羟基在苯酚亲电取代反应中的定位作用
苯酚硝化反应的产物

苯酚硝化反应的产物
苯酚硝化反应是一个典型的亲电芳香取代反应,其产物主要取决于反应的条件,尤其是温度和硝酸与硫酸的比例。
苯酚分子上有一个羟基,这个羟基是一个吸电子基团,能够增强苯环上的电子密度,使得苯酚比苯更容易发生亲电取代反应。
在温和的硝化条件下(如低温和低浓度的硝酸),苯酚的硝化反应主要产生单硝基苯酚(C6H5ONO2)。
单硝基苯酚有两种异构体,即邻位(2-硝基苯酚)和对位(4-硝基苯酚)。
由于羟基的定向效应(羟基是一个吸电子基团,但同时也是一个共振稳定的基团),它倾向于引导硝酸根进入邻位或对位,因此在温和条件下主要产生4-硝基苯酚。
当反应条件变得更加剧烈时,如提高温度或使用更浓的硝酸和硫酸混合物,苯酚可以进一步硝化形成二硝基苯酚或三硝基苯酚。
二硝基苯酚有三种异构体,即2,4-二硝基苯酚、2,5-二硝基苯酚和2,6-二硝基苯酚,其中2,4-二硝基苯酚最为稳定。
在极端条件下,苯酚可以硝化为三硝基苯酚(C6H2(NO2)3OH),但这种产物不稳定,易发生爆炸。
苯酚硝化反应的一般方程式如下:
C6H5OH + HNO3→C6H4(ONO2)OH + H2O
苯酚硝化反应需要在酸性介质中进行,通常使用硫酸作
为催化剂和吸收剂。
反应过程中,硝酸根(NO2+)作为亲电试剂攻击苯环,形成硝基苯酚,并释放出水分子。
由于硝基苯酚类化合物具有较高的毒性和环境危害性,因此在处理和合成过程中需要采取严格的安全措施。
同时,硝化反应的副产品和未反应的硝酸、硫酸需要妥善处理,避免对环境造成污染。
苯环上亲电取代反应的定位规律
定位规律的理论解释
H 是交替极化, 是交替极化,即使甲基的邻位和对位上 H C H 电子云密度增加的更多些, 电子云密度增加的更多些,量子化学计 甲苯中各碳上电子云密度分布如图。 算,甲苯中各碳上电子云密度分布如图。 O 甲苯中各碳上电子云密度分布如图 O 所以亲电试剂主要进攻邻位和对位。 所以亲电试剂主要进攻邻位和对位。 从反应历程和σ 络合物的稳定性看: 从反应历程和σ-络合物的稳定性看: O H H H 慢 δ E E E +E
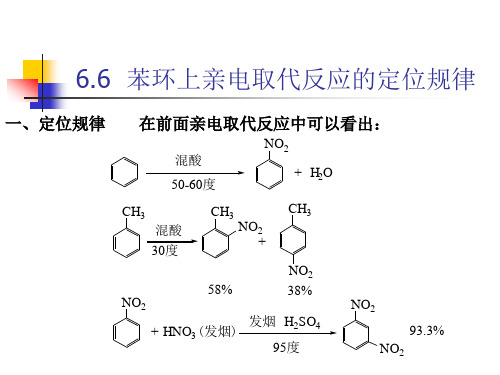
一、定位规律
由此可以看出,当芳环上已有取代基时,新引入基团是否容易, 由此可以看出,当芳环上已有取代基时,新引入基团是否容易, 进入哪个位置,主要由芳环原来取代基的性质所决定。 进入哪个位置,主要由芳环原来取代基的性质所决定。我们把 芳环上原来的取代基叫做定位基 定位基。 芳环上原来的取代基叫做定位基。而把定位基支配新引入基团 进入芳环的位置和定位能力的大小称为定位规律或定位效应 定位规律或定位效应。 进入芳环的位置和定位能力的大小称为定位规律或定位效应。 第一类定位基称为邻 对位定位基, 第一类定位基称为邻、对位定位基,它使新引入的基团主要进入 定位基的邻、对位。除卤素之外,它们都是供电子基, 定位基的邻、对位。除卤素之外,它们都是供电子基,使芳环 上电子云密度增加,活化芳环,亲电取代活性大于苯。 上电子云密度增加,活化芳环,亲电取代活性大于苯。它们定 位能力强弱的次序为: >-OH> 位能力强弱的次序为:-O->-N(CH3)2>-NH2>- > >-NHCOCH3>-R>- >-OCOCH3>- 6H5>-F> >-C -OCH3>- >- > >-Br>- -Cl>- >- >- >-I 可以看出,这些基团与芳环相连的原子(除 和 外 都有未共用 可以看出,这些基团与芳环相连的原子 除R和Ph外)都有未共用 电子对,可以芳环发生P- 超共轭,使芳环上电子云密度增加。 电子对,可以芳环发生 -π超共轭,使芳环上电子云密度增加。
请用共振论解释羟基对苯环上亲电取代反应定位作用的影响

请用共振论解释羟基对苯环上亲电取代反应
定位作用的影响
1 羟基对苯环上亲电取代反应定位作用
共振理论是一种电化学和化学反应的理论,它是由Friedel一家在20世纪50年代提出的,它解释了苯环芳香环中取代反应中定位作用的影响。
羟基可以在芳香环中影响取代反应的定位,从而使反应更有效地发生。
