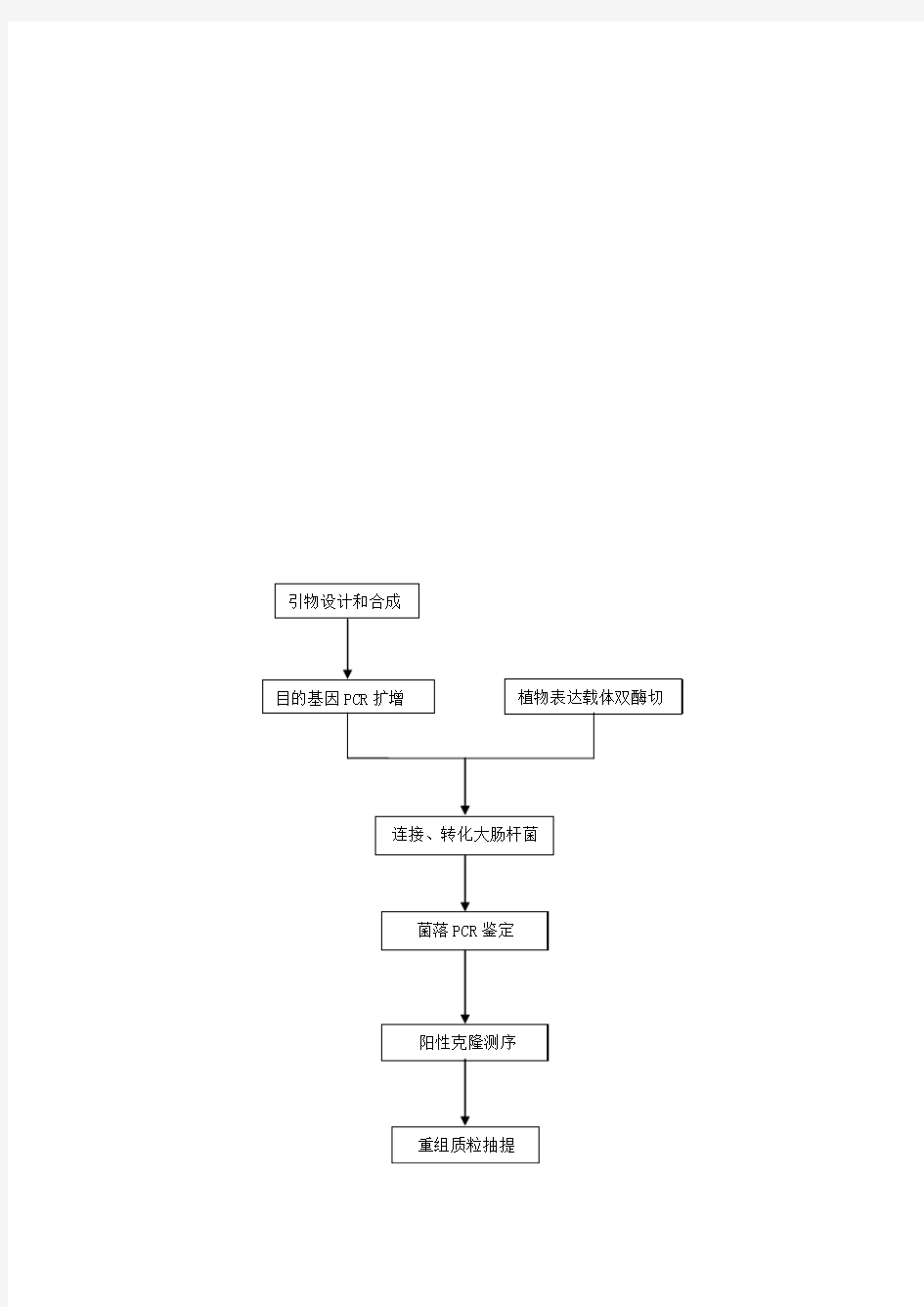
载体构建流程图
表达载体的构建方法及步骤
表达载体的构建方法及步骤 令狐采学 一、载体的选择及如何阅读质粒图谱 目前,载体主要有病毒和非病毒两大类,其中质粒DNA 是一种新的非病毒转基因载体。 一个合格质粒的组成要素: (1)复制起始位点Ori 即控制复制起始的位点。原核生物DNA 分子中只有一个复制起始点。而 真核生物DNA 分子有多个复制起始位点。 (2)抗生素抗性基因可以便于加以检测,如Amp+ ,Kan+ (3)多克隆位点MCS 克隆携带外源基因片段 (4)P/E 启动子/增强子 (5)Terms 终止信号 (6)加poly(A)信号可以起到稳定mRNA 作用 选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。如果构建的目 的是要表达一个特定的基因,则要选择合适的表达载体。 载体选择主要考虑下述3点: 【1】构建DNA 重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。 【2】.载体的类型:
(1)克隆载体的克隆能力-据克隆片段大小(大选大,小选小)。如<10kb 选质粒。 (2)表达载体据受体细胞类型-原核/真核/穿梭,E.coli/哺乳类细胞表达载体。 (3)对原核表达载体应该注意:选择合适的启动子及相应的受体菌,用于表达真核蛋白质时注意克服4个困难和阅读框错位;表达天然蛋白质或融合蛋白作为相应载体的参考。 【3】载体MCS 中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接,不能产生阅读框架错位。 综上所述,选用质粒(最常用)做载体的5点要求: (1)选分子量小的质粒,即小载体(1-1.5kb)→不易损坏,在细菌里面拷贝数也多(也有大载 体); (2)一般使用松弛型质粒在细菌里扩增不受约束,一般10个以上的拷贝,而严谨型质粒<10个。 (3)必需具备一个以上的酶切位点,有选择的余地; (4)必需有易检测的标记,多是抗生素的抗性基因,不特指多位Ampr(试一试)。 (5)满足自己的实验需求,是否需要包装病毒,是否需要加入荧光标记,是否需要加入标签蛋白,是否需要真核抗性(如Puro、G418)等等。 无论选用哪种载体,首先都要获得载体分子,然后采用适当的限制酶将载体DNA 进行切割,获得线性载体分子,以便于与
载体构建流程
载体构建SOP流程: GenBank查询目的基因序列→根据ORF序列利用引物设计软件设计引物→表达目的基因的组织或细胞总RNA提取→RT-PCR获取目的基因→酶切目的基因和载体→分别纯化酶切的目的基因和载体并建立连接反应→转化→初步筛选阳性克隆→阳性克隆测序→测序正确的质粒保种并重提质粒 I.获取目的基因/序列片段 一.获取序列信息 通过GENBANK数据和生物信息的方法设计目的基因或目的片段引物(shRNA、miRNA)。 PCR引物的设计原则: ①引物应用核酸系列保守区内设计并具有特异性。 ②产物不能形成二级结构。 ③引物长度一般在15~30碱基之间。 ④G+C含量在40%~60%之间。 ⑤碱基要随机分布。 ⑥引物自身不能有连续4个碱基的互补。 ⑦引物之间不能有连续4个碱基的互补。 ⑧引物5′端可以修饰。 ⑨引物3′端不可修饰。
⑩避免在引物的3’端使用碱基A。 在实际设计引物中由于ORF两末端序列本身的限制,不能完全按照上述理想的设计原则,但也切记引物不能过长或过短。过长的引物不容易打开其二级结构,与模板结合缓慢,也容易形成引物二聚体,通常不超过35bp(不包括酶切位点和保护碱基)。过短的引物特异性差,扩出其它不相关片段,最终很难得到目的片段,通常不短于18bp(不包括酶切位点和保护碱基)。要将目的基因定向克隆至相应载体,需要在上下游引物两端设计不同的酶切位点,由于酶切位点位于线性末端时酶对其识别切割能力大大降低,需依据NEB目录添加相应保护碱基,酶切时可相应增加时间。 二.制备模板 1.分离高质量RNA:成功的cDNA合成来自高质量的RNA。高质量的RNA至少应保证全长并且不含逆转录酶的抑制剂,如EDTA或SDS。RNA的质量决定了能够转录到cDNA上的序列信息量的最大值。现在实验室通常使用Trizol试剂法提取总RNA,可以从多种组织和细胞中提取高质量的非降解RNA。Trizol试剂法可以从最少100个细胞或1mg组织中提取RNA。在逆转录反应中经常加入RNase抑制剂以增加cDNA合成的长度和产量。RNase抑制剂要在第一链合成反应中,在缓冲液和还原剂(如DTT)存在的条件下加入,因为cDNA合成前的过程会使抑制剂变性,从而释放结合的可
载体构建的基本步骤
载体构建 一.原理 依赖于限制性核酸内切酶,DNA连接酶和其他修饰酶的作用,分别对目的基因和载体DNA进行适当切割和修饰后,将二者连接在一起,再导入宿主细胞,实现目的基因在宿主细胞内的正确表达。 二.操作步骤 1.摇菌 取装有液体培养基的3ml试管两支(依情况而定),每管加40-100μl菌种,过夜摇。2.提质粒 依照提质粒试剂盒中的说明书操作(根据情况最后一步洗脱时可以多洗1-2次)。 3.酶切 按下表加入试剂。 反应所需试剂体积(单位:ul) 质粒10 所需内切酶反应缓冲液 2 所需限制性内切核酸酶 1 H2O 7 将加好的EP管置于37℃保温1-2h。(依照提酶切的具体步骤操作;为了达到最佳酶切的效果,最好根据所选用的酶确定所需要的反应温度) 4.电泳检测 将酶切产物进行琼脂糖凝胶电泳,检测酶切是否成功。 5.连接 如果电泳检测酶切成功的话,则仔细将所需的片段切割下来,将胶体回收(依照胶回收试剂盒说明书操作);之后将回收的片段和载体连接,连接体系如下: 双蒸水5μL 10×T4 DNA连接缓冲液1μL 载体2μL 酶切后的目的基因1μL T4DNA连接酶1μL 总体积10μL 置于温箱,12-16℃,保温8-16h 6.转化 依照转化具体操作步骤做感受态,将上述连接产物进行转化实验,涂板培养,37℃,12-16h。 7.单克隆检测 (1)挑单克隆
