2011我国药品注册审批数据解读

2011年我国药品注册审批数据解读
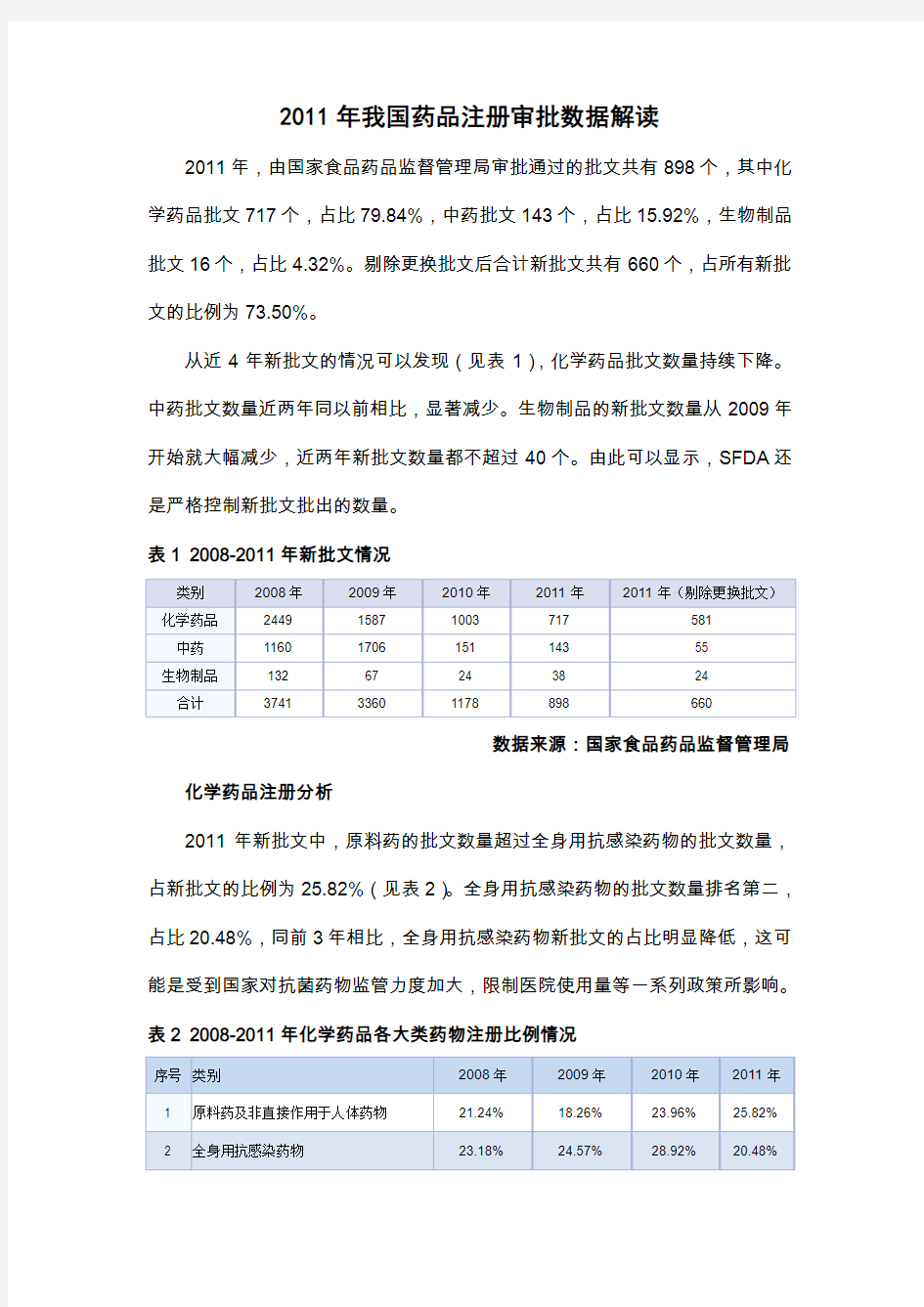
2011年,由国家食品药品监督管理局审批通过的批文共有898个,其中化学药品批文717个,占比79.84%,中药批文143个,占比15.92%,生物制品批文16个,占比4.32%。剔除更换批文后合计新批文共有660个,占所有新批文的比例为73.50%。
从近4年新批文的情况可以发现(见表1),化学药品批文数量持续下降。中药批文数量近两年同以前相比,显著减少。生物制品的新批文数量从2009年开始就大幅减少,近两年新批文数量都不超过40个。由此可以显示,SFDA还是严格控制新批文批出的数量。
表1 2008-2011年新批文情况
数据来源:国家食品药品监督管理局化学药品注册分析
2011年新批文中,原料药的批文数量超过全身用抗感染药物的批文数量,占新批文的比例为25.82%(见表2)。全身用抗感染药物的批文数量排名第二,占比20.48%,同前3年相比,全身用抗感染药物新批文的占比明显降低,这可能是受到国家对抗菌药物监管力度加大,限制医院使用量等一系列政策所影响。表2 2008-2011年化学药品各大类药物注册比例情况
注:只统计新批文,不包含更换批文情况,如无特别说明,下同其它类别方面,血液和造血系统药物2011年新批文比例同2008年相比,有较明显的上升。心血管系统药物近年新批文比例呈下降的趋势,由2008年的10.18%下降到2011年的7.57%。
2011年有5个1类化学药品获批,其中4个为1.1类新药(见表3),分别是艾瑞昔布片、盐酸埃克替尼片、艾拉莫德片、赛洛多辛胶囊,前3个制剂产品的原料药批文同时获批。这4个1.1类新药中,艾瑞昔布片和艾拉莫德片用于肌肉-骨骼系统,盐酸埃克替尼片用于抗肿瘤,赛洛多辛胶囊为泌尿系统药品。湘北威尔曼制药的注射用头孢曲松钠舒巴坦钠属于1.5类新药,为第三代头孢类抗生素。帕珠沙星的滴眼剂获得批准上市,该产品属于2类新药,在此之前帕珠沙星均为注射剂剂型。另外还有多个新复方药品获批上市,包括吡格列酮二甲双胍
片、坎地氢噻片、青蒿琥酯阿莫地喹片、色甘萘甲那敏鼻喷雾剂、替米沙坦氢氯噻嗪片,其中坎地氢噻片和替米沙坦氢氯噻嗪片为抗高血压的新型复方药品。表3 2011年化学药品部分新药情况
2011年批文数量最多的产品是葡萄糖注射液和注射用头孢地嗪钠(见表4),葡萄糖注射液有4个厂家,共16个生产批文获批;注射用头孢地嗪钠有8个厂家,共16个生产批文获批。在注册数量排名前列的化学药品中,全身用抗感染药物的产品最多,共有5个,且都是注射剂产品。注射用盐酸头孢替安,2011年之前只有4个厂家获得生产批文,2011年再有2个厂家获得新生产批文。
