)仪器分辨率高,干扰少,检出限低的特点,采用乙醇增强氢化物发生系统与ICP-MS仪器联用(见图1),实现了在折衷条件下同时测定土壤试样中较难测定的砷等5个元素,其灵敏度比直接溶液雾化法提高4~17倍。并详细研究了硼氢化钠浓度,酸度,载气流速对分析元素的影响,以及乙醇增强效应的最佳浓度和过渡元素对氢化物元素的化学干扰。
图1 氢化物发生系统示意图
3 试剂和材料
3.1 盐酸(ρ 约1.19g/mL)。
3.2 硝酸(ρ 约1.42g/mL)。
3.3 氢氟酸(ρ 约1.15g/mL)。
3.4 高氯酸(ρ 约1.67g/mL)。
3.5 过氧化氢(30%)。
3.6 无水乙醇。
3.7 碘化钾。
3.8 硫脲。
3.9 抗坏血酸。
3.10 硼氢化钠。
3.11 还原剂:硼氢化钠[5g/L,内含0.5g/L氢氧化钠和乙醇(4+96),现用现配]。
3.12 预还原剂:硫脲+抗坏血酸(80g/L+100g/L,现用现配)。
3.13 亚沸蒸馏水。
4 仪器
4.1 电感耦合等离子体-质谱(ICP-MS)仪器,仪器参数列于表1。
4.2 氢化物发生系统。
采用仪器原有的溶液雾化系统的雾化室作为气液分离器,用一个经改进的同心雾化器取代原气动雾化器,将引入硼氢化钠的毛细管与原有的试样引入管通过Y型三通管与一根混合线圈相连,硼氢化钠与试样溶液分别由多道蠕动泵送入,经过混合线圈并由改进的雾化器引
入雾室进行气液分离,气态物由氩气送入炬管激发,废液经蠕动泵排出(见图1)。
表1 HG-ICP-MS仪器工作参数
项目参数项目参数射频发生器质谱仪及测量条件
输出功率1350W 采样锥(Ni) 1.2mm
反射功率<5W 截取锥(Ni) 1.0mm
冷却气(Ar) 15L/min 分辨率0.8μ
辅助气(Ar) 1.5L/min 分析方式脉冲计数
载气(Ar) 0.8L/min 扫描次数 5 氢化物发生条件测量点/峰 5 蠕动泵速度100r/min 扫描时间/u2s
样品提升量 1.8mL/min 测量次数 3
还原剂提升量 1.8mL/min 校准方式外标法
还原剂浓度0.5% 内标元素Ge
5 试样的制备
将样品粉碎至粒度100μm~74μm,在干净的房间风干。称样测定时,另称取一份试样测定吸附水,最后换算成烘干样计算结果。
6 操作步骤
6.1 试样溶液的制备
实验了王水水浴溶解和微波高压密封消解两种方法。微波炉为实验室微波炉系统,HPV80高压消解罐。
方法1:称取0.25g风干土样(精确至0.0001g)于25mL比色管中,加入5mL新配制的王水,在水浴中加热溶解2h。将溶液冷却至室温,取出试管用亚沸蒸馏水稀释至刻度,备用。分取上述制备好的溶液2mL到10mL比色管中,加入1mL盐酸,锗内标500ng,5mL预还原剂,然后用亚沸蒸馏水稀释至刻度,室温下放置1h。此溶液用于测定砷,锑,铋。
方法2:称取0.25g风干土样(精确至0.0001g)于HPV80消解罐中,加入4mL硝酸,0.5mL 过氧化氢,0.5mL氢氟酸,1mL高氯酸,用紧盖工具拧紧盖。按以下程序重复消解2~3次;10%功率10min,25%功率10min,40%功率10min,50%功率20min。取出消解罐并冷却,用开盖工具打开盖,将罐内溶液全部倒入聚四氟乙烯坩埚中,在电炉上加热至刚冒白烟,取下加入1mL硝酸,继续加热至刚冒白烟,重复加硝酸2~3次,使有机物完全破坏,最后加入1mL硝酸,用亚沸水冲入到25mL比色管中备用。分取5mL溶液于10mL比色管中,加入锗内标500ng,4mL盐酸,用亚沸水稀释至刻度并在水浴中加热30min,此溶液用于测定硒、碲。
6.2 标准溶液的制备
首先用光谱纯金属或化合物制备单个元素的储备液。然后稀释成浓度为0,10ng/mL的混合元素的标准溶液,并含内标锗500ng/mL,酸度和预还原剂等介质严格与试样一致。
6.3 分析元素同位素及干扰信息列表2。
表2 分析同位素及干扰信息
元素质荷比(m/z) 相对丰度(%) 干扰信息
40Ar 35Cl+
100
As 75
100
Bi 209
57.25
Sb 121
123Te(0.87)
42.75
Sb 123
40Ar 37Cl+,40Ar36ArH+
Se 77
7.50
70Ar,38Ar,78Kr(0.35)
23.61
Se 78
82Kr(11.56)
8.84
Se 82
6.99
Te 125
126Xe(0.09)
18.71
Te 126
128Xe(1.92)
31.79
Te 128
130Xe(4.08),130Ba(0.10)
Te 130
34.49
7 结果计算
将测出各元素的结果,按下式进行水份校正。
ρ
w(x)=
m×
K
w(x)——某个被测出元素的质量分数,μg/g。
ρ——测出元素的质量浓度,μg/g。
m——测定样品的质量,g。
K——水份系数。
8 精密度
取一个试样重复测定10次,其RSD%为:As 1.21%,Bi 1.60%,Sb 2.16%,Se 2.44%,
Te 2.26%。
注1:本法首先将乙醇作增强剂用于HG-ICP-MS中,使砷,锑,硒,碲的灵敏度提高4~17倍,比AFS法的检出限低1~2个数量级。
注2:本法选择能形成氢化物的锗为内标元素,不仅显著地改善了精密度,而且也提高了测定的准确度。
9 参考文献
[1] 李冰等. 乙醇增强氢化物发生ICP-MS法测定砷锑铋硒和碲的研究. 一九九七年度岩矿
测试科研及试验成果汇编.地矿部岩矿测试技术研究所,1998,12~21.