先将AMP从冰箱中取出,待融化后,在3ml装有LB液体培养基的试管中加入3μL的AMP,用枪头混匀;取1.5 mlEP管5支(依情况可以多挑几管),给每支管中加500μL上述培养液,然后用接种环(或黄枪头)挑单克隆,挑完后用枪吹打;之后,将挑好的菌摇4-5小时,至混浊即可。 (2)单克隆检测 以每管摇好的菌液为模板,以原有的引物进行PCR,然后将PCR产物跑电泳,观察电泳图像中那几管的条带正确,将正确条带相对应管的菌液再抽取100μL,加到3ml(有LB液体培养液,AMP+)试管中,过夜摇;第二天重摇,将摇好的菌取1ml于1.5mlEP管中送测序,并保种。 注:①挑单克隆时,一定要挑单一圆润的菌落,有卫星斑的不挑。 ②别忘记往培养基中加AMP。 ③用接种环挑菌后,要在酒精灯上反复灼烧,然后再进行下一次挑菌。 三.注意事项 1.连接产物可短时间在-20℃保存,使用时可以取出进行后续实验; 2.在细胞转化时,冰浴和热激要严格控制好时间; 3.连接反应是DNA重组过程中的关键步骤,其成败的重要参数之一就是温度,因此要控制好连接温度。 4.进行黏末端连接时,会产生一定数量的载体自身环化分子,导致转化菌中过高的假阳性克隆背景。针对这一问题,常采用牛小肠碱性磷酸酶(CIP)去掉载体的5’-磷酸以抑制DNA 片段的自身环化。 参考: 1.刘进元,常智杰,赵广荣,等.分子生物学实验指导[M].北京:清华大学出版社,2002 2.周俊宜.分子生物学基本技能和策略[M].北京:科学出版社,2003:117-120 3.李海英,杨峰山,邵淑丽,等.现代分子生物学与基因工程[M].北京:化学工业出版社,2008:138-142 4.朱旭芬.基因工程实验指导[M].北京:高等教育出版社,2006:134-139
实验室常用细胞株
ATCC? Number: CRL-2865? Price: $329.00 Designations: T47D-KBluc Depositors: VS Wilson Biosafety Level: 1 Shipped: frozen Medium & Serum: See Propagation Growth Properties: adherent Organism: Hom o sapi ens (human) Morpholog y: epithelial Source: Organ: mammar y gland; breas t Tissue: duct Disease: ductal carcinoma Derived from metastatic site: pleural effusion Cell T y p e: epithelialtransfected with reporter plas mid Permits/F orms: In addition to the MTA menti oned above, other ATCC and/or regulatory per mits may be required for the transfer of this ATCC material. Any one purchasing ATCC material is ultimatel y r esponsible for obtaining the permits. Please click here for information regarding the s pecific requirements for shipment to your loc ation. Reverse Transcript: N Age: 54 years adult HeLa Mar kers: N
慢病毒载体包装构建过程
慢病毒载体包装构建过程 原理:慢病毒载体可以将外源基因或外源的shRNA有效地整合到宿主染色体上,从而达到持久性表达目的序列的效果。在感染能力方面可有效地感染神经元细胞、肝细胞、心肌细胞、肿瘤细胞、内皮细胞、干细胞等多种类型的细胞,从而达到良好的的基因治疗效果。对于一些较难转染的细胞,如原代细胞、干细胞、不分化的细胞等,使用慢病毒载体,能大大提高目的基因或目的shRNA的转导效率,且目的基因或目的shRNA整合到宿主细胞基因组的几率大大增加,能够比较方便快捷地实现目的基因或目的shRNA的长期、稳定表达。 概念:慢病毒载体是指以人类免疫缺陷病毒-1 (H IV-1) 来源的一种病毒载体,慢病毒载体包含了包装、转染、稳定整合所需要的遗传信息,是慢病毒载体系统的主要组成部分。携带有外源基因的慢病毒载体在慢病毒包装质粒、细胞系的辅助下,经过病毒包装成为有感染力的病毒颗粒,通过感染细胞或活体组织,实现外源基因在细胞或活体组织中表达。 辅助成分:慢病毒载体辅助成分包括:慢病毒包装质粒和可产生病毒颗粒的细胞系。 慢病毒载体包含了包装、转染、稳定整合所需要的遗传信息。慢病毒包装质粒可提供所有的转录并包装RNA 到重组的假病毒载体所需要的所有辅助蛋白。为产生高滴度的病毒颗粒,需要利用表达载体和包装质粒同时共转染细胞,在细胞中进行病毒的包装,包装好的假病毒颗粒分泌到细胞外的培养基中,离心取得上清液后,可以直接用于宿主细胞的感染,目的基因进入到宿主细胞之后,经过反转录,整合到基因组,从而高水平的表达效应分子。 基本原理:慢病毒载体系统由两部分组成,即包装成分和载体成分。