羟基对介导亲电取代反应的定位作用是通过共振效应发挥的。
羟基可以与亲电基的电子对形成共振来增强取代反应的机会。
因为羟基可以促进电子的移动,增加取代反应发生的机会,所以反应会变得更加有利。
共振效应是一种通过分子结构之间的电子色散来影响取代反应定位作用的情况。
羟基对于亲电取代反应的定位作用非常重要,它使取代反应变得更加高效。
然而,由于羟基存在大量电子,它也会使化合物稳定受到影响,所以反应位点的位置非常重要。
从这一点来看,羟基可以改变取代反应的定位和产物的稳定性。
总的来说,共振理论有助于解释羟基对苯环上亲电取代反应定位作用的影响。
由于它可以增加取代反应发生的机会,从而提高反应的效率。
但也有可能改变取代反应的定位和产物的稳定性,因此在应用这些反应时要特别注意。
苯环定位规则

苯环上原有的取代基对新导入取代基有影响,这种影响包括反应活性和进入位置两个方面。
通常,苯环上原有的第一取代基称为定位基,从大量实验事实的分析总结中发现,定位基的定位作用遵循一定的规律,这一规律称为苯环上亲电取代反应定位规律(又称定位规则)。
下面分别讨论定位基的类型;定位规则的理论解释;二元取代苯的定位规律;定位规律的应用。
(一)定位基的类型1.邻、对位定位基。
这类定位基的结构特征是定位基中与苯环直接相连的原子不含不饱和键(芳烃基例外),不带正电荷,且多数具有未共用电子对。
常见的邻、对位定位基及其反应活性(相对苯而言)如下:强致活基团:―NH2(―NHR,―NR2),―OH中致活基团:―OCH3(―OR),―NHCOCH3(-NHCOR)弱致活基团:―ph(―Ar),―CH3(-R)弱致钝基团:―F,―Cl,―Br,―I这类定位基多数使亲电取代反应较苯容易进行,但卤素例外。
2.间位定位基。
这类定位基的结构特征是定位基中与苯环直接相连的原子一般都含有不饱和键(-CX3例外)或带正电荷。
常见的间位定位基及其定位效应从强到弱顺序如下:―N+H3,―N+R3,―NO2,―CF3,―CCl3,―CN,―SO3H,―COH,―COR,―COOH,―COOR,―CONH2等。
这类定位基属致钝基团,通常使苯环上亲电取代反应较苯难进行,且排在越前面的定位基,定位效应越强,反应也越难进行。
(二)定位规则的理论解释苯环上的取代反应是亲电取代反应。
因此,从反应活性的角度分析,凡有助于提高苯环上电子云密度的基团,就能使苯环活化,反应活性提高;反之,凡是使环上电子云密度降低的基团,就能使苯环钝化,反应活性降低。
从反应位置的角度分析,当苯环上没有取代基时,环上六个碳原子的电子云密度是均等的;但当苯环上有取代基时,由于取代基的电子效应沿着苯环共轭体系传递。
在环上出现了出现了电子云密度的疏密交替分布现象。
第二个取代基总是进入苯环上电子云密度相对较大的部位,从而使这些碳原子上的取代物占了多数。
苯环上亲电取代反应的定位规律

之阳早格格创做苯环上亲电与代反应的定位程序基础观念:定位基:正在举止亲电与代反当令,苯环上本有与代基,不但是效率着苯环的与代反应活性,共时决断着第二个与代基加进苯环的位子,即决断与代反应的位子.本有与代基称干定位基.一、二类定位基正在一元与代苯的亲电与代反应中,新加进的与代基不妨与代定位基的邻、间、对于位上的氢本子,死成三种同构体.如果定位基不效率,死成的产品是三种同构体的混同物,其中邻位与代物 40%(2/5)、间位与代物 40%(2/5)战对于位与代物 20%(1/5).本量上惟有一种或者二种主要产品.比方百般一元与代苯举止硝化反应,得到下表所示的截止:排正在苯前里的与代硝化产品主假如邻位战对于位与代物,除卤苯中,其余与代苯硝化速率皆比苯快;排正在苯后里与代硝化产品主假如间位与代物,硝化速率比苯缓得多.归纳洪量真验截止,根据苯环上的与代基(定位基)正在亲电与代反应中的定位效率,普遍分为二类:第一类定位基又称邻对于位定位基:—O-,—N(CH3)2,—NH2,—OH,—OCH3,—NHCOCH3,—OCOCH3,—F,—Cl,—Br,—I,—R,—C6H5等.