表4 2011年注册数量排名前列的化学药品
消化系统及代谢药类
2011年,在消化系统及代谢药物的新批文中,抗酸药及治疗消化性溃疡和胃肠胀气用药占比最高,达到30.77%(见表5、表6),其中兰索拉唑片和奥美拉唑肠溶胶囊各有6个和4个新批文。
表5 2011年消化系统及代谢药类注册产品比例表
表6 2011年消化系统及代谢药类药物注册(前10位)
在新产品方面,止吐药甲磺酸多拉司琼注射液获批为3.1类新药,生产厂家为辽宁海思科制药有限公司,商品名立必复,规格有1ml:12.5mg和5ml:100mg 两个,该产品属于国内首次批准上市。江苏德源药业有限公司的吡格列酮二甲双胍片(15mg/500mg)2011年也获批上市,为新型糖尿病复方药物,这是继中美华东制药2010年首次获得生产批文后,第二个获批的生产厂家。深圳赛诺菲巴斯德生物制品有限公司的多烯磷脂酰胆碱注射液获批上市,该产品本来只有成都天台山制药有限公司一个企业生产,现在将迎来外资企业产品的竞争。
血液和造血系统药物
2011年血液和造血系统药物新批文以血浆代用品和输注液的产品为主,包括葡萄糖注射液、氯化钠注射液等(见表7、表8)。抗血栓形成药的批文只有4个,分别是奥扎格雷钠注射液、注射用比伐芦定、西洛他唑片。
表7 2011年血液和造血系统药物注册产品比例分布表
表8 2011年血液和造血系统药物注册(前5位)
注射用比伐芦定生产厂家为信立泰药业,商品名泰加宁,规格250mg,同时获得原料药批文,在我国首次获批上市。该产品为抗凝血酶药物,适用于择期经皮冠状动脉介入治疗(PCI)的抗凝。
心血管系统药物
2011年心血管系统药物的新批文中以抗高血压药物批文较多,其次是心脏病治疗药物批文(见表9、表10)。抗高血压药物批文中,有多个复方药品出现,包括:坎地氢噻片、替米沙坦氢氯噻嗪片、厄贝沙坦氢氯噻嗪片、颉沙坦氢氯噻嗪分散片、氨氯地平贝那普利片(I)。
表9 2011年心血管系统药物注册产品比例分布表
表10 2011年心血管系统药物注册(前9位)
新产品方面,坎地氢噻片为国内首次获批上市,生产企业是江苏德源药业有限公司,每片含坎地沙坦酯16mg,氢氯噻嗪12.5mg。替米沙坦氢氯噻嗪片由湖北丝宝药业有限公司生产,每片含替米沙坦40mg和氢氯噻嗪12.5mg,2011年之前上海欣生源药业有限公司获得过替米沙坦氢氯噻嗪胶囊的国内生产批文,勃林格殷格翰获得替米沙坦氢氯噻嗪片进口批文。2011年,江苏万邦生化医药的匹伐他汀钙片获批,有两个规格,是第二个国内厂家获得该品种的生产批文,
第一个获得国内生产批文的厂家是北京双鹤药业,另外日本Kowa公司也于2008年获得匹伐他汀钙片(商品名:力清之)的进口批文。
皮肤病用药