包装成分:由HIV-1基因组去除了包装、逆转录和整合所需的顺式作用序列而构建,能够反式提供产生病毒颗粒所必需的蛋白。包装成分通常被分开构建到两个质粒上,一个质粒表达Gag和Pol蛋白,另一个质粒表达Env蛋白,其目的也是降低恢复成野生型病毒的可能。将包装成分与载体成分的3个质粒共转染细胞(如人肾293T细胞),即可在细胞上清中收获只有一次性感染能力而无复制能力的、携带目的基因的HIV-1载体颗粒。 载体成分:与包装成分互补,即含有包装、逆转录和整合所需的HIV顺式作用序列,同时具有异源启动子控制下的多克隆位点及在此位点插入的目的基因。 为降低两种成分同源重组恢复成野生型病毒的可能,需尽量减少二者的同源性,如将包装成分上5′LTR换成巨细胞病毒(CMV)立即早期启动子、3′LTR换成SV40 polyA等。 一、实验流程(1和2为并列步骤) 1.慢病毒过表达质粒载体的构建 设计上下游特异性扩增引物,同时引入酶切位点,PCR(采用高保真KOD酶,3K内突变率为0%)从模板中(CDNA质粒或者文库)调取目的基因CDS区(coding sequence)连入T载体。将CDS区从T载体上切下,装入慢病毒过表达质粒载体。 2.慢病毒干扰质粒载体的构建 合成siRNA对应的DNA颈环结构,退火后连入慢病毒干扰质粒载体 3. 慢病毒载体的包装与浓缩纯化 制备慢病毒穿梭质粒及其辅助包装原件载体质粒,三种质粒载体分别进行高纯度无内毒素抽提,共转染293T细胞,转染后6 h 更换为完全培养基,培养24和48h后,分别收集富含
实验室细胞株管理规定
细胞株管理规定 一、什么是细胞株? 原代代培养物经第一次传代培养后的细胞,常被称为细胞系(cell line)。由某一细胞系分离出来的、在性状上与原细胞系有一定差异的细胞系,常被称为该细胞系的亚系(subline)。 从一个经过鉴定的细胞系采用选择法或单细胞克隆进一步所得到的细胞群,常被称细胞株(cell strain)。由原细胞株进一步分离培养出的与原株性状不同的细胞群,又被称为亚株(substrain)。 二、细胞株管理规定 对已建立和经过鉴定的细胞株要进行严格管理,规定如下: 1.制度管理规定 A.细胞株应有专人负责保管,并由实验室负责人监督检查。更换保管人时应提 前做好工作交接,确保使用安全。 B.建立细胞株使用登记制度,记录细胞株的编号、名称、来源、冻存数量及日 期、取用数量及日期、剩余数量及保存方法。 C.细胞株长期保存应放置于液氮罐中,短时保存或过渡保存可置于-80℃冰箱 中。 D.保存的细胞株应定期复活、保种,即每半年对没有复苏过的细胞株进行维护, 复苏、培养和保存,并详细记录细胞株状态,如形态、生长速度、抗体分泌情况等。 E.细胞株不得外借,不得随便带出实验室。 F.细胞株的使用、保存、转移和销毁应符合相关部门的有关规定。 2.使用流程规定 A.如有新细胞株进入实验室,需告知实验室负责人并进行冻存保种; B.实验室人员需复苏细胞时,需提前向实验室负责人申请,经批准后方可复苏; C.设存取记录表,实验人员存取细胞时做好记录并把细胞复苏情况反馈至实验
室负责人,实验室负责人需监督实验人员做好细胞株使用记录情况; D.若复苏细胞人员复苏同一株细胞株两次均未成活,需通知负责人进行检查和 复苏,如贮存细胞确实出现问题,则及时清理。 三、细胞保存 A.冻存条件:90%FBS+10%DMSO,4℃冰箱预冷; B.冻存方法: 4 ℃(30-40min)→-20 ℃(1-2h)→-80 ℃(过夜/24 h以上,可短时保存 1-3个月)→液氮(永久保存; C.填写冻存记录表 四、细胞复苏 A.水浴锅37℃预热 B.从细胞冻存管理表中查找并记录所需细胞在液氮罐中的位置。 C.液氮罐中快速取出细胞,迅速放入水浴锅中融化,并持续晃动,避免因受热 不均产生冰晶,损伤细胞,融化时间为1~2min; D.将细胞悬液移至离心管中,加入10倍体积的细胞培养液,混匀; E.1200rpm/min,离心5min F.弃去上清液,加入新的培养液重悬,移至细胞培养瓶中,显微镜下观察细胞 形态,37℃培养箱培养。 G.次日,更换培养液继续培养,待其铺满培养瓶面积80%及以上,可尽心传代。 四、液氮罐的使用 A.开启液氮罐时,戴好防冻棉手套,防止冻伤; B.提前确定细胞保存位置,避免提篮在外停留时间过长; C.填写复苏记录表,负责人做好整理保藏记录,禁止短期内使用同一株细胞的 多管细胞,复苏后使用细胞的同时需补充细胞株库存,写明细胞名称、冻存时间、冻存人等细节。 五、附件 1.细胞冻存记录表 以50L液氮罐所配的6个提篮,每个提篮6层抽屉柜,每个抽屉柜5*5个冻存管。
体外培养细胞的种类和命名