土壤中总砷的分光光度法测定
土壤中总砷的分光光度法测定 相关背景:砷是世界卫生组织确定的高毒致癌物质,从上世纪初就开始受到科学家们的广泛关注。在农业生产中,砷主要是通过工业“三废”、农业利用等方式进入土壤,施用含砷的农药、化肥、有机肥等是土壤中砷的重要来源之一。砷进入土壤后,可被土壤胶体吸附固定,使其有效性降低。有机态砷进入土壤后,不仅被土壤吸附固定,也可在土壤微生物的作用下,并通过一系列的土壤过程,发生形态和价态的转化。农业生产与人类生活息息相关,研究不同形态砷在土壤中的转化及对植物砷有效性的影响,对提高农产品质量,预防设施土壤中砷含量超标等具有很重要的意义。由环保部牵头制定的《全国土壤环境保护“十二五”规划》已进入国务院审批程序,国家发改委批准了“‘十二五’重金属污染防治规划”,将“土壤与场地污染治理与修复”列入“十二五”社会发展科技领域国家科技计划项目指南。 依据标准:1997年12月8日,国家环境部发布GB/T 17135-1997 《土壤质量总砷的测定硼氢化钾-硝酸银分光光度法》。 检测方法简介: 土壤样品经氧化分解后,使不同形式的砷转化为可溶态砷离子,硼氢化钾(钠)在酸性的溶液中产生新生态氢,使五价砷还原为三价砷,三价砷还原成气态砷化氢,再用硝酸-硝酸银-聚乙烯醇-一算溶液为吸收液,银离子被砷化氢还原成单质银,使溶液成黄色,在400nm 分光光法测定。(10mm光程) 赛默飞世尔科技有限公司(ThermoFisher)的紫外可见分光光度计产品完全能够满足上述检测需要,并且可以为客户提供方法建立的工作,以方便有此需求的客户快速使用仪器,达到单位检测要求。请您联系赛默飞世尔科技有限公司(8008105118,4006505118),或者咨询我们当地的代理商。
《玻璃纤维中铅、汞、镉、砷及六价铬的限量指标与测定方法》编制说明
国家标准《玻璃纤维中铅、汞、镉、砷及六价铬的限量指标与测 定方法》编制说明 草案稿 一、工作简况 欧盟RoHS《关于限制在电子电器设备中使用某些有害成分的指令》标准已于2006年7月1日开始正式实施。该标准规定了电机电子产品中的铅、汞、镉、六价铬、多溴联苯和多溴二苯醚共6项物质,并重点规定了铅的含量不能超过0.1%。2009年IMO 在香港外交大会上通过了《2009年香港国际安全与无害环境船舶拆解公约》(“香港公约”),对船舶设计/建造/营运、拆船设施、有害材料的控制及人员保护等提出了要求。公约对铅、汞、镉、六价铬的含量进行了明确限定。 玻璃纤维是重要的工业原材料,是制造电脑、手机主版等电子器件的印刷电路板的主要增强材料,在机械船舶、石油化工以及市政工程上也有大量的应用。玻璃纤维中的有毒有害物质主要为玻璃澄清过程中引入的砷、矿物杂质引入的铅、汞、镉、六价铬等。 目前无论是出口还是内销都对玻璃纤维产品提出了要求,为了人体健康和环保要求,要控制产品种有毒有害物质的含量。本标准的编制可以规范行业对于产品的技术要求以及试验方法,可以规范产业的进步和发展,可以更好的使我国的产品与国际接轨,为玻纤产业与国际接轨提供技术支持,对规范产业的发展具有积极意义。 国家标准化管理委员会于2015年4月30下达2015年第一批国家标准制修订计划,下达了计划编号为20150380-T-609的《玻璃纤维中铅、汞、镉、砷及六价铬的限量指标与测定方法》国家标准制定计划。标准负责起草单位在接到标准编制计划任务后组成了标准起草小组。起草小组根据申报时的情况,对国内外相关行业、相关技术方法进行了收集与分析,拟出了标准草案稿。 二、标准编制原则和主要内容 1. 编制原则
回收还原沉金后液中硒碲铂钯的方法研究
回收还原沉金后液中硒碲铂钯的方法研究 方法研究概述: 通过 SO2 直接还原回收硒碲和捕集铂钯的方法研究,以沉金后液为原料。采用 X 射线衍射仪(XRD)和扫描电镜(SEM) 对还原产物的物相、微观形貌进行表征,结果表明:当反应温度为 85 ℃,SO2 流量为 0.2 L/min,反应时间为 4 h,H + 浓度为 3.3 mol/L 和 Cl ?浓度为 0.72 mol/L 时,Se 和 T e 回收率分别为 99.5%和 96.64%,Pt 和 Pd 回收率均到了 100%,所得黑色还原产物中硒、碲、铂和钯的质量分数分别为 28.06%、52.3%、0.084%和 0.588%。产物中硒和碲均以单质态形式存在,其形貌为球状体和柱状体。 铜阳极泥是电解精炼粗铜时所产生的不溶物,它的产率一般为电解铜产量的 0.2%~1.0%,因其中含有大量的贵金属和稀有元素而成为提取稀贵金属的重要原料。目前,国内采用湿法流程处理铜阳极泥的工厂已达 40%以上。铜阳极泥经预处理脱铜(硒碲)之后,采用亚硫酸钠或氨水浸出银、氯酸钠浸出金工艺分别得到分银液和分金液,分银液用水合肼或甲醛还原得到银粉,分金液用亚硫酸钠或草酸还原得到金粉和沉金后液。沉金后液含有大量的硒碲铂钯,目前一般在沉金后液中加入锌粉置换铂钯得到铂钯精矿,其工艺简单,操作方便;但是,锌粉置换法得到的铂钯精矿中铂钯含量低,回收铂钯时损失大,造成硒碲流失,锌粉用量大,生产成本高。此外,有报道采用铜粉或铜片置换沉淀溶液中的硒碲,不过沉淀物中的硒和碲是以硒化铜和碲化铜的形式存在,不利于后续硒碲分离,延长了工艺流程。本研究
采用 SO2 直接还原沉金后液回收硒碲和捕集铂钯,该处理工艺使硒和碲得到回收,所得还原产物硒碲品位高,有利于硒碲分离回收;另外,铂、钯等贵金属得到完全回收并高度富集,为含稀贵金属溶液的有效综合利用提供一条可行途径。 1 实验 1.1 实验步骤 实验取某铜冶炼厂草酸还原分金液得到的沉金后液,其成分如表 1 所列。 表 1 沉金后液主要化学成分 准确量取一定量的沉金后液,倒入容量为 1 L的三颈瓶中,启动搅拌,使用硫酸调整原料酸度,使用氯化钠增加溶液 Cl ?浓度,并在电热套中加热至一定温度,通入恒定流量的 SO2 还原,反应一段时间后过滤、洗涤、烘干得到硒碲精矿用于分别回收硒碲和铂钯,滤液返回用于调整酸度。其工艺流程如图 1 所示。 1.2 分析与检测 采用美国热电元素公司的 I ntrepid II XSP 型电感耦合等离子体发射光谱仪(ICP)分析溶液化学成分;X射线荧光光谱仪(XRF)分析固体物质成分;日本理学D/max-TTR III 型 X 射线衍射仪(XRD)分析固体物质物相;日本电子株式会社 JSM?6300 型扫描电镜(SEM) 观察固体产物微观形貌。