第二类定位基又称间位定位基:—N+(CH3)3,—NO2,—CN,—SO3H,—CHO,—COCH3,—COOH,—COOCH3,—CONH2,—N+H3等.二类定位基的结构特性:第一类定位基与苯环间接贯串的本子上惟有单键,且普遍有孤对于电子或者是背离子;第二类定位基与苯环间接贯串的本子上有沉键,且沉键的另一端是电背性大的元素或者戴正电荷.二类定位基中每个与代基的定位本领分歧,其强度序次近似如上列程序.苯环上亲电与代反应的定位程序二、定位程序的电子表里阐明正在一与代苯中,由于与代基的电子效力沿着苯环共轭链传播,正在环上出现了电子云稀度较大战较小的接替分集局里,果而环上诸位子举止亲电与代反应的易易程度分歧,出现二种定位效率.也不妨从一与代苯举止亲电与代反应死成的中间体σ络合物的相对于宁静性的角度举止观察,当亲电试剂 E+打击一与代时,死成三苯σ络合物:Z 分歧,死成的三种σ 络合物碳正离子的宁静性分歧,出现了二种定位效率.1.第一类定位基对于苯环的效率及其定位效力以甲基、氨基战卤素本子为例证明.甲基正在甲苯中,甲基的碳为 sp3纯化,苯环碳为 sp2纯化,sp2纯化碳的电背性比 sp3纯化碳的大,果此,甲基表示出供电子的诱导效力(A).其余,甲基 C—H σ 键的轨讲与苯环的π 轨讲产死σ—π 超共轭体系(B).供电诱导效力战超共轭效力的截止,苯环上电子稀度减少,更加邻、对于位减少得更多.果此,甲苯举止亲电与代反应比苯简单,而且主要爆收正在邻、对于位上.亲电试剂 E+打击甲基的邻、间、对于位子,产死三种σ 络合物中间体,三种σ 络合物碳正离子的宁静性可用共振纯化体表示:打击邻位:打击对于位:打击间位:亲电试剂打击苯死成的σ 络合物的碳正离子也不妨用共振纯化体表示:苯环上亲电与代反应的定位程序隐然,共振纯化体Ⅰ战Ⅱ比Ⅲ宁静,果为Ⅰc战Ⅱb的正电荷正在有供电基的叔碳上,较分别.而正在Ⅲ中,正电荷皆分集正在仲碳上,不宁静.所以甲基是邻对于位定位基.共振纯化体Ⅲ比Ⅳ宁静,虽然正在Ⅲ战Ⅳ中的共振极限结构式皆是正电荷分集正在仲碳上,但是甲基有供电性,使Ⅲ的正电荷不妨分别正在环战甲基上,果此,甲基活化了苯环. 从共轭效力战共振论二种瞅面分解、观察甲苯的亲电与代反应,皆得出甲基是第一类定位基、有活化苯环效率的普遍论断.氨基正在苯胺中,N—C 键为极性键,N有吸电子的诱导效力(C),使环上电子稀度缩小;但是共时氮本子有孤对于电子,与苯环产死供电的p—π共轭效力(D),使环上电子稀度减少:正在那里,共轭效力大于诱导效力,所以概括效力使是环上电子稀度减少,更加是氨基的邻位战对于位减少更多.果此,苯胺举止亲电与代反应比苯更简单,且主要爆收正在氨基的邻、对于位上.观察死成的中间体σ络合物碳正离子的宁静性也得到共样的论断.(3)卤本子卤本子比较特殊,是一类使苯环钝化的第一类定位基.以氯苯为例,正在氯苯中氯本子是强吸支电子基,强的吸电子诱导效力使苯环电子稀度落矮,比苯易举止亲电与代反应.但是氯本子与苯环又有强的供电的 p-π 共轭效力(C的2p轨讲与 Cl 的 3p 轨讲产死 p-π 共轭体系,不 C 的 2p 轨讲与 N 的 2p 轨讲产死的 p-π 共轭体系灵验),使氯本子邻、对于位上电子稀度缩小得已几,果此表示出邻对于位定位基的本量.2.第二类定位基对于苯环的效率及其定位效力以硝基苯为例证明.正在硝基苯中,硝基存留着吸电子的诱导效力(E),还存留着吸电子的π-π共轭效力(F):那二种电子效力皆使苯环上电子稀度落矮,亲电与代反应比苯易;共轭效力的截止,使硝基的间位上电子稀度落矮得少些,表示出间位定位基的效率.亲电试剂打击硝基苯时,产死邻、间、对于三种σ 络合物中间体:打击邻位:打击对于位:打击间位:共振纯化体Ⅲ比Ⅰ战Ⅱ宁静,果为正在Ⅰ战Ⅱ中有正电荷分集正在有强吸电子基团的叔碳上的极限结构式Ⅰc战Ⅱb不宁静.果此,硝基是第二类定位基,与代反应爆收正在间位上.共振纯化体Ⅲ有强吸电子基团,与相映的苯的共振纯化体相比,Ⅲ不宁静.果此,硝基表示出钝化苯环的效率.苯环上亲电与代反应的定位程序三、对于邻、对于位产品比率的效率果素1.空间效力环上有邻对于位定位基存留时,死成邻位战对于位产品的比率与定位基战新加进基团的体积有闭系.那二种基大众积越大,空间位阻越大,邻位产品越少.