2011年,皮肤病用药新批文共有20个,其中皮肤病用抗真菌药占比最大,达到50%(见表11)。产品中特比萘芬制剂获批数量最多,分别有盐酸特比萘芬乳膏、盐酸特比萘芬凝胶、盐酸特比萘芬喷雾剂,其次是鬼臼毒素制剂产品。表11 2011年皮肤病用药注册产品比例分布表
生殖泌尿系统和性激素类药物
2011年,生殖泌尿系统和性激素类药物新批文共有10个,其中赛洛多辛胶囊是唯一的1个1.1类新药。赛洛多辛胶囊由第一三共制药(北京)有限公司生产,商品名优利福,规格4mg,该产品为α1-肾上腺素受体拮抗剂,用于治疗良性前列腺增生(也称为前列腺肥大,BHP)。另外赛洛多辛原料药也在2011年获批进口。
全身用激素类制剂(不含性激素)
2011年,全身用激素类制剂(不含性激素)药物新批文只有3个,是成都
力思特制药的鲑降钙素注射液和上海复旦复华药业的比卡鲁胺片。
全身用抗感染药物
2011年,在全身用抗感染药物新批文中,头孢类产品占比最大,超过47%(见表12、表13),第二是青霉素类产品,占比16.81%,第三是喹诺酮类产品,占比11.76%,第四是大环内酯类和林可胺类产品和全身用抗病毒药产品,占比都是10.08%,这5类产品共占有超过95%的新批文。
表12 2011年全身用抗感染药物注册产品比例分布表
表13 2011年全身用抗感染药物注册(前10位)
在新产品方面,1.5类新药注射用头孢曲松钠舒巴坦钠获批,该产品由湘北威尔曼制药有限公司生产,规格3.0g。安徽贝克生物制药的齐多拉米双夫定片获批上市,从而打破葛兰素史克在齐多拉米双夫定制剂市场的垄断。浙江南洋药业获得乙胺吡嗪利福异烟片(II)的生产批文,这是继沈阳红旗制药之后,第二家国内生产厂家获得该产品的批文。2011年第4个注射用比阿培南生产批文出现,批文由山东鲁抗辰欣药业获得,商品名华劲,比阿培南属于碳青霉烯类药物,研究发现某些方面的抗菌活性比亚胺培南和美罗培南要强2-8倍。
抗肿瘤和免疫调节剂
2011年,抗肿瘤和免疫调节剂新批文共有24个,其中有21个批文属于抗肿瘤药物(见表14)。盐酸埃克替尼片属于1.1类新药,由浙江贝达药业独立开发,商品名凯美纳,规格125mg,原料药同时获批。埃克替尼是“十一五”期间我国在新药研发上获得的重大突破,标志我国的小分子靶向抗癌药将不再依赖外国进口,从而打断外资市场的垄断地位。埃克替尼片用于晚期非小细胞肺癌二线治疗。
表14 2011年抗肿瘤和免疫调节剂注册情况
石药集团中奇制药的盐酸多柔比星脂质体注射液获批,是第二家获得生产批文的国内厂家。
肌肉-骨骼系统
2011年,肌肉-骨骼系统药物新批文有16个,其中以抗炎药和抗风湿药占比最多,有62.5%。2011年肌肉-骨骼系统药物出现两个1.1类新药,分别是艾瑞昔布片和艾拉莫德片(见表15)。