铅镉砷汞铜检测法
铅、镉、砷、汞、铜测定法 一、原子吸收分光光度法 本法系采用原子吸收分光光度法测定中药中的铅、镉、砷、汞、铜,所用仪器应符合使用要求(附录V D)。除另有规定外,按下列方法测定。 1.铅的测定(石墨炉法) 测定条件参考条件:波长283. 3nm,干燥温度100~120 ℃,持续20秒;灰化温度400~750℃,持续20~25秒;原子化温度1700~2100℃,持续4~5秒。 铅标准贮备液的制备精密量取铅单元素标准溶液适量,用2%硝酸溶液稀释,制成每lml含铅(Pb)lug 的溶液,即得(0~5℃贮存)。 标准曲线的制备分别精密量取铅标准贮备液适量,用2%硝酸溶液制成每lml分别含铅0ng、5ng、20ng、40ng、60ng、80ng的溶液。分别精密量取lml,精密加含1%磷酸二氢铵和0.2%硝酸镁的溶液0 .5 ml,混匀,精密吸取20ul注人石墨炉原子化器,测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。 供试品溶液的制备A法取供试品粗粉0.5g,精密称定,置聚四氯乙烯消解罐内,加硝酸3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内,进行消解(按仪器规定的消解程序操作)。消解完全后,取消解内罐置电热板上缓缓加热至红棕色蒸气挥尽,并继续缓缓浓缩至2~3ml,放冷,用水转入25ml量瓶中,并稀释至刻度,摇勻,即得。同法同时制备试剂空白溶液。 B法取供试品粗粉1g , 精密称定,置凯氏烧瓶中,加硝酸-高氣酸(4:1 )混合溶液5~10ml,混勻,瓶口加一小漏斗,浸泡过夜。置电热板上加热消解,保持微沸,若变棕黑色,再加硝酸-髙氣酸(4:1)混合溶液适量,持续加热至溶液澄明后升高温度,继续加热至冒浓烟,直至白烟散尽,消解液呈无色透明或略带黄色,放冷,转入50ml量瓶中,用2%硝酸溶液洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,即得。同法同时制备试剂空白溶液。 C法取供试品粗粉0 .5g,精密称定,置瓷坩埚中,于电热板上先低温炭化至无烟,移人高温炉中,于500℃灰化5~6小时(若个别灰化不完全,加硝酸适童,于电热板上低温加热,反复多次直至灰化完全),取出冷却,加10%硝酸溶液5ml使溶解,转人25ml量瓶中,
48硫硒碲(7页32题)
硫硒碲 A组 1.在人体所需的十多种微量元素中,有一种称为“生命元素”的R元素,对延长人类寿命起着重要的作用。已知R元素的原子有四个电子层,其最高价氧化物分子式为RO3,则R元素的名称为 A 硫 B 砷 C 硒 D 硅 2.不能说明氧的非金属性比硫强的是 A 在通常状况下,氧的单质为气体,硫的单质为固体 B 氢硫酸溶液在空气中置露会变浑浊 C 硫化氢在加热的条件下既可分解成氢气和硫,而水在强高温时很少分解 D 铜与硫反应生成硫化亚铜,与氧气反应生成氧化铜 3.下列说法不正确的是 A 硫是一种淡黄色的不能溶于水的晶体 B 硫的化合物常存在于火山喷出的气体中和矿泉水里 C 硫与氧属于同一主族 D硫在空气中的燃烧产物是二氧化硫,在纯氧中的燃烧产物是三氧化硫 4.下列哪些事实是由于氯的非金属性比硫强的结果 A 次氯酸的酸性比硫酸弱 B 氯能置换硫化氢中的硫 C 硫离子的还原性比氯离子强 D 硫能在空气中燃烧,而氯则不能 5.下列结论正确的是 ①微粒半径:S2->Cl>S>F ②氢化物稳定性:HF>HCl>H2S>H2Se ③离子还原性:S2->Cl->Br->I-④氧化性:Cl2>S>Se>Te ⑤酸性:H2SO4>HClO4>H2SeO4⑥非金属性:F>Cl>S>Se A ②④⑥ B ①③④ C 只有① D 只有⑥ 6.今有A、B、C、D四种短周期元素,它们的核电荷数依次增大,A与C、B与D 分别是同族元素,B、D两元素的质子数之和是A、C两元素质子数之和的两倍,这四种元素中有一种元素的单质易溶于二硫化碳,则这四种元素分别是 A 、 B 、 C 、 D 。 B组 7.已知2.1g KOH和1.6g硫粉混合后加热,恰好相互完全反应:aKOH+bS=cK2S x +dK2SO3+eH2O则x值为 A 0 B 1 C 2 D 3 8.元素X的气态氢化物的分子式为H2X,这种元素的最高价氧化物的水化物的化学式可能是 A H2XO3 B X(OH)2 C H2XO4 D H6XO6 9.常温下单质硫主要以S8形式存在。加热时,S8会转化为S6、S4、S2等。当温度达到750℃时,硫蒸气主要以S2形式存在(占92%)。下列说法中正确的是 A S8转化为S6、S4、S2属于物理变化
实验三 食品中硒的测定-原子荧光光谱法
光谱技术在食品分析中的应用 实验三食品中硒的测定-原子荧光光谱法 一、实验目的 1、了解原子荧光光度计仪器的基本结构和原理; 2、学会原子荧光光度计的操作技术; 3、了解食品中硒的测定意义; 4、学会湿法消化样品的操作。 二、基本原理 利用硼氢化钠作为还原剂,将四价硒在盐酸介质中还原为硒化氢(SeH2),由载气带 入原子化器中进行原子化,在硒特制空心阴极灯照射下,基态硒原子被激发至高能态,再 去活化回到基态时,发射出特征波长的荧光,其荧光强度与硒含量成正比,从而定量硒在 食品中的含量。 三、仪器和试剂 1、仪器: AFS-230E型双道原子荧光光谱仪、硒特制空心阴极灯、可调式电热板 2、试剂 除非另有规定,本方法所使用试剂均为分析纯,水为GB/T 6682 规定的三级水;所用玻璃仪器均需以硝酸(1+5)浸泡过夜,用水反复冲洗,最后用纯水冲冼干净。 硝酸(优级纯)、盐酸(优级纯)、氢氧化钠(5g/L,优级纯)、硼氢化钠溶液(8g/L)、铁氰化钾溶液(100g/L)、硒标准储备液(100μg/mL,光谱纯)、盐酸(6 mol/L)、混合酸:将硝酸与高氯酸按9:1 体积混合等。 硒标准储备液制备(100μg/mL):称取0.100g高纯硒粉于1000mL容量瓶中,溶于少 量硝酸中,加入2mL高氯酸,置沸水浴中加热3h~4h冷却后再加8.4mL盐酸,再置沸水浴 中煮2min,用蒸馏水准确稀释至1000mL,摇匀。 硒标准应用液制备:取100μg/mL硒标准储备液1.0mL,定容100mL,摇匀备用。 硼氢化钠溶液(8g/L)制备:称取8.0g硼氢化钠(NaBH4),溶于氢氧化钠溶液(5g/L)中,然后定容至1000mL。 铁氰化钾溶液(100g/L)制备:称取10.0g铁氰化钾(K3Fe(CN)6),溶于100mL容量 瓶中,摇匀。 载流溶液:5%盐酸水溶液。