烷基苯的硝化反应随着烷基的体积删大,邻位硝基苯的比率缩小.烷基硝化反当令同构体分集苯环上本有定位基稳定,随着加进基大众积删大,邻位同构体的比率也缩小.如表所示.甲基苯烷基化时同构分集2.反应温度的效率反应温度分歧,邻、对于位同构体的比率分歧.如3.催化剂的效率利用新颖催化技能,不妨统造与代基的定位效率,如使用有择型催化效率的分子筛催化乙苯的乙基化,不妨得到下采用性的对于二乙苯.工业上便是用分子筛催化合成对于二乙苯.后者催化脱氢,得到接联散苯乙烯的共散单体对于二乙烯基苯:甲苯与丙烯烷基化反应,使用分歧孔径的分子筛催化剂,分别得到间甲基同丙苯战对于甲基同丙基苯.已应用于工业死产拆置上.间甲基同丙基苯战对于甲基同丙基苯分别是造备间甲基苯酚战对于甲基苯酚的本料.四、二元与代苯的定位程序当苯环上有二个与代基时,第三个与代基加进苯环的位子,主要由本去的二个与代基的本量决断.大概上道,苯环上有二个与代基时,有三种定位情况.苯环上亲电与代反应的定位程序1.苯环上本有二个与代基对于引进第三个与代基的定位效率普遍,第三个与代基加进苯环的位子便由它们共共定位.比方,下列化合物引进第三个与代基时,第三个与代基主要加进箭头所示的位子:2.苯环上本有二个与代基,对于加进第三个与代基的定位效率纷歧致,二个与代基属共一类定位基,那时第三个与代基加进苯环的位子主要由定位效率强的与代基所决断.如果二个与代基定位效率强度较小时,得到二个定位基定位效率的混同物:3.苯环上本有二个与代基对于引进第三个与代基的定位效率纷歧致,二个与代基分歧类定位基时,那时第三个与代基加进苯环的位子主要由第一类定位基定位:正在思量第三个与代基加进苯环的位子时,除思量本有二个与代基的定位效率中,还该当思量空间位阻,如3-乙酰氨基苯甲酸的 2 位与代产品很少.五、定位程序正在有机合成上的应用应用定位程序不妨采用可止的合成门路,得到较下的产率战预防搀纯的分散历程.比方由甲苯合成间硝基苯甲酸,应采与先氧化后硝化的步调:由对于硝基甲苯合成2,4-二硝基苯甲酸,其合成门路犹如下二条:隐然第一条合成门路较合理,不妨简化分散步调,共时硝化一步反应较第二条门路的硝化一步反应易举止,果为二个与代基(—CH3,—NO2)的定位效率是普遍的. 定位程序只适用于能源教统造的反应.比方,叔丁苯正在FeCl3 催化下,与叔丁基氯反应死成对于二叔丁基苯:苯环上亲电与代反应的定位程序那与定位程序普遍,但是用过量的AlCl3为催化剂,则死成1,3,5-三叔丁基苯:那是果为正在过量强酸效率下,烷基化战脱烷基化完毕仄稳,邻、对于位烷基化快,脱烷基化也简单;间位烷基化缓,脱烷基化也较易,末尾形成热力教上宁静的均三叔丁基苯.六、闭键词汇定位基,定位程序,二类定位基,定位程序的本量阐明,二元与代苯的定位程序,定位程序的应用。
定位效应的解释

同学们,大家好。
今天要讲的是定位效应的解释。
通过上节课的学习,我们已经知道,有些基团会使苯环的亲电取代反应活性增大,称为活化基;有些基团会使苯的亲电取代反应活性减小,称为钝化基;苯环上的基团还会影响取代位置,根据定位效果分为邻对位定位基和间位定位基。
苯环上原有基团为什么会影响亲电取代活性和取代位置呢?今天我们就来分析并解释这一问题。
大家都知道,苯亲电取代时,亲电试剂靠近苯环生成σ-络合物是整个反应的决速步骤。
同样,取代苯反应的决速步骤也生成σ-络合物,如图,决速步骤中苯与亲电试剂的成键能力与苯环上电子密度有关。
若原有基团是供电子基,苯环电子密度增大,容易受到亲电试剂的进攻,则亲电取代活性增大,该基团就是活化基。
若原有基团是吸电子基,会使苯环电子密度减小,吸引亲电试剂的能力减小,则反应活性减小,该基团是钝化基。
因此判断一个基团是活化基还是钝化基,只需要分析基团与苯环间的电子效应(包括诱导效应和共轭效应)来确定该基团是供电子基还是吸电子基即可。
那么如何分析判断一个基团是邻对位定位基还是间位定位基呢?从反应式可以看出,决速步骤中生成了三种σ-络合物:邻位、间位、对位,这三个平行反应的相对速度决定了最终产物的多少,即决定了取代位置。
这三个平行反应的相对速度可以从两个角度比较。