艾瑞昔布片有恒瑞医药生产,商品名恒扬,规格0.1g。艾瑞昔布属于选择性的COX-2抑制剂,由中国医科院药物研究所进行化合物的设计合成和初步活性评价,恒瑞医药则负责动物评价试验及后续研发工作,从研发、临床、获批历时超过12年。
表15 2011年肌肉-骨骼系统药物注册产品比例分布表
艾拉莫德片,先声药业有限公司产品,商品名艾得辛,规格25mg。据介绍,该产品是全球第一个上市的艾拉莫德制剂,也是一个全新结构类型的DMARDS (Disease Modifying Anti-rheumatic Drugs,疾病修饰抗风湿药)药物,其主要
适应症为活动性类风湿性关节炎,可显著改善类风湿性关节炎患者的疾病症状和炎症指标,减轻患者的痛苦。
神经系统药物
2011年,神经系统药物新批文共有27个,其中以精神安定药和精神兴奋药占比最多,两者合计占有50%的新批文(见表16)。
表16 2011年神经系统药物注册产品比例分布表
新产品方面,盐酸右美托咪定注射液又有2家生产厂家获得生产批文,分别是江苏恩华药业股份有限公司和四川国瑞药业有限责任公司,加上恒瑞在2009年获得的生产批文,令盐酸右美托咪定注射液累计有3家厂家生产。该产品用于全身麻醉手术患者气管插管和机械通气时的镇静。
枸橼酸芬太尼注射液有第二个国内厂家获得生产批文,由江苏恩华药业获得,有两个规格获批。
厦门金日制药有限公司的盐酸多奈哌齐口腔崩解片获批,相比于以前的剂型,这次批准的产品是属于口腔崩解片剂型,为新剂型。
抗寄生虫药、杀虫剂和驱虫剂
2011年,抗寄生虫药、杀虫剂和驱虫剂新批文共有9个,桂林南药股份有限公司的青蒿琥酯阿莫地喹片获得其中3个批文。青蒿琥酯阿莫地喹片,有3
个规格,国内首次上市,属于3.2类新药,适应症为用于治疗对阿莫地喹和青蒿琥酯敏感的恶性疟原虫引起的非重症疟疾(单纯性疟疾发作)。
呼吸系统用药
2011年,呼吸系统药物新批文有16个,其中以咳嗽和感冒用药占比最多(见表17、表18)。产品中氨溴索制剂批文最多,有6个,分别有盐酸氨溴索注射液、盐酸氨溴索口腔崩解片、盐酸氨溴索口服溶液。
新产品中,山东天顺药业股份有限公司的色甘萘甲那敏鼻喷雾剂属于3.2类新药,每瓶10ml,含色甘酸钠0.1g、盐酸萘甲唑啉0.0025g、马来酸氯苯那敏0.025g;每喷0.1ml,含色甘酸钠1.0mg、盐酸萘甲唑啉0.025mg、马来酸氯苯那敏0.25mg。该产品用于治疗过敏性鼻炎。
表17 2011年呼吸系统药物注册产品比例分布表
表18 2011年呼吸系统药物注册
感觉系统药物
2011年,感觉系统药物新批文共有11个,全部为眼科用药。其中甲磺酸帕
珠沙星滴眼液和曲尼司特滴眼液都属于新药。
甲磺酸帕珠沙星滴眼液属于2类新药,生产厂家为浙江莎普爱思药业股份有限公司,规格0.3%(以帕珠沙星计)。帕珠沙星制剂产品剂型一直都是注射剂,这次改为滴眼液属于改变给药途径。