原子荧光法测定土壤中砷和汞的实验研究
原子荧光法测定土壤中砷和汞的实验研究 发表时间:2016-11-10T09:44:29.837Z 来源:《低碳地产》2016年13期作者:张雅静陈佳亮 [导读] 【摘要】本文对原子荧光法测定土壤中砷和汞的实验展开了研究,应用王水水浴消解-原子荧光法对土壤样品中的砷和汞进行了测定,并对本次实验的结果及精密度、准确度、回收率等进行了分析。通过本次实验结果得出,该方法可以准确测定土壤中的砷和汞,可在土壤中砷和汞的检测中广泛应用。 1.广东省环境监测中心广东广州 510308; 2.广东省环境科学研究院广东广州 510045 【摘要】本文对原子荧光法测定土壤中砷和汞的实验展开了研究,应用王水水浴消解-原子荧光法对土壤样品中的砷和汞进行了测定,并对本次实验的结果及精密度、准确度、回收率等进行了分析。通过本次实验结果得出,该方法可以准确测定土壤中的砷和汞,可在土壤中砷和汞的检测中广泛应用。 【关键词】原子荧光法;砷;汞;土壤 引言 土壤中的砷和汞等重金属元素,可以通过迁移以及食物链进入人体中,严重危害到食品的安全及人体的健康。因此,了解土壤中砷和汞的含量,对土壤中的砷和汞进行准确的测定具有十分重要的意义。但由于砷和汞的复杂化学性质以及土壤基质的复杂性,如何准确测定土壤中的砷和汞是一个难题。当前,测定土壤中砷和汞的常用方法有原子吸收法、分光光度法、原子荧光法等,其中原子荧光法具有比较高的准确度及灵敏性被广泛采用。 1 实验部分 1.1 主要仪器及试剂 仪器:AFS-2202E型双道原子荧光光度计,砷、汞空心阴极灯,电热恒温水浴锅HHS-11-2,电子天平BT224S。 试剂:砷还原剂(2.0%硼氢化钾+0.5%氢氧化钠),载液(5%盐酸溶液),10%硫脲溶液,10%抗坏血酸溶液。(1+1)王水,汞还原剂(0.05%硼氢化钾+0.5%氢氧化钠),汞保存液(0.05%重铬酸钾+5%硝酸溶液),汞稀释液(0.02%重铬酸钾+2.8%硫酸溶液),砷标准贮备液(100mg/L,环保部标样所),汞标准贮备液(100mg/L,环保部标样所),土壤标准样品ESS-2(GSBZ500012-87,中国环境监测总站)。试验所用试剂均为优级纯。 1.2 样品保存与消解 土壤样品干燥后,过0.149mm孔径筛,于棕色广口玻璃瓶中保存,备用。 砷的消解:分别称取6份0.5000g上述土壤样品于50mL比色管中,分别加10mL水润湿样品,再加入10mL(1+1)王水,加塞用纱布捆紧摇匀,于沸水浴中消解2h,20min摇动1次,冷却后用水定容至刻度,摇匀[1]。 吸取10mL消解试液于50mL比色管中,加3mL5%的盐酸、5mL10%的硫脲溶液、5mL10%的抗坏血酸溶液,用水稀释至刻度,摇匀静置,取上清液待测。同法做2个空白试验。 汞的消解:分别称取6份0.5000g上述土壤样品于50mL比色管中,分别加10mL水润湿样品,再加入10mL(1+1)王水,加塞用纱布捆紧摇匀,于沸水浴中消解2h,20min摇动1次,消解完毕后冷却。在消解液(视情况可做稀释)中加入10mL汞保存液,用汞稀释液定容至刻度,摇匀后放置,取上清液待测,同法做2个空白试验。参照《土壤干物质和水分的测定重量法》(HJ613—2011)进行土壤含水量的测定。 1.3 标准溶液配制 将100mg/L砷标准贮备液配制成1.00mg/L的砷标准工作溶液,准确吸取0.00,0.50,1.00,2.00,3.00和4.00mL砷标准工作液置于6个100mL的容量瓶中,加入5mL5%的盐酸、5mL10%的硫脲溶液和5mL10%的抗坏血酸溶液,用纯水定容,摇匀,得0.0,5.0,10.0,20.0,30.0和40.0μg/L的砷标准溶液系列。 将100mg/L汞标准贮备液配制成100μg/L的汞标准工作液,准确吸取0.00,0.4,0.8,1.2,2.0和4.00mL汞标准工作液于6个100mL的容量瓶中,加入20mL汞保存液,用汞稀释液定容,得0.0,0.4,0.8,1.2,2.0和4.0μg/L的汞标准溶液系列。 1.4 仪器测定条件 原子荧光光度计工作参数见表1。 1.5 样品测定 土壤中重金属元素含量ω以质量比计(单位:mg/kg),按下式计算。 错误!未找到引用源。, 式中:c——重金属元素的质量浓度,μg/L;c0——试剂空白溶液重金属元素的质量浓度,μg/L;V2——测定时分取样品溶液稀释定容体积,mL;V总——样品消解后定容总体积,mL;V1——测定时分取样品消解液体积,mL;m——试样质量,g;f——土壤含水量,%。 2 结果与讨论 2.1 还原剂质量分数的选择 现以硼氢化钾为还原剂,20.0μg/L的砷标液和0.80μg/L的汞标液为研究对象,考察硼氢化钾质量分数对砷汞测定结果的影响,结果见图1。
土壤中砷汞的测定
土壤中总砷/总汞的测定 1主要仪器 AFS-9700 9700-214561型原子荧光光度计 2测定 2.1分析条件 光电倍增管负高压290V 空心阴极灯电流砷60mA 汞25mA 原子化高度8mm 载气(高纯氩)400mL/min;屏蔽气800mL/min 2.2样品消解: 称取经风干,研磨并过筛(100目)的土壤样品0.5g于50mL比色管中,加少量水润湿样品,加(HNO3:HCl=1:3)王水10mL,加塞摇匀过夜,于沸水中消解4个小时,冷却后加入2.5mL盐酸,10mL5%硫脲+5%抗坏血酸溶液,定容待测。 2.3校准曲线 砷标准曲线:分别准确吸取砷标准工作溶液(1.00mg/L)0.00、0.50、1.00、2.00、3.00、4.00、5.00mL置于100mL容量瓶中,分别加入5mL盐酸,10mL5%硫脲+5%抗坏血酸溶液,定容,此时得砷含量分别为:0.00、5.00、10.0、20.0、30.0、40.0、50.0ng/mL的标准溶液系列。 汞标准曲线:分别准确吸取汞标准工作溶液(20ng/mL)【标100mg/L=100ng/L,稀释1-100,10-500】0.00、0.50、1.00、2.00、3.00、5.00、10.00mL置于50mL容量瓶中,用5%盐酸定容,此时得汞含量分别为:0.00、0.20、0.40、0.80、1.20、2.00、4.00ng/mL的标准溶液系列。 2.4样品分析 将仪器调节至工作条件,在还原剂(2%硼氢化钾+0.5%氢氧化钾)和载液(5%盐酸)的带动下,测定标准系列和空白及试样。 3结果计算 含量(mg/kg)=c×V×0.01×n/m c----砷/汞的浓度,ng/ml;V----样品消解后定容体积,mL n----稀释倍数 m ---样品取样量,g;