一方面可以从反应物中邻、间、对三个位置上碳的电子密度相对大小分析。
基团与苯环间的电子效应使邻间对位碳上电子密度不同,电子密度大的碳自然容易受到亲电试剂的进攻而表现出较大的反应活性。
另一方面也可以从三个σ-络合物的稳定性比较。
σ-络合物越稳定,能量越低,生成时经历的过渡态能量越低,反应的活化能越小,反应速度快,相应的σ-络合物生成的就多。
通过以上讲解,大家脑海中要有这样几个概念:第一,分析基团与苯环间的电子效应可以判断基团是供电子基还是吸电子基,从而来确定基团使苯环活化还是钝化;第二,分析基团与苯环间的电子效应可以比较邻间对位碳的电子密度大小,以此判断基团的定位效果;第三,分析σ-络合物的稳定性也可以判断基团的定位效果。
苯环上亲电取代反应的定位规律

HNO3 , H2SO4 30℃ ~60℃
NO2
•磺化 低温(80℃)与浓硫酸生成﹣萘磺酸;高温(165℃)
生成﹣萘磺酸。 ﹣萘磺酸与硫酸共热,也转变为﹣萘磺酸。
+ H 2SO4
80℃ 165℃
SO3H (>95 %)
SO3H (>85 %)
原因:磺酸基体积大,与8位氢原子间距小于其范德华半径之和
•分子式:C10H8;由两个苯环共用两个碳原子并联而成。
•结构:
0.142 0.136
0.1390.140
( 单 位n:m)
8α 7β
6β 5α
1α 2β
3β 4α
——两个苯环共平面;C—C键长介于C—C单键和C = C双键之间 (碳碳单键154pm长,双键134pm长);C—C键键长并不完全相同;
,故﹣萘磺酸稳定性小于﹣萘磺酸。
空间作用大(不稳定) H SO3H
空间作用小(稳定) H
SO3H
H
﹣萘磺酸动力学,﹣萘磺酸热力学控制。﹣萘磺酸是重要的
有机合成中间体,可转化为﹣萘酚、 2020/3/11
﹣萘胺等(合成偶氮染料13的
中间体)。
•酰基化(傅﹣克反应)
——概况 萘的酰基化反应产物与温度和溶剂的极性有关。低温和
NO2 还原
氧化
NO2 COOH COOH
2020/3/11
17
(c)还原反应
用金属钠在液氨和乙醇的混合液中还原生成1,4﹣二氢萘。产物中 的一个孤立双键不被还原。
Na , C2H5OH NH3 (液 )
催 化 加 氢 时 , 可 生 成 1,2,3,4 四 氢 化 萘 ( 又 称 萘 满 ) 或 十 氢 化 萘 (又称萘烷)。
苯环上亲电取代反应的定位规律

O 2 + 7O2
V2 O5 , K SO4 2 385℃~390℃
2 O
O + 4CO 2 + 4H2O
取代萘氧化时环破裂的规律: 取代基为邻对位定位基,使所在
环活化,氧化时同环破裂;取代基为间位定位基,使所在环钝化, 氧化时异环破裂。
H2 / Ni 150℃ H2 / Ni 200℃
应用:四氢化萘和十氢化萘是两种良好的高沸点溶剂。
2003年9月27日(15-16到此止) 思考题:P864:8,9
2013-6-30
作业:P866习题1的5的c,d;习题3的4,5
18
2013-6-30
10
(2)萘的性质 物理性质
萘为无色片状晶体,熔点80.2℃,沸点218℃,易升华。萘有特 殊的气味,不溶于水,溶于乙醇、乙醚及苯中。
2013-6-30
11
化学性质 ——概况 与苯相似,但芳香性比苯差,更易发生亲电取代反应。 位电子云密度比位高,亲电取代首先在位。但1与8
萘的离域能
254.98kJ•mol –1,稳定,但比两个单独苯环离域能 的总和(300kJ•mol‐1)小,故芳香性比苯差,比苯活泼。
萘衍生物的命名
与多官能团取代苯的母体优先选择次序相同。 常见官能团的优先次序为: ﹣COOH , ﹣SO3H , ﹣COOR , ﹣COX , ﹣CONH2 , ﹣CN , ﹣CHO,﹣CO﹣R,﹣OH,﹣NH2,﹣C≡C﹣, C=C ,﹣OR, ﹣X,﹣NO2。 排在前面的官能团优先选择为母体,后三个官能团以苯为母体:
原因:电子云密度较高的环,较活泼,易被氧化破裂。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