曲尼司特滴眼液也是国内首次获批,生产厂家为中国药科大学制药有限公司,以前曲尼司特制剂给药途径都是口服。
中药注册分析
2011年,中药新批文只有55个,其中消化系统疾病用药和心脑血管疾病用药均占比20%。消化系统疾病用药批文比例近年变化不大,都在16%-20%之间波动(见表19)。
表19 2008-2011年中药各大类药物新批文比例情况
同以往批文比例情况对比可以发现,泌尿系统疾病用药和神经系统疾病用药的占比有显著增长。泌尿系统疾病用药新批文比例同比增长10个百分点。神经系统疾病用药前几年的比例都在5%以下,2011年则超过9%,而妇科用药的占比则有明显下降。
2011年成都百裕科技制药的银杏内酯注射液获批,属于中药5类新药(见表20),规格每支装2ml(含萜类内酯10mg),功效为活血化瘀,通经活络。用于淤血阻络所致的缺血性中风病中经络,症见头晕目眩,口舌歪斜,言语蹇涩,肢体麻木、头痛,半身不遂。适用于急性期脑梗死和恢复期脑梗死见上述表现者。表20 2011年部分中药新药列表
其它中药新药以6类新药为主,涉及消化系统疾病用药、泌尿系统疾病用药等多个大类,剂型以胶囊剂为主。
贵州渝生制药的柏花草胶囊,曾用名痔比舒胶囊,用于便血、脱出、肛门肿痛、坠胀、肛门潮湿等痔疮症状的治疗,产品并获得国家发明专利。
康恩贝制药的黄莪胶囊,主要成分为黄芪、桃仁、莪术、大黄等,用于I、II期良性前列腺增生症气虚血瘀、湿热阻滞症的治疗。
舒筋除湿胶囊,由湖南敬和堂制药有限公司研发,是一种由天然药物制成的新中药复方制剂,用于治疗类风湿性关节炎和骨关节炎。
小儿益麻颗粒,由武汉健民药业集团股份有限公司生产,适用于治疗小儿遗尿肾气不足。据公开资料显示,小儿益麻颗粒处方在北京市儿童医院儿科临床应用三十多年,疗效确切。
生物制品注册分析
2011年,生物制品新批文只有24个,以全身抗感染药物居多(见表21),由7个厂家分别获得。按药物大类划分,24个批文属于5个大类。单品种中,狂犬病人免疫球蛋白的新批文最多,有4个。
表21 2008-2011年生物制剂各大类药物新批文比例情况
新产品有注射用重组人尿激酶原、重组抗CD25人源化单克隆抗体注射液、重组戊型肝炎疫苗(大肠埃希菌)、重组人干扰素α1b喷雾剂(见表22)。
表22 2011年部分生物制剂新药列表
注射用重组人尿激酶原,由上海天士力药业有限公司生产,商品名普佑克,规格5mg(50万IU)/支,属于生物1类新药,适应症为急性ST段抬高性心肌梗死的溶栓治疗。
重组抗CD25人源化单克隆抗体注射液是单抗产品,由上海中信国健药业股份有限公司生产,商品名健尼哌,规格25mg/5ml/瓶,属于生物2类新药,适用于预防器官移植后急性排斥反应的发生。