体外培养细胞的种类和命名 体外培养细胞的名称,随培养细胞技术的发展和细胞种类的增多而演变。最早采用的名称为细胞株(Cell strain),以后又出现细胞系(Cell Line)一词,两者曾一度混用致概念不明确,导致文献中也很混乱。我国也曾有类似情况,在我国尚未制定出统一名词前,本书用的名词基本参考Schaeffer,W.I.(1979)和国内有关会议、以及国内外杂志常用名词为准。 (一)初代培养 初代培养又称原代培养,即直接从体内取出的细胞、组织和器官进行的第一次的培养物。一旦已进行传代培养(Subculture)的细胞,便不再称为初代培养,而改称为细胞系。 (二)细胞系 初代培养物开始第一次传代培养后的细胞,即称之为细胞系。如细胞系的生存期有限,则称之为有限细胞系(Finite Cell Line);已获无限繁殖能力能持续生存的细胞系,称连续细胞系或无限细胞系(Infinite Cell Line)。无限细胞系大多已发生异倍化,具异倍体核型,有的可能已成为恶性细胞,因此本质上已是发生转化的细胞系。无限细胞系有的只有永生性(或不死性),但仍保留接触抑制和无异体接种致癌性;有的不仅有永生性,异体接种也有致瘤性,说明已恶性化。这两种不同性质的无限细胞系,在国内外文献中对这些名词的应用上也常不十分严格。为概念上的明确,本书中对有恶性的无限细胞系采用“恶性转化细胞系”一词表示可能更妥。而对那些只具永生性而无恶性的细胞系,则用无限细胞系或转化细胞系即可。当前流传的NIH3T3、Rat-1、10T1/2等均属这类细胞系。 由某一细胞系分离出来的、在性状上与原细胞系不同的细胞系,称该细胞系的亚系(Subline)。 (三)克隆细胞株 从一个经过生物学鉴定的细胞系用单细胞分离培养或通过筛选的方法,由单细胞增殖形成的细胞群,称细胞株。再由原细胞株进一步分离培养出与原株性状不同的细胞群,亦可称之为亚株(Substrain) (四)二倍体细胞 细胞群染色体数目具有与原供体二倍细胞染色体数相同或基本相同(2n细胞占75%或80%以上)的细胞群,称二倍体细胞培养。如仅数目相同,而核型不同的即染色体形态有改变者为假二倍体。二倍体细胞在正常情况下具有限生命期,故属有限细胞系。但随供体年龄和组织细胞的不同,二倍体细胞的寿命长短各异。人胚肺成纤维细胞可传50代土10代,人胚肾只有8~10代,人胚神经胶质细胞15~30代;如从老龄个体取可传50则细胞生存期更短。由不同年龄供体取材建立的二倍体细胞系可供研究衰老之用。为保持二倍体细胞能长期被利用,一般在初代或2~5代即大量冻存作为原种(Stook Cells),用时再进行繁殖,用后再继续冻存,可供长期使用或延缓细胞的衰老。 (五)遗传缺陷细胞 从有先天遗传缺陷者取材(主要为成纤维细胞)培养的细胞,或用人工方法诱发突变的细胞,都属遗传缺欠细胞。这类细胞可能具有二倍体核型,也可呈异倍体。 (六)肿瘤细胞系或株 这是现有细胞系中最多的一类,我国已建细胞系主要为这类细胞。肿瘤细胞系多由癌瘤建成,多呈类上皮型细胞,常已传几十代或百代以上,并具有不死性和异体
表达载体的构建方法及步骤
表达载体的构建方法及步骤 一、载体的选择及如何阅读质粒图谱 目前,载体主要有病毒和非病毒两大类,其中质粒DNA 是一种新的非病毒转基因载体。一个合格质粒的组成要素: (1)复制起始位点Ori 即控制复制起始的位点。原核生物DNA 分子中只有一个复制起始点。而 真核生物DNA 分子有多个复制起始位点。 (2)抗生素抗性基因可以便于加以检测,如Amp+ ,Kan+ (3)多克隆位点MCS 克隆携带外源基因片段 (4)P/E 启动子/增强子 (5)Terms 终止信号 (6)加poly(A)信号可以起到稳定mRNA 作用 选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。如果构建的目 的是要表达一个特定的基因,则要选择合适的表达载体。 载体选择主要考虑下述3点: 【1】构建DNA 重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。【2】.载体的类型: (1)克隆载体的克隆能力-据克隆片段大小(大选大,小选小)。如<10kb 选质粒。(2)表达载体据受体细胞类型-原核/真核/穿梭,E.coli/哺乳类细胞表达载体。
(3)对原核表达载体应该注意:选择合适的启动子及相应的受体菌,用于表达真核蛋白质时注意克服4个困难和阅读框错位;表达天然蛋白质或融合蛋白作为相应载体的参考。【3】载体MCS 中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接,不能产生阅读框架错位。 综上所述,选用质粒(最常用)做载体的5点要求: (1)选分子量小的质粒,即小载体(1-1.5kb)→不易损坏,在细菌里面拷贝数也多(也有大载 体); (2)一般使用松弛型质粒在细菌里扩增不受约束,一般10个以上的拷贝,而严谨型质粒<10个。 (3)必需具备一个以上的酶切位点,有选择的余地; (4)必需有易检测的标记,多是抗生素的抗性基因,不特指多位Ampr(试一试)。(5)满足自己的实验需求,是否需要包装病毒,是否需要加入荧光标记,是否需要加入标签蛋白,是否需要真核抗性(如Puro、G418)等等。 无论选用哪种载体,首先都要获得载体分子,然后采用适当的限制酶将载体DNA 进行切割,获得线性载体分子,以便于与目的基因片段进行连接。 如何阅读质粒图谱 第一步:首先看Ori 的位置,了解质粒的类型(原核/真核/穿梭质粒) 第二步:再看筛选标记,如抗性,决定使用什么筛选标记。 (1)Ampr 水解β-内酰胺环,解除氨苄的毒性。 (2)tetr 可以阻止四环素进入细胞。 (3)camr 生成氯霉素羟乙酰基衍生物,使之失去毒性。 (4)neor(kanr)氨基糖苷磷酸转移酶使G418(长那霉素衍生物)失活
细胞株和细胞系的区别