羟基在苯酚亲电取代反应中的定位作用*邝代治1 张志坚1,2 冯泳兰1 张复兴1 王剑秋1(1衡阳师范学院化学与材料科学系,2衡阳师范学院生命科学系,衡阳 421008)摘要 苯酚的羟基在空间的位置影响苯酚的能量和原子电荷密度,从而影响苯酚的亲电取代反应。
苯酚的O —C 键旋转形成不同构象之间的最大与最小体系能量值之差ΔE 仅ΔE =0.002508a.u .(1.5738kcal•mol -1)。
当O —H 与苯环共平面时体系能量[E (0)=E (180)= -305.4412692a.u ]最低,为较稳定的优势构象。
苯酚中羟基虽然表现为吸电基,但它的作用使苯环的邻、对位碳原子的负电荷比苯环碳原子高,成为亲电取代反应中心,在邻、对位碳原子上较容易被亲电试剂进攻。
亲电试剂Me +与苯酚反应形成邻、对位的碳正离子中间体比间位碳正离子中间体稳定,有利于生成邻、对位取代产物,因此,羟基为邻、对位定位基。
关键词 苯酚电子结构,羟基的定位效应苯酚是常见且重要的有机中间体,苯酚的亲电取代反应是一类重要有机反应,苯酚的羟基在苯酚的亲电取代反应中起着重要作用。
有机化学教材和教学中用定位法则解释苯酚在亲电取代反应时羟基的定位效应[1-3]。
认为羟基向苯环供电,即活化苯环,又由于羟基的供电子使苯环的邻、对位电荷密度相对较大,亲电取代反应新引入基团主要进入羟基的邻、对位,所以羟基为邻、对位定位基。
其实芳烃的亲电取代反应较复杂,主要与电子效应和空间效应有关,教材和教学中涉及到基团的空间效应主要是基团的空间位阻,很少注意基团本身的空间效应和空间效应影响电子效应的关系。
我们认为,苯酚通过它的C —O 键可自由旋转,形成各种构象,最终以主要的稳定构象存在。
一旦C —O 键旋转,空间效应影响电子效应,从而羟基的定位作用(能力)受到影响。
因此,对定位效应作用的认识还须考虑空间效应与电子效应相互影响。
所以,教学中主要从三个方面理解羟基在苯酚亲电取代反应中的定位作用。
低能稳定构象是参与化学反应的主要物种;原子电荷分布,尤其是苯环碳原子所带电荷是发生亲电取代反应和反应位置的重要条件;亲电取代反应中间体的稳定性是影响反应途径或反应位置的重要因素。
1 分子构象与体系能量E为了阐明这个问题,我们进行了分子建模,以C2-C1-O7-H13的二面角为零时的构象为初始构象(Scheme 1,原子符号略),记作α(0),羟基绕O —C 键逆时针方向旋转(设分子随O —C键旋转时其它键参数不变),寻找旋转角度与分子体系能量的关系,每旋转10度选取一构象计算单点能量和原子电荷。
苯酚的-OH 基绕O —C 键空间旋转,分子体系能量变化如图1所示,发现体系能量E 与旋转角α有关。
当C1、O7和H13三个原子构成的平面与苯环垂直时的构象,体系能量[E (90)=E (270)= -305.4387612a.u .]最高,与苯环共平面时体系能量[E (0)=E (180)= -305.4412692a.u ]最低,为较稳定的优势构象。
但它们之间的能量差[ΔE =0.002508a.u ]很小,O7—C1单键可自由旋转。
Scheme 12 分子构象与原子电荷分布苯酚分子中O 、H 和C 三种元素的电负性不同而原子带电荷不同。
苯酚-OH 基绕O —C 键旋转,形成不同的构象中O 、H 和C 三种原子所带电荷应有变化,将影响亲电取代反应。
* 湖南省高校教改研究资助项目(湘教通[2008]263号148) 12346713812111092.1 苯酚构象与-OH基的原子电荷分布在所有构象中,与O原子相连的H13总是带正电荷,O带负电荷,-OH基的两个原子所带电荷之和q(O7+H13)为负值,说明连在苯环上-OH基拉苯环的电子,为吸电子基。
另一方面,q(O7+H13)值呈规律变化,当α(50)、α(130)、α(230)和α(310)时,q(O7+H13)值负电荷最大,为-0.37421,此时应为一个孤立电子对与苯环共平面,另一个孤立电子对与苯环平面成100°或80°的平面上。
当α(0)和α(180)时,q(O7+H13)值负电荷最小,为-0.37237,此时,两个孤立电子对与苯环平面分别成50°的平面上,相对-OH基拉苯环的电子最弱,苯环容易发生亲电取代反应的构象,与体系能量相一致。