72 土壤中总砷的测定原子荧光法GBT22105.2-2008演示教学
新项目试验报告 项目名称:土壤中总砷的测定 原子荧光法 GB/T 22105.1-2008 项目负责人:杨刚 项目负责人: 审批日期:
一、新项目概述 砷(As)是人体非必需元素,元素砷的毒性较低,而砷的化合物均有剧毒,砷通过呼吸道、消化道和皮肤接触进入人体,如摄入量超过排泄量,砷就会在人体多数器官中蓄积,从而引起砷中毒。砷还有致癌作用,能引起皮肤癌。在一般情况下,土壤、水、空气、植物和人体都含有微量的砷,对人体不会构成危害。但是工业生产中大部分是三价砷的化合物,因此,土壤砷污染是当今全球十分严重的环境与健康问题之一。 GB/T 22105的部分规定了土壤中总砷的原子荧光光谱测定方法。 本部分方法检出限为0.01mg/kg。 二、检测方法与原理 检测方法:原子荧光法 原理:样品中的砷经加热消解后,加入硫脲使五价砷还原为三价砷,再加入硼氢化钾将其还原为砷化氢,由氩气导入石英原子化器进行原子化分解为原子态砷,在特制砷空心阴极灯的发射光激发下产生原子荧光,产生的荧光强度与试样中被测元素含量成正比,与标准系列比较,求得样品中砷的含量。 三、主要仪器和试剂 1.仪器 1.1 氢化物发生原子荧光剂。 1.2 砷空心阴极灯。 1.3 水浴锅。 2.试剂 2.1 盐酸:1.19 g/ml,优级纯 2.2 硝酸:1.42 g/ml,优级纯 2.3 氢氧化钾:优级纯
2.4 硼氢化钾:优级纯 2.5 硫脲:分析纯 2.6 抗坏血酸:分析纯 2.7 三氧化二砷:优级纯 2.8(1+1)王水:取1份硝酸和3份盐酸混合均匀,然后用水稀释一倍。 2.9 还原剂(1%硼氢化钾+0.2%氢氧化钾溶液):称取0.2g氢氧化钾放入烧杯中,用少量水溶解,称取1g硼氢化钾,放入氢氧化钾溶液中溶解后用水稀释至100ml,此溶液用时现配。 2.10 载液:(1+9)盐酸溶液 2.11 硫脲溶液(5%):称取10g硫脲,溶解于200ml水中,摇匀。用时现配。 2.12 抗坏血酸(5%):称取10g抗坏血酸,溶解于200ml水中,摇匀。用时现配。 2.13 砷标准贮备液:称取0.6600g三氧化二砷(在105℃烘2h)于烧杯中,加入10ml 10% 氢氧化钠溶液,加热溶解,冷却后移入500ml容量瓶中,并用水稀释至刻度,摇匀。此溶液砷浓度为1.00mg/ml。 2.14 砷标准中间溶液:吸取10.00ml砷标准贮备液注入100ml容量瓶中,用(1+9)盐酸溶液稀释至刻度,摇匀。此溶液砷浓度为100ug/ml。 2.15 砷标准工作溶液:吸取1.00ml砷标准中间溶液注入100ml容量瓶中,用(1+9)盐酸溶液稀释至刻度,摇匀。此溶液砷浓度为1.00ug/ml。 四、采样要求和/或样品预处理技术 从野外采回的土壤样品要及时放在样品盘上,摊成薄薄的一层,置于室内通风处自然风干,严禁暴晒。风干过程中药经常翻动。将风干土壤剔除石块,用木棍或塑料碾压。压碎的土壤要最终通过0.149mm孔径筛。 五、检测步骤
50-铅、镉、砷、汞、铜测定法标准操作规程
目的:建立铅、镉、砷、汞、铜测定法检验标准操作规程,保证操作正确,确保检品质量。 范围:本标准规定了铅、镉、砷、汞、铜测定法的检验方法和操作要求;适用于本公司检品铅、镉、砷、汞、铜测定。 职责:QC执行,QC主任、质量部经理监督执行。 依据:《中国药典》2010年版一部附录Ⅸ B及中国药品检验标准操作规范 内容: 1、原子吸收分光光度法:本法系采用原子吸收分光光度法测定中药中的铅、镉、砷、汞、铜,所用仪器应符合使用要求(附录Ⅴ D)。除另有规定外,按下列方法测定。 . 铅的测定(石墨炉法)。 1.1.1. 测定条件参考条件:波长,干燥温度100~120℃,持续20秒;灰化温度400~750℃,持续20~25秒;原子化温度1700~2100℃,持续4~5秒。 1.1. 2. 铅标准储备液的制备:精密量取铅单元素标准溶液适量,用2%硝酸溶液稀释,制成每1ml含铅(Pb)1μg的溶液,即得(0~5℃贮存)。 1.1.3. 标准曲线的制备:分别精密量取铅标准储备液适量,用2%硝酸溶液制成每1ml分别含铅0ng、5ng、20ng、40ng、60ng、80ng的溶液。分别精密量取1ml,精密加含1%磷酸二氢铵和%硝酸镁的溶液,混匀,精密吸取20μl注入石墨炉原子化器,测定吸光度,以吸光度为纵光标,浓度为横坐标,绘制标准曲线。 1.1.4. 供试品溶液的制备。 1.1.4.1. A法取供试品粗粉0.5g,精密称定,置聚四氟乙烯消解罐内,加硝 第1页共6页
酸3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内,进行消解(按仪器规定的消解程序操作)。消解完全后,取消解内罐置电热板上缓缓加热至红棕色蒸气挥尽,并继续缓缓浓缩至2~3ml,放冷,用水转入25ml 量瓶中,并稀释至刻度,摇匀,即得。同法同时制备试剂空白溶液。 1.1.4. 2. B法取供试品粗粉1g,精密称定,置凯氏烧瓶中,加硝酸-高氯酸(4:1)混合溶液5~10ml,混匀,瓶口加一小漏斗,浸泡过夜。置电热板上加热消解,保持微沸,若变棕黑色,再加硝酸-高氯酸(4:1)混合溶液适量,持续加热至溶液澄明后升高温度,继续加热至冒浓烟,直至白烟散尽,消解液呈无色透明或略带黄色,放冷,转入50ml量瓶中,用2%硝酸溶液洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,即得。同法同时制备试剂空白溶液。 