细胞株和细胞系的区别 摘要“细胞株”和“细胞系”是动物细胞工程中常用的两个名词,但由于概念不清,使用混乱,为中学生物教学带来不必要的困惑。本文拟就这两个名词的历史渊源和现实应用进行讨论。 关键词细胞株细胞系 1 名词的由来: 细胞株(cell strain)最初为W.Earle(1940)所采用,他把自己建立的小鼠结缔组织L系称为“株”,1951年,George Gey把其建立的人体宫颈癌细胞Hela 系称为“株”。1954年,White另外使用了“系”来称呼A Carrel培养的鸡胚心脏的细胞群,细胞系(cell line)由此产生,之后这两个名词长期混用。即使在1979年国际组织培养协会专业术语委员会颁布“专业名词建议”和1984年在宁波召开的全国动物组织和细胞培养会议通过“动物细胞、组织和器官培养技术中的一些术语的译名和解释”之后,到目前为止国内对这两个名词的使用仍是混乱的,不仅在商品名中混用,在专业文献中亦十分严重。 2 名词使用的现实情况 2.1 中学教材定义在人教版高中生物(选修)全一册第70页中,细胞株被定义为经原代培养的组织细胞,培养到第10代左右,度过第一次死亡危机后的细胞群,这种细胞的遗传物质没有发生改变;细胞系则被定义为可以度过第二次死亡危机,传代培养到40~50代的细胞,这部分细胞的遗传物质发生了部分改变并且细胞带有癌变的特点,这种细胞在适宜条件下无限传代。 2.2 文献定义由新鲜组织制成的细胞培养物称原代细胞,这种细胞经首次传代成功后的细胞称做细胞系,可泛指一般可能传代的细胞,由原先存在于原代培养物中的细胞世系(lineages of cells)所组成,这种细胞世系含有多种细胞类型。若用胰蛋白酶等将原代细胞消解分散后,再培养一代,称次代培养。如果不能继续传代或传代有限,可称为“有限细胞系(finite cell line)”或“已建立的细胞
质粒构建流程
质粒构建流程 一、引物设计 1)取得目的基因序列,可选用数据库NCBI 2)软件分析目的基因可用酶切位点。使用primer5分析出序列不包含的酶切位点,即为可用没切位点。 3)选择载体。根据转染细胞和实验室资源,选择合适载体。如pcDNA3.1(+), 4)选择酶切位点。对照目的基因可用酶切位点和载体上的酶切位点,选择二者共有的作为备选。载体上两个酶切位点的距离应有几十bp以上,选实验室常用酶切位点。 5)使用primer5设计目的基因引物,目的产物应包含从启动子到终止子全部碱基。 6)根据选择的酶切位点,查找对应的酶切位点保护碱基,将对应片段添加到设计的引物两端,注意酶切位点的前后顺序。一般选择三个保护碱基。 7)引物设计完成,送公司合成。 二、目的片段获取 1. RNA提取 试剂盒:Bioteke 高纯总RNA快速提取试剂盒离心柱型(裂解液RL 4℃、漂洗液RW -20℃保存) 准备:冰盒、4℃预冷离心机、EP管2套、吸附柱RA一套 操作步骤: 1)将1000μl裂解液RL加入细胞中,混合5min。 2)加200μl氯仿混合,震荡15s,室温孵育3min。 3)4℃,12000rpm离心10min。 4)最上层水相转移至新EP管中(体积约550μl) 5)加入1倍体积(550μl)70%乙醇,混匀 6)全部转移到套收集管的吸附柱RA中,4℃,10000rpm离心45s 7)弃废液,重套收集管,加500μl去蛋白液RE,12000rpm离心45s 8)弃废液,重套收集管,加700μl去漂洗液RW,12000rpm离心60s 9)弃废液,重套收集管,加500μl去漂洗液RW,12000rpm离心60s 10)弃废液,重套收集管,12000rpm空离2min 11)吸附柱放入新EP管,加50μl RNase free water于膜上,室温放置2min 12)4℃,12000rpm离心60s 13)点样:5μl RNA+ 1μl 10×buffer,1.5%琼脂糖凝胶电泳,100V,3min,可见3条亮带。14)-20℃保存 2.RNA反转录 试剂盒:TaKaRa primescript RT reagent kit with gDNA eraser(-20℃保存) 准备:冰盒,②④⑤⑥取出解冻,①③为酶不可取出,预冷离心管 操作步骤: 1)基因组DNA去除(10μl体系) ② 5×gDNA eraser buffer 2μl ① gDNA eraser 1μl
常用细胞库
中国典型培养物保藏中心(也称武汉大学保藏中心)CCTCC https://www.360docs.net/doc/864169286.html,/cctcc/ https://www.360docs.net/doc/864169286.html,/ 中国科学院上海生命科学研究院细胞资源中心 https://www.360docs.net/doc/864169286.html,/ 要国产的,请查一查国内的细胞库 武汉细胞库 https://www.360docs.net/doc/864169286.html,/fzlbc/cell.doc 上海细胞库 https://www.360docs.net/doc/864169286.html,/xibao/catalogue.html 昆明细胞库 https://www.360docs.net/doc/864169286.html,/CELL.HTM 协和医科大学基础医学细胞中心 https://www.360docs.net/doc/864169286.html,/pumc/jiaoxue_hj/zhongdian_sys.htm 成都基因治疗肿瘤药物工程技术研究中心细胞及载体服务 