图1 苯酚分子体系能随C—O键旋转变化图2 苯环H原子电荷随C—O键旋转变化2.2 苯环的氢原子电荷变化如图2所示,苯环H都带正电荷,但各H原子所带正电荷量受构象影响且呈规律性,分别出现明显的极大和极小值。
其中邻位两个H8和H12受OH基的空间影响最大,当O—C 键旋转到OH基的氢与苯环邻位H靠近形成共平面时,该邻位H原子带正电荷最少(+0.197561),而处于另一邻位H带最大正电荷(+0.225343),从空间位阻和电荷分布均预期到与OH基反方向的邻位上亲电取代反应的活性较大;对位H10带正电荷明显低于邻、间位氢;间位H9和H11原子电荷变化较小,且对位和间位氢原子电荷受OH基的影响不明显。
2.3 苯酚中苯环碳原子总电荷与旋转角α有关在苯分子中C和H原子分别处于相同的结构环境,它们的原子电荷分别相等,用相同方法计算得苯环碳原子电荷为-0.198944,六个碳原子的总电荷∑Q = -1.193664。
羟基取代苯环一个氢后,苯酚分子的环碳原子电荷发生明显变化,如图3所示。
当O—H键与苯环共平面时[α(0)、α(180)],苯环碳原子总负电荷最少,∑Q(0) = ∑Q(180)= - 0.67175,此时OH基通过O—C的σ键拉苯环电子,当O—H键与苯环垂直时[α(90)、α(270)],苯环碳原子的总负电荷最大,∑Q(90) = ∑Q(270) = -0.68367,都达不到苯环碳原子总电荷∑Q,表现OH基为吸电子基团,与前面的结论吻合。
图3 苯环C原子总电荷随C—O键旋转变化2.4 苯环中各碳原子电荷随α变化苯酚中环碳原子总电荷密度虽不如苯,但各个碳原子的电荷分布差异较大,且各环碳原子电荷分布有规律。
①与OH基直接相连的碳C1带较多正电荷,如图4,图4与图3相似但所带原子电荷正负相反。
②如图5,邻、对位碳原子集中较多负电荷,且两个邻位碳原子电荷受α影响比较大,从α(0)开始,邻位C2带有最大负电荷,q C2(0) = -0.2476,有利于亲电取代试剂在C2位发生反应。
随着α增大,邻、对位碳的原子负电荷分别以不同速率下降,当α=90时,邻、对位碳原子负电荷分别出现极值,q C2(90) =q C6(90) = -0.1980,此时构象,邻位比苯环碳的原子负电荷少,不利于亲电取代反应,对位q C4(90) = -0.2180,稍有利于亲电取代反应。
随着α的增大,邻、对位碳原子带的负电荷分别以不同速率递增,当α达180时,邻、对位三个碳原子负电荷分别达最大值:q C2(180) = -0.2067,q C6(180) =-0.2477,q C4(180) = -0.2180,尤其是C6碳原子明显高于苯环碳原子负电荷,最有利于亲电取代反应的构象。
③间位碳原子均带负电荷,但两个间位碳原子带负电荷随α变化不大,变化规律与邻、对位相反,且均低于苯环碳的原子所带负电荷,特别是在稳定构象时所带负电荷最小,不利于亲电取代反应。
因此,苯酚的羟基表现为邻、对位定位基。
图4 C1原子电荷随C—O键旋转变化图5 邻、间和对位碳原子电荷随C—O键旋转变化3 亲电反应中间体的稳定性与反应途径当α为0或180时分子体系能量最低,为最稳定构象,此时OH基的邻、对位电荷密度最大,尤其是与O—H共平面且与苯环的二面角为0时的邻位碳最可能优先发生亲电取代反应。
以此结构与亲电试剂(R+)反应,理论上将在邻、间和对位发生取代反应,产生(Ⅱ)~(Ⅵ)五种中间体(M+),如Scheme 2所示,现以R=Me为例,它们的中间体M+和产物P的能量(a.u.)分别为:II III IV V VIM+ -344.7509562 -344.7389407 -344.7507651 -344.7375737 -344.7571365P -344.4450042 -344.4508108 -344.4494633 -344.4510245 -344.4504317结果发现它们的中间体的能量关系:E(Ⅵ)<E(Ⅳ) <E(Ⅱ) <E(Ⅲ) <E(Ⅴ),可见,间位中间体能量最高,稳定性最差,亲电取代反应主要沿着生成较稳定的邻、对位中间体进行。