1.1.4.3. C法:取供试品粗粉0.5g,精密称定,置瓷坩埚中,于电热板上先低温炭化至无烟,移入高温炉中,于500℃灰化5~6小时(若个别灰化不完全,加硝酸适量,于电热板上低温加热,反复多次直至灰化完全),取出冷却,加10%硝酸溶液5ml使溶解,转入25ml量瓶中,用水洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,即得。同法同时制备试剂空白溶液。 1.1.5. 测定法:精密量取空白溶液与供试品溶液各1ml,精密加含1%磷酸二氢铵和%硝酸镁的溶液,混匀,精密吸取10~20μl,照标准曲线的制备项下的方法测定吸光度,从标准曲线上读出供试品溶液中铅(Pb)的含量,计算,即得。 . 镉的测定(石墨炉法)。 1.2.1. 测定条件参考条件:波长,干燥温度100~120℃,持续20秒;灰化温度300~500℃,持续20~25秒;原子化温度1500~1900℃,持续4~5秒。 1.2.2. 镉标准储备液的制备精密量取镉单元素标准溶液适量,用2%硝酸溶液稀释,制成每1ml含镉(Cd)1μg的溶液,即得(0~5℃贮存)。 1.2.3. 标准曲线的制备分别精密量取镉标准储备液适量,用2%硝酸溶液稀释制成每1ml分别含镉0ng、、、、、的溶液。分别精密吸取10μl,注入石墨炉原子化器,测定吸光度,以吸光度为纵光标, 第2页共6页
原子荧光法测定食品中的硒
【摘要】采用原子荧光法测定食品中的硒,硒的相对标准偏差为 1.2%,检出限为0.40mg/ml。线性范围为0~400μg/L,相关系数为0.9999.回收率为95.1%~103.8%。【关键词】硒;原子荧光;食品硒是人体必需的微量元素,但是摄入过多对人体健康造成危害。近年来,在食品加工方面有任意在一些食品中强化硒的倾向。为了保障人体健康,因此要加强硒在食品、水中含量的监测力度。常用方法有荧光法、原子吸收法和比色法等,荧光法虽然准确但繁琐,并且所用试剂DAN毒性大且需进口。原子吸收法火焰法灵敏度低,石墨炉法有严重的基体干扰,比色法简便、灵敏度低。原子荧光法测定硒克服以上方法的缺点,是一种简便、灵敏度高的方法。 1 实验部分 1.1 试剂与仪器单光道原子荧光分光光度计(北京瑞利分析仪器公司AF-610),配有计算机处理系统,硒空心阴极灯:北京真空电子技术研究所,硝酸(优级纯),高氯酸(优级纯),盐酸(优级纯),混合酸:硝酸+高氯酸(4+1),氢氧化钾(分析纯),硼氢化钾溶液(15g/L)称取2g KOH溶于约200ml纯水中,加入15g硼氢化钾并使之溶解后,用脱脂棉过滤,用纯水稀释至1000ml,摇匀。宜现配现用。铁氰化钾(100g/L):称取10.0g铁氰化钾(K3Fe(CN)6)溶于100ml蒸馏水中,混匀。硒标准贮备液:国家标准物质GBW(E)080215 100μg/ml,硒标准应用液:取100μg/ml 硒标准贮备液1ml,定容至100ml,此应用液浓度为1μg/ml。载流:20%(V/V)HCl本方法中,除特殊规定外,所用试剂分析纯,实验用水为去离子水。 1.2 仪器工作条件PMT电压:340V,HCl主阴极电流:100mA。HCl辅助阴极电流:0mA;原子化器温度:室温(档)。原子化器高度:7mm。载气流量:600ml/min。测量方式:标准曲线法,进样体积:0.5ml。分析信号:峰面积,读数延时:1.0sec。读数时间:12.0sec。流动程序方式:断续流动。采样泵速:100r/min,采样时间:5sec,停泵时间:4sec,注入泵速:100r/min,注入时间:16sec,停泵时间:5sec。 1.3 样品处理及消化粮食:样品用水洗3次,60℃烘干,用不锈钢磨粉碎,储于塑料瓶内,备用。蔬菜及其他植物性食物:取可食部分用水洗净后用纱布吸去水滴,打成匀浆后备用。称取0.5~2.0g样品于150ml 高筒烧杯内,加10ml混合酸及几粒玻璃珠,盖上表面器冷消化过夜。次日于电热板上加热,并及时补加混合酸。当溶液变为清亮无色并伴有白烟时,再继续加热至剩余体积2ml左右,切不可蒸干。冷却,再加5ml 6mol/L盐酸,继续加热至溶液变为清亮无色并伴有白烟出现,以完全将六价硒还原成四价硒。冷却,转移定容至50ml容量瓶中,同时做空白实验。吸取10ml样品有消化液于15ml离心管中,加浓盐酸2ml,铁氰化钾溶液1ml,混匀待测。 1.4 标准曲线制作分别取0.0,0.5,1.0,2.0,4.0,5.0ml标准应用液于50ml比色管中,再分别加浓盐酸5ml,铁氰化钾1ml,混匀,用去离子水定容至50ml制成标准工作曲线。[!--empirenews.page--] 1.5 测定[1]开机,预热20~30min,输入必要的参数:样品量(g或ml)、稀释体积(ml)、进样体积(ml)、结果的浓度单位、测定点重复测量次数、标准系列点数及各点的浓度值。先测定空白溶液,然后转入标准系列测量,绘制标准曲线后,再测定两个空白溶液的空白值。随后测定样品,测定完毕,打印出测定结果。2结果与讨论 2.1 标准曲线线性范围及回归方程测定0~100μg/L的标准曲线,相关系数为0.9999,回归方程Y=33.807-24.515X。标准曲线线性范围0~400μg/L。 2.2 干扰及消除经实验1000倍Al3+、Fe2+、Fe3+、Ca2+、Mg2+、K+及Cl-、F-、NO3-、SO42-、PO43-不干扰测定。氢化物发生原子吸收法中观察到Sn对Se测定的严重干扰,而氢化物原子荧光法中未发现。对于基体成分复杂,而Se的含量较低的样品,可采用萃取、疏基棉吸附、离子交换等分离高集方法来消除干扰[2]。 2.3 相对标准偏差和检出限对40ng/ml的硒进行11次测定得相对标准偏差1.2%。连续测定11次空白荧光值11次。空白值的标准偏差SD。根据工作曲线的斜率K和检出限D=3δ/K。算出硒的检出限0.4ng/ml。按分析方法测定高中低浓度样品,各测定6次得平均值及相对标准偏差。平均值分别为0.013、0.156、0.456mg/kg,其相对标准偏差分别为5.0、4.2、3.2%。 2.4 方法准确度用
土壤中砷检测方法-砷化氢原子吸收光谱法