https://www.360docs.net/doc/864169286.html,/cell_catalog.asp 湖南远泰生物技术有限公司生物资源库 https://www.360docs.net/doc/864169286.html,/Source.asp 上海午立生物技术有限公司细胞株 https://www.360docs.net/doc/864169286.html,/cpjs-4.htm 中国医学科学院基础医学研究所细胞中心保藏细胞目录 https://www.360docs.net/doc/864169286.html,/1652-1.doc 要进口的,请查美国和欧洲细胞库: ATCC(细胞株及胚胎干细胞库) 1)下载细胞库目录:https://www.360docs.net/doc/864169286.html,/pdf/tcl.pdf 2)查询细胞株:https://www.360docs.net/doc/864169286.html,/SearchCatalogs/CellBiology.cfm 2)胚胎干细胞库:https://www.360docs.net/doc/864169286.html,/products/cells.cfm CABRI https://www.360docs.net/doc/864169286.html,/HyperCat/cells/all.htm
实验室常用细胞表征及处理方法(实战版)
实验室常用细胞表征及处理方法 一、细胞培养 1.1细胞培养基配方 普通细胞所需要的培养基配方主要为:购买的培养基+10 %血清(小牛或者胎牛)+1%双抗。(常用的双抗及胰酶建议直接购买) 1.2细胞传代 1.2.1洗涤:将培养瓶内的旧培养液吸出,加入2~3ml PBS缓冲液洗涤2-3次,摇晃使其均匀铺满整个平面,清洗细胞,随后吸弃PBS液以除去残留的血清和死细胞。 1.2.2消化:加入适量(盖满细胞层表面即可,25cm2的培养瓶加入0.5-0.6mL 即可,75 cm2加入1.0mL)的0.25%胰蛋白酶液,轻轻摇晃,普通细胞一般需要60s左右,特别难以消化的细胞可以放在37℃孵箱孵育适当的时间,如Hela细胞大约需要30s-40s,翻转培养瓶。然后在倒置相差显微镜下观察细胞消化情况,待细胞开始收缩成圆形、细胞间的连接明显消失,细胞间出现明显的空隙,即可终止消化(若细胞成片收缩,倒去消化液)。若存在细胞团,可以轻轻拍打培养瓶侧壁,尽量消化完全,不然传代后会出现较多的细胞团,再次消化会非常困难。 1.2.3终止消化:在培养瓶中加入1-3ml培养液(胰酶遇血清会终止其活性)以终止消化。用吸管反复吹打洗瓶壁上的细胞,边角部分特别注意多次吹打。可在倒置相差显微镜下观察细胞吹打的程度,(轻轻摇动培养瓶,观察细胞是否完全脱壁),最后形成单细胞悬浮液。 将单细胞悬液移入15ml离心管内,离心1000r×5min,弃去上清液,加入5ml 培养基进行吹打重悬(也可不用离心,直接把单细胞悬液分装于新的培养瓶里,补加适量培养基)。小培养瓶需5ml左右培养基,直径60mm培养皿5-7ml,以上均应视细胞的数目而定,细胞多,可适当加多些培养基)。 1.2.4.传代:取上述步骤所得单细胞悬液,进行细胞悬液计数,然后按所需密度接种到其它培养瓶中。标注上代数、日期及操作人。 注:本实验所需的PBS液、DMEM细胞培养液及0.25%胰蛋白酶液均需37℃预热处理。
常用细胞类型
BHK21细胞:BHK-21细胞即乳仓鼠肾细胞(Baby Hamster Syrian Kidney)。BHK细胞是指幼年叙利亚地鼠肾细胞(BabyHamster Syrian Kidney),1961年建 株。原始的细胞株是成纤维细胞,贴壁依赖性。1963年获得单细胞克隆细胞。后经无数次传代后细胞可悬浮生长,它广泛用于增殖各种病毒,生产兽用疫苗。最常用的是BHK21的一个亚克隆细胞,即克隆13或C13 CHO细胞:(Chinese Hamster Ovary,CHO) 中国仓鼠卵巢细胞 该细胞具有不死性,可以传代百代以上,是生物工程上广泛使用的细胞。另外CHO细胞在基因工程使用中还有一个优点,该细胞属于成纤维细胞(fibroblast),是一种非分泌型细胞,它本身很少分泌CHO内源蛋白,因此对目标蛋白分离纯化工作十分有利。可形成有活性的二聚体(如白介素2),具有糖基化的功能(如EPO),CHO为表达复杂生物大分子的理想宿主。 由于该细胞存在遗传缺陷,无脯氨酸合成基因,不能将谷氨酸转变为谷氨酸--半醛,培养过程中需在培养基中添加L-脯氨酸才能生长。并且由于该细胞已经霍乱毒素适应,形态学有所改变。最初细胞为贴壁型细胞,经多次传代筛选后,也可悬浮生长。 工业生产上应用较多的是CHO-K1细胞,为转化细胞系,细胞染色体分布频率是2n=22,系亚二倍体细胞。ATCC保存CHO-K1细胞株,编号为CCL-61,被广泛地用于重组DNA蛋白的表达。 MDCK细胞:(Madin-Daby canine cells)犬肾上皮连续细胞系 病毒感染率高,增值快,不易变易,被公认为最适于甲乙型流感病毒疫苗生产的三种细胞系之一 MDBK细胞:牛肾细胞 VERO细胞:(Verda Reno)非洲绿猴肾细胞 1. 用于检查大肠杆菌毒素。大肠杆菌毒素最初就因此细胞系而被命名为Vero毒素,后来被称为志贺菌素样毒素,因为它与在痢疾志贺菌中分离出来的志贺菌素很相似。 2.作为培养病毒的细胞宿主。比如:测定某种药物对病毒复制速度的影响,检验是否存在狂犬病毒或者为了研究目的培养病毒。 3.作为真核寄生虫的宿主细胞,尤其是锥体虫属。 4.用于疫苗生产中的病毒培养基质细胞,是在限定代次内不致癌的异倍体细胞,WHO批准可以用于人类疫苗的生产基质。 EB66细胞:鸭胚胎干细胞 基因稳定,无菌,易接种,全悬浮,无血清培养,规模化
