autodock中文版使用说明

AUTODOCK 4.0 使用中文版
国立东华大学生物技术研究所宣戴维教授实验室制作
版面作者:林世浤如有问题请回信指教e-mail: jolin31912811@https://www.360docs.net/doc/f23884562.html,
AutoDock 是一套做docking的软件,主要是做小分子与大分子(receptor)键结的预测,使用autodock4.0图形接口,需安装四项程序:
PS.找机会会把安装程序放上去
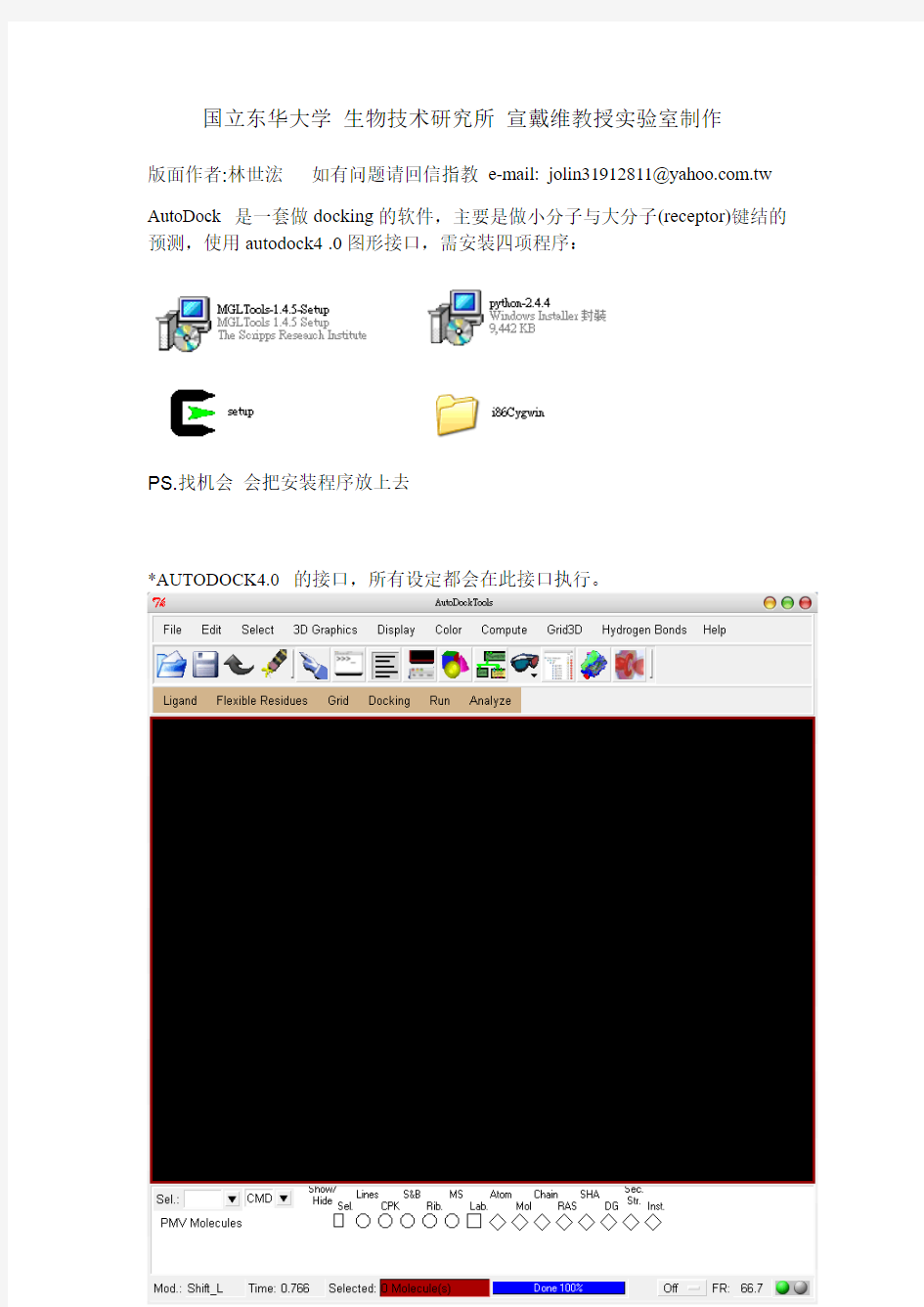
*AUTODOCK4.0 的接口,所有设定都会在此接口执行。
(一)、
1. File -> Read Molecule (选择分子)
2. Select -> Select From String(要标定水分子) -> Add
3. Edit -> Delete -> Delete AtomSet -> Continue(将水分子消除)
4. Edit -> Hydrogen -> Add -> OK
5.File -> Save -> Wrtie PDB
6. Display -> Show/Hide Molecule (先隐藏起来,接下来要设定ligand)
(二)、/* Ligand */
1.Ligand -> input -> open -> (檔名.pdb)
2.Ligand -> Output -> Save as pdbqt
(三) autodock4.0新增的东西(Flexible Residues)
A:文献上没有提到,接合位置的重要胺基酸,就直接跳过
File -> Save -> Write PDBQT
B:文献上提到,接合位置的重要胺基酸,就直接寻找
1.Flexible Residues -> Input -> Choose Macromolecule
2.Edit -> Hydrogens -> Merge Non-Polar -> CONTINUE
3.Select -> Select From String /* Residue打上ARG8 -> Add */
4.Flexible Residues -> Choose Torsions in Currently Select Resdues…
5.Flexible Residues -> Oput -> Save Rigid pdbqt
(四) /* Grid */
1.Grid -> macromolecule -> (檔名.pdbqt) (protein)
2.Grid -> Set Map Type -> (檔名.pdbqt) (ligand)
3. Grid -> Grid Box (把所有参数调整好) ->
4. File(Grid Options) -> Close saving current
5. Grid -> Output -> Save GPF (檔名.gpf)
6. /* Edit GPF (是在看档案.gpf,的文件) */
(五) /* Run */
1.Run -> Run AutoGrid
https://www.360docs.net/doc/f23884562.html,umch (autodcok3要改成autodcok4才能执行)
PS.执行结束后,有几个档案出来,之后要执行分子间的力场。
(六) /* Docking */
1.Docking -> Mocromolecule -> (檔名.pdbqt) (protein)
2.Docking -> Ligand -> (檔名.pdbqt) (ligand)
3. Docking -> Output -> (看你要选择哪一个算法,存取它)
4. /* Edit DPF (是在看档案.dpf,的文件) */
(七) /* Run */
1.Run -> Run AutoDock
https://www.360docs.net/doc/f23884562.html,umch (autodcok3要改成autodcok4才能执行)
(八) /* Analyze */
A:
1.Analyze -> Docking -> (檔名.dlg)
2.Analyze -> Conformations -> Play (可让ligand变更位置,ps.已对结好的位置)
3.Macromolecule -> (檔名.pdbqt) (protein)
(八) /* Analyze */
B:
1. Analyze -> Grids -> Open Other [选择步骤(五)中的九个由autogrid 产生的文件,每一个文件都代表不同的力场]
2.Analyze -> Conformations -> Play (ligand可配合任一力场,做autodock所产生出的九个docking位置,并判断它们的关系)
(八) /* Analyze */
C:
1. Analyze -> Conformations -> Extract Histogram… (计算分子对接,ligand与protein bind site的分数高低并由高到低)