土壤中砷檢測方法-砷化氫原子吸收光譜法 NIEA S310.63C 一、方法概要 土壤樣品以過氧化氫氧化分解有機質後,以9.6 M鹽酸萃取,經碘化鈉或碘化鉀還原為三價砷,再經氫硼化鈉還原為砷化氫,此砷化氫經由氣體載送至原子吸收光譜儀,於波長193.7 nm處定量之。 二、適用範圍 本方法適用於檢測土壤中砷濃度。 三、干擾 由於砷及其化合物均具揮發性,在前處理時應小心,以免揮發而漏失。使用連續式氫化物產生器,應小心樣品交叉污染的干擾。 四、設備 (一) 加熱板:能控溫在100至120℃者。 (二) 水平振盪器:能控制振盪頻率在120至180 rpm者。 (三) 原子吸收光譜儀附有氫化物產生器之裝置(註1)。光源使用砷中空陰極燈管或無電極放 電式燈管,偵測波長選在193.7 nm處。在原子吸收光譜儀燃燒器上端設置排氣罩,抽氣以除去火焰中的薰煙及蒸氣。 (四) 烘箱:自動控溫,附排氣設備,可維持溫度105 5℃者。 (五) 密閉乾燥器。 (六) 分析天平:可精秤至0.1 mg者。 (七) 濾紙:Whatman No.40,或同級品。 五、試劑 所有檢測時使用的試劑化合物除非另有說明,否則必須是分析級試藥。 (一) 試劑水:比電阻≧ 16 MΩ – cm不含砷之去離子水。
(二) 過氧化氫(H2O2),30%。 (三) 濃鹽酸。 (四) 9.6 M鹽酸溶液:將400 mL濃鹽酸加入約80 mL的試劑水中,再以試劑水稀釋至500 mL。 (五) 碘化鈉溶液(NaI),10%:溶解10 g碘化鈉於試劑水中,定容至100 mL。 (六) 碘化鉀溶液(KI),10%:溶解10 g碘化鉀於試劑水中,定容至100 mL。 (七) 維他命C(Ascorbic acid),5 %:溶解5 g維他命C於試劑水中,定容至100 mL。 (八) 硼氫化鈉溶液(NaBH4),4%:溶解4 g硼氫化鈉於10%(w/v)氫氧化鈉溶液中,並定 容至100 mL,使用前配製。 (九) 砷儲備溶液:購買經濃度確認之標準品或精秤1.320 g純度99.9%以上之三氧化二砷 (As2O3),放入1,000 mL量瓶內,加入100 mL 4%(w/v)氫氧化鈉溶液,使之溶解後,再以20 mL之濃鹽酸酸化後,以試劑水稀釋至刻度;1.00 mL=1.00 mg As。 (十) 砷中間溶液:精取1.0 mL砷儲備溶液置於100 mL量瓶內,加入1.5 mL濃鹽酸後以試劑 水稀釋至刻度;1.00 mL=10.0 μg As。 (十一) 砷標準溶液:精取1.0 mL砷中間溶液置於100 mL量瓶內,加入1.5 mL濃鹽酸後,以試劑水稀釋至刻度;1.00 mL=0.1 μg As。 六、採樣及保存 (一) 土壤樣品之採樣與保存依據「土壤採樣方法」採集具有代表性之土壤樣品,並經樣品處理 後貯存於塑膠或玻璃瓶中密封以備分析,最長保存期限6個月。 (二) 土壤樣品處理參考「土壤污染物檢測方法總則」,為使土壤樣品均勻化,應再研磨樣品使 通過0.250 mm篩網(60 mesh)過篩(必要時可使通過0.150 mm(100 mesh)篩網)。 七、步驟 (一) 操作步驟 1、秤取經程序六、採樣及保存處理過之適量風乾土壤樣品,依據「土壤水分含量測定方法-重量法」測定土壤水分含量。 2、另取相同之風乾土壤約1 g,置於100 mL三角錐瓶中,加入5 mL 30%過氧化氫後,將三角瓶置於加熱板上,加熱至近乾,然後冷卻至室溫。
铅、镉、砷、汞、铜测定规程
1.目的:建立铅、镉、砷、汞、铜测定法操作规程,规范铅、镉、砷、汞、铜测定法的操作。 2.范围:本公司产品铅、镉、砷、汞、铜的检验。 3.责任:QC检验员。 4.内容: 4.1 原子吸收分光光度法: 本法系采用原子吸收分光光度法(附录Ⅴ D)测定中药材中的铅、镉、砷、汞、铜,除另有规定外,按下列方法测定。 4.1.1铅的测定(石墨炉法) 4.1.1.1测定条件 4.1.1.1.1参考条件:波长 283.3nm,干燥温度 100~120℃,持续 20 秒;灰化温度 400~750℃,持续 20~25 秒;原子化温度 1700~2100℃,持续 4~5秒;背景校正为氘灯或塞曼效应。 4.1.1.2铅标准储备液的制备 精密量取铅单元素标准溶液适量,用 2%硝酸溶液稀释,制成每 1ml 含铅(Pb)1μg 的溶液,即得(0~5℃贮存)。 4.1.1.3标准曲线的制备 分别精密量取铅标准储备液适量,用 2%硝酸溶液制成每1ml 分别含铅 0ng、5ng、20ng、40ng、60ng、80ng 的溶液。分别精密量取 1ml,精密加含 1%磷酸二氢铵和 0.2%硝酸镁的溶液 1ml,混匀,精密吸取20μl 注入石墨炉原子化器,测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。 4.1.1.4供试品溶液的制备 4.1.1.4.1 A 法 取供试品粗粉 0.5g,精密称定,置聚四氟乙烯消解罐内,加硝酸3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内,进行消解(按仪器规定的消解程序操作)。消解完全后,取消解内罐置电热板上缓缓加热至红棕色蒸气挥尽,并继续缓缓浓缩至2~3ml,放冷,用水转入 25ml 量瓶中,并稀
Han原子荧光测硒2015-11-7
氢化物原子荧光测硒实验方案 1. 主要仪器和试剂: 1.1主要仪器:A FS-922 型氢化物发生双道原子荧光光谱仪(北京吉天仪器有限公司);硒空心阴极灯(北京真空电子技术研究所)、SH220石墨消解仪(型号SH220;济南海能仪器有限公司)、优普超纯水机、电子天平(赛多利斯;Sartorious BS2245)、数显鼓风干燥箱(上海博迅实业有限公司医疗设备厂)、25ml容量瓶、消解管、小烧杯、漏斗、移液枪、离心管等常用实验仪器。 1.2主要试剂:盐酸(优级纯;武汉市中天化工有限公司)、硝酸(优级纯;国药集团化学试剂有限公司)、30%过氧化氢(优级纯;天津市光复科技发展有限公司);氢氧化钾(优级纯)、硼氢化钾(优级纯);硒标准储备液(100ug/ml)(GSB07 – 1253)(由国家环境保护总局标准样品研究所研制)。实验用水为超纯水。 1.3仪器工作条件(仪器设置及药品) B道硒灯; 光电倍增管负高压:300V; 原子化器高度:10mm; 灯电流:80mA 载气流量:400ml/min 屏蔽气流量:8000mL/min 分析信号:峰面积 读数时间:12s 延迟时间:1.2s 注入量:1.5ml 测定次数:3次 载流:5%HCl;还原剂:1%硼氢化钾(钠)