载体构建的基本步骤
载体构建的基本步骤 Company Document number:WUUT-WUUY-WBBGB-BWYTT-1982GT
载体构建 一、原理 依赖于限制性核酸内切酶,DNA连接酶和其他修饰酶的作用,分别对目的基因和载体DNA进行适当切割和修饰后,将二者连接在一起,再导入宿主细胞,实现目的基因在宿主细胞内的正确表达。 二、操作步骤 1、摇菌(制作感受态细胞备用) 取装有液体培养基的3ml试管两支(依情况而定),每管加40-100μl菌种,过夜摇。 2、提质粒(也就是载体) 依照提质粒试剂盒中的说明书操作(根据情况最后一步洗脱时可以多洗1-2次)。 3、酶切(双酶切产生粘性末端) 反应所需试剂体积(单位:ul) 质粒 10 所需内切酶反应缓冲液 2 所需限制性内切核酸酶 1 H2O 7 将加好的EP管置于37℃保温1-2h。(依照提酶切的具体步骤操作;为了达到最佳酶切的效果,最好根据所选用的酶确定所需要的反应温度) 4、电泳检测 将酶切产物进行琼脂糖凝胶电泳,检测酶切是否成功。 回收胶:琼脂糖与缓冲液一比一制胶,经过切胶回收目的产物,也有目的产物纯化的功能; 检测胶:琼脂糖与缓冲液一比二制胶,为了检测目的条带与预期是否相符。
切胶回收与产物纯化是差不多的过程,所达到的目的是一样的:切胶回收也是一种纯化过程,它能去除非目的片段,然后用回收试剂盒进行纯化,能将很不纯的DNA溶液纯化;产物纯化是将较纯的DNA溶液进一步除去多余的杂质,用纯化试剂盒,你会发现纯化试剂盒和回收试剂盒的步骤几乎一样。 5、载体与目的基因连接 如果电泳检测酶切成功的话,则仔细将所需的片段切割下来,将胶体回收(依照胶回收试剂盒说明书操作);之后将回收的片段和载体连接。置于温箱,12-16℃,保温8-16h 6、转化(连接产物转化到感受态细胞中) 依照转化具体操作步骤做感受态,将上述连接产物进行转化实验,涂板培养,37℃, 12-16h。 7、单克隆检测 (1)挑单克隆 先将AMP从冰箱中取出,待融化后,在3ml装有LB液体培养基的试管中加入3μL的 AMP,用枪头混匀;取 mlEP管5支(依情况可以多挑几管),给每支管中加500μL上述培养液,然后用接种环(或黄枪头)挑单克隆,挑完后用枪吹打;之后,将挑好的菌摇4-5小时,至混浊即可。 (2)单克隆检测 以每管摇好的菌液为模板,以原有的引物进行PCR,然后将PCR产物跑电泳,观察电泳图像中那几管的条带正确,将正确条带相对应管的菌液再抽取100μ
细胞株和细胞系
细胞株和细胞系 摘要“细胞株”和“细胞系”是动物细胞工程中常用的两个名词,但由于概念不清,使用混乱,为中学生物教学带来不必要的困惑。本文拟就这两个名词的历史渊源和现实应用进行讨论。 关键词细胞株细胞系 1 名词的由来 细胞株(cell strain)最初为W.Earle(1940)所采用,他把自己建立的小鼠结缔组织L系称为“株”,1951年,George Gey把其建立的人体宫颈癌细胞Hela 系称为“株”。1954年,White另外使用了“系”来称呼A Carrel培养的鸡胚心脏的细胞群,细胞系(cell line)由此产生,之后这两个名词长期混用。即使在1979年国际组织培养协会专业术语委员会颁布“专业名词建议”和1984年在宁波召开的全国动物组织和细胞培养会议通过“动物细胞、组织和器官培养技术中的一些术语的译名和解释”之后,到目前为止国内对这两个名词的使用仍是混乱的,不仅在商品名中混用,在专业文献中亦十分严重。 2 名词使用的现实情况 2.1 中学教材定义在人教版高中生物(选修)全一册第70页中,细胞株被定义为经原代培养的组织细胞,培养到第10代左右,度过第一次死亡危机后的细胞群,这种细胞的遗传物质没有发生改变;细胞系则被定义为可以度过第二次死亡危机,传代培养到40~50代的细胞,这部分细胞的遗传物质发生了部分改变并且细胞带有癌变的特点,这种细胞在适宜条件下无限传代。 2.2 文献定义由新鲜组织制成的细胞培养物称原代细胞,这种细胞经首次传代成功后的细胞称做细胞系,可泛指一般可能传代的细胞,由原先存在于原代培养物中的细胞世系(lineages of cells)所组成,这种细胞世系含有多种细胞类型。若用胰蛋白酶等将原代细胞消解分散后,再培养一代,称次代培养。如果不能继续传代或传代有限,可称为“有限细胞系(finite cell line)”或“已建立的细胞
载体构建
载体构建
载体构建分为以下步骤: 1.载体的选择和引物设计以扩增目标基因 2.目标片段的PCR扩增 3.载体和目标片段的限制性酶切 4.连接转化 5.挑取克隆提质粒 6.单克隆的验证及送样测序。 载体的选择 实验室常用的载体有pUC19(扩繁目标片段),pet28a(原核表达载体Kna+),pGEX-4T-1(原核表达载体Amp+),pCI-NEO(真核表达载体Amp+),pCDNA3.1(真核表达载体Amp+),pLKO.1(shRNA构建Amp+)。根据所要构建的质粒目的的差异以及载体和目标片段的酶切位点分析结果来选择载体。举例如下: 实验目的是将MYC基因插入到载体中,转染进细胞中表达。应选用真核表达载体pCI-NEO 或pCDNA3.1。 引物设计 引物设计的目的是将目标片段扩增出来以便与载体连接,所以在引物的两端应人为加上酶切位点,酶切位点前加上保护碱基。