中间体脱去一个H+,形成取代产物(Ⅱp)~(Ⅵp),产物的体系能量分别为:E(Ⅴp)<E(Ⅲp) <E(Ⅵp) <E(Ⅳp) <E(Ⅱp),可见五个产物的能量差ΔE较小,最高与最低能量差0.0060 a.u.,预计在有限的反应时间内间位取代产物应是少量,邻、对位产物为主。
Scheme 2参考文[1] 曾昭琼主篇,有机化学[M].北京:高等教育出版社,第四版(上册),2004.166-172.[2] 夏淑真,罗一鸣主篇,Organic Chemistry[M].武汉:华中科技大学出版社,2006.90-92.[3] R.T.Morrison and R.N. Boyd,Organic Chemistry,fourth edition.,Allyn and Bacon,Inc.1983.598-601Orientation Effect of the hydroxy group on the Reaction of Electrophilic Aromatic SubstitutionKUANG Dai-Zhi1Zhang Zhijian1,2FENG Yong-Lan1ZHANG Fu-Xing1WANG Jian-Qiu1(1Department of Chemistry and Materials Science, Hengyang Normal University, Hengyang421008;2 Department of Life Sciences, Hengyang Normal University, Hengyang 421008, Hunan;China)The atomic charge of phenol is effected by the space location of the hydroxy, so that the orientation effect of the hydroxy on the reaction of electrophilic aromatic substitution will also be effected.The molecular conformation is formed by the O—C bond rotation , the 0.002508a.u (1.5738kcal•mol-1) barrier is not a very high one.even at room temperature with sufficient energy is large enough that a rapid interconversion between conformations, that the potential energy of the molecule is at a minimum E(0)=E(180)= -305.4412692a.u for the eclipsed is stabilized. The hydroxy is a drawing electronic group by σ and p-π bond. The hydroxy group is a ortho-para director,thereby increasing the negative electric charges of the ortho-para of benzene and activating the benzene reaction. The carbocations was formed via the reaction of phenol with Me+, the carbocation dehydrogenation gave substituted products that of ortho-para is to be more stable than that of meta position.keywords: electronic structure of the phenol, orientation effect of the hydroxyl。