2、实验步骤:本次实验共有7个样品,14分血样,一份空白对照,分别做两个重复。分1次消解定容,每次消解16个。 2.1. 药品准备(注意:配制药品所用到的所有器皿均需要用1:1的硝酸浸泡过夜,第二天,将侵泡好了的仪器用清水冲洗,再用超纯水润洗三遍,烘干备用。)所需器皿:1000ml容量瓶1个,烧杯,离心管,25ml容量瓶,枪头,500ml容量瓶,移液管,玻璃棒,量筒,集气瓶) 2.1.1配制6ml/L的盐酸溶液200ml。 配方:量取50ml盐酸缓慢加入40ml水中,冷却后定容至100ml。(配制200ml)(※注意:盐酸的配制循序,先加水,再加盐酸,防止盐酸溅出受伤) 2.1.2 配制5%盐酸溶液 配方:50ml定容至1000ml(配2L) 2.1.3 配制标准硒溶液(硒标准溶液最好手工配制,其剃度为40ug/L、20 ug/L、10 ug/L、5 ug/L、2.5 ug/L) 配方:吸取0.025ml 40ug/mL 硒标准溶液,以5%盐酸溶液定容至25mL。制成20ug/L 储备液。再吸取12.25ml硒标准溶液,以5%盐酸溶液定容至25mL,同理配制10 ug/L、5 ug/L、2.5 ug/L硒标准溶液。(仪器可测定的最高浓度为20-30ug/L) 2.1.4配制1%硼氰化钾溶液 配方:将1.0g KBH4(优级纯)溶于100ml 0.5% KOH(优级纯)溶液中,混匀。该溶液易失效,所以需用前配制,放冰箱内可保存10天。(配200ml) 2.2样品处理 2.2.1取20支消解管、20个10ml容量瓶,20个15ml离心管,置于1:1的硝酸液中侵泡(至少一夜)。第二天,将侵泡好了的仪器用清水冲洗,再用超纯水润洗三遍,烘干备用。 2.2.2微波消解 ⑴浸酸:取5ml鳝鱼鲜血于消解管中,加8mL的浓硝酸,2mL的双氧水,浸酸一小时,每个土洋做两个个重复,分别记为A1、A2、并做一组对照 ⑵消解:至微波消解仪中,按下列表格设置好程序,
土壤中砷含量的测定
土壤中砷含量的测定(二乙基二硫代氨基甲酸银分光光度法)
砷不是生物所必需的元素,但砷在自然界分布广,尤其是砷的用途多,主要用于制造砷酸和砷的化合物、应用于熔剂砷合金,杀虫剂、除草剂和其他农药,在医药、木材防腐、制革、制乳白色玻璃、军用毒药及烟火方面也有广泛用途。因此砷进入农田和生态系统的可能性很大.元素砷毒性极低,而砷的化合物均有剧毒,三价砷化合物比其他砷化物毒性更强,如As2O3(砒霜)毒性最大。工业生产中大部分是三价砷的化合物,因此, 土壤砷污染是当今全球十分严重的环境与健康问题之一。本实验主要讲诉了测定土壤砷的一系列步骤及方法。 关键词 砷土壤采样分析方法
第一章土壤样品的采集与处理··························· 1.1 土壤样品的采集······························ 1.2 土壤样品的处理与贮存························第二章土壤砷含量的测定······························· 2.1采用原理···································· 2.2仪器、试剂及其配制方法······················· 2.3分析步骤···································· 2.4校准曲线的绘制······························ 2.5结果计算···································· 2.6结论········································主要参考文献···········································致谢·················································附录···················································
铅镉砷汞铜检测法
铅镉砷汞铜检测法公司标准化编码 [QQX96QT-XQQB89Q8-NQQJ6Q8-MQM9N]
铅、镉、砷、汞、铜测定法 一、原子吸收分光光度法 本法系采用原子吸收分光光度法测定中药中的铅、镉、砷、汞、铜,所用仪器应符合使用要求(附录V D)。除另有规定外,按下列方法测定。 1.铅的测定(石墨炉法) 测定条件参考条件:波长283. 3nm,干燥温度100~120 ℃,持续20秒;灰化温度400~750℃,持续20~25秒;原子化温度1700~2100℃,持续4~5秒。 铅标准贮备液的制备精密量取铅单元素标准溶液适量,用2%硝酸溶液稀释,制成每lml含铅(Pb)lug 的溶液,即得(0~5℃贮存)。 标准曲线的制备分别精密量取铅标准贮备液适量,用2%硝酸溶液制成每lml分别含铅0ng、5ng、20ng、40ng、60ng、80ng的溶液。分别精密量取lml,精密加含1%磷酸二氢铵和%硝酸镁的溶液0 .5 ml,混匀,精密吸取20ul 注人石墨炉原子化器,测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。 供试品溶液的制备 A法取供试品粗粉,精密称定,置聚四氯乙烯消解罐内,加硝酸3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内,进行消解(按仪器规定的消解程序操作)。消解完全后,取消解内罐置电热板上缓缓加热至红棕色蒸气挥尽,并继续缓缓浓缩至2~3ml,放冷,用水转入25ml量瓶中,并稀释至刻度,摇匀,即得。同法同时制备试剂空白溶液。 B法取供试品粗粉1g , 精密称定,置凯氏烧瓶中,加硝酸-高气酸(4:1 )混合溶液5~10ml,混匀,瓶口加一小漏斗,浸泡过夜。置电热板上加热消解,保持微沸,若变棕黑色,再加硝酸-髙气酸(4:1)混合溶液适量,持续加热至溶液澄明后升高温度,继续加热至冒浓烟,直至白烟散尽,消解液呈无色透明或略带黄色,放冷,转入50ml量瓶中,用2%硝酸溶液洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,即得。同法同时制备试剂空白溶液。 C法取供试品粗粉0 .5g,精密称定,置瓷坩埚中,于电热板上先低温炭化至无烟,移人高温炉中,于500℃灰化5~6小时(若个别灰化不完全,加硝