在选择酶切位点是应注意,选择的应该是目的基因片段中没有而载体上有的限制性内切酶,防止目标片段被切不完整,也便于和载体连接。 载体构建之后我们的目的是要表达,所以应该加入标签以便western blot检测蛋白是否表达。常用的标签有Flag标签、6XHis标签、HA标签和Myc标签。 Flag标签: D Y K D D D D K GAT TAC AAG GAT GAC GAC GAT AAG 6XHis标签: H H H H H H CAC CAC CAC CAC CAC CAC HA标签: Y P Y D V P D Y A TAC CCA TAC GAT GTT CCA GAT TAC GCT Myc标签: E Q K L I S E E D L 这些标签被表达成氨基酸后,特定的氨基酸序列会与相应抗体结合,在western blot时可被检测。因此,在设计引物时,这些核酸序列应位于起始密码子ATG之后。
细胞培养实验室的设备配置及用途
细胞培养实验室的设备配置及用途 1. 超净工作台 目前绝大部分细胞实验室使用超净工作台实现无菌操作,具有操作简单、安装方便、占用空间小且净化效果很好。安徽人和净化为您介绍两种主要超净工作台——侧流式(垂直式)和外流式(水平层流式)。工作原理一般是将室内空气经粗过滤器初滤,由离心风机压入静压箱,再经高效空气过滤器精滤,由此送出的洁净气流以一定的均匀的断面风速通过无菌区,从而形成无尘无菌的高洁净度工作环境。 (1)侧流式工作台:空气净化后的气流由左或右侧通过工作台面流向对侧,也有从上向下或从下向上流向对侧,形成气流屏障保持工作区无菌,工作台结构为封闭式; (2)外流式工作台:净化后的空气面向操作者流动,因而外方气流不致混入操作,工作台结构为开放式,但进行有害物质实验操作对操作者不利。超净工作台应需要定期请有关部门检查洁净度,符合要求的超净工作台其洁净度应达到100级,用尘埃粒子计数仪检测粒径≤5μm的尘埃粒子数量不应超过3.5个/L;空气流量应控制在0.75-1.0m3/s;细菌菌落数平均<1个,根据无菌状况必要时需要置换过滤器。 2. 显微镜 倒置式显微镜是细胞培养实验室日常工作常规必备设备,主要用于日常了解细胞的生长情况并观察有无污染发生。如资金允许,建议选用配置有照相系统的高品质相差显微镜、解剖显微镜、荧光显微镜、录像系统或缩时电影拍摄装置等,可随时拍摄并记录细胞生长情况。 3. 培养箱 体外培养的细胞和体内细胞一样,需要在恒定的温度下生存,一般最适生长温度为37℃,温差变化不应超过±0.5℃。温度升高2℃时,变不利于细胞生存,温度达到40℃以上细胞将很快死亡。因此,可精确控温的恒温培养箱、CO2培养箱是最佳选择。
CRISPRCas9基因敲入试验步骤(三)donor载体构建载体构建
CRISPRCas9基因敲入试验步骤(三)donor载体构建载体构 建 1、质粒骨架的选择 CRISPR/Cas9质粒按编辑细胞类型可分为八种,Mammalian 、Bacteria 、Drosophila 、Plant、C. elegans 、Yeast 、Zebrafish 、Xenopus,根据质粒所携带的编辑基因的不同,可分为野生型的cas9、突变型的cas9、cpf1、C2c2(可剪切RNA)。当然,还有一些筛选标记,如puro、GFP、RFP、mcherry、潮霉素等等,我们可以根据自己的需求选择自己合适的质粒。这里故意还掉了一种,最具有争议的Ngago,本楼主也重复过这个试验,也和大多数重复的人一样,GFP验证试验时,看到荧光显著减弱(本人还特意构建了一种Ngago载体,效果比韩老师的效果好很多),但是当时没有验证这个减弱是切割还是敲低,非常遗憾自己想法太简单,以先入为主认为是切割而没有去验证;最终结果是刘东老师使用Ngago发现了有表型(斑马鱼的眼睛上有缺陷),这个就是很强的提示,需要去做下一步,尽管基因组上没有被切割,有表型,那势必会去看该基因表达的数据,最后才有这篇文章。刘东老师运气也很好,knock down 有表型,如果没表型,估计也会放弃。这里就说到这,有不懂的可以留言询问。。具体分类在addgene上有,网址/。
2、酶切位点的选择 插入sgRNA的酶切位点,质粒基本为这两种,BsmbI和Bbsi,具体可以参见该质粒的protocol,后续的连接、转化、验证protocol里面有,我就不啰嗦了。哦还忘了一件事,使用snapgene这个软件可以很好地看质粒图谱,非常方便,强烈推荐!!以慢病毒载体V2为例具体说明3、同源臂的设计 同源臂我们一般都是在切割位点的两端各选一段,长度大约1kb-2kb,效率还可以。当然了,有人也用40-100bp做了也做成功了,但基本原则是越长效率越高。1kb-2kb效率已经很高,所以这个最合适。有相关文献支持,自己Pubmed查看。 4、启动子、目的基因及其终止子的扩增 这里也没什么可说的,启动子在我们选择质粒的时候就决定了,当然我们还可以改造以提高sgRNA的表达效率,这个时候我们可以通过方法筛选出该物种的启动子,替换上去就ok了。 目的基因的话,如果我们要做敲除,基本就是根据基因组,在外显子区域设计sgRNA序列,切割后以非同源性末端接合(Non-homologous end joining, NHEJ),造成变异而使该基因失去功能。当然还有一种修复方式,同源性重组(Homologous recombination,HR)同源性重组修复是利用细胞内的染色体两两对应的特性,若其中一条染色体上的