重组戊型肝炎疫苗(大肠埃希菌),获批生产厂家是厦门万泰沧海生物技术有限公司,商品名:益可宁,规格30μg/0.5ml/支。该产品是国家1类新药,由厦门大学、养生堂万泰公司联合研制,是世界上第一个用于预防戊型肝炎的疫苗。
重组人干扰素α1b喷雾剂,获批生产厂家是北京三元基因工程有限公司,商品名:运德素,规格25万IU(25μg)/5ml/支。三元基因原有重组人干扰素α1b 的注射剂产品,这次喷雾剂获批,属于新剂型。
进口产品注册分析
2011年,有多个治疗糖尿病的药物首次获批进口,分别是诺和诺德的利拉鲁肽注射液、百时美施贵宝的沙格列汀片、诺华的维格列汀片(见表23)。
表23 2011年首次进口的新药
利拉鲁肽注射液(商品名:诺和力)是GLP-1受体激动剂,其工作原理是在人体血糖水平过高时刺激胰腺β细胞释放胰岛素,从而降低血糖,2010年获得FDA批准上市。2011年接近60亿丹麦克朗,同比增长158.57%。
沙格列汀片(商品名:安立泽)和维格列汀片(商品名:佳维乐)都属于DPP-4抑制剂。百时美施贵宝的沙格列汀片,2009年获得FDA批准上市,2011年全球销售额6.84亿美元,同比增长超过200%。诺华的维格列汀片2011年全球销售额6.77亿美元,同比增长73.15%,不过维格列汀暂时未获FDA批准上
市。在2010年之前,我国进口的DPP-4抑制剂只有默沙东的磷酸安格列汀片,2011年增加了2个新产品,令DPP-4抑制剂市场竞争越趋激烈。
礼来的特立帕肽注射液(商品名:复泰奥),用于治疗绝经后妇女的骨质酥松,2011年全球销售额接近9.5亿美元,同比增长14.46%。2002年已获FDA 批准上市,美国英文名为FORTEO。
雷珠单抗注射液(英文名:Lucentis),诺华制药进口产品,中文商品名:诺适得,规格10mg/ml,每瓶装量0.20ml。2011年首次获批进入中国。雷珠单抗主要用于治疗湿性老年性黄斑病变。这种疾病会造成黄斑部(眼睛视网膜中心的一部分)损伤,可能导致失明和严重的视力损失。雷珠单抗,2006年获FDA批准上市。根据诺华公司年报公布,雷珠单抗注射液上市后,销售额持续强劲增长,2011年已超过20亿美元,同比增长超过33%。
度他雄胺软胶囊(商品名:安福达)属于5α-还原酶抑制剂,用于抗前列腺增生,为葛兰素史克产品,2011年首次获批进口我国,2011年全球销售额7.48亿英镑,同比增长18.92%。该产品2001年已获FDA批准。
阿戈美拉汀片(商品名:维度新),首个褪黑素受体激动剂,具有良好的抗抑郁效果。主要用于抗抑郁、抗焦虑、调整睡眠节律及调节生物钟作用。进口厂家是施维雅。
左乙拉西坦口服液属于新剂型进口,之前进口剂型均为片剂。
药品注册管理办法20200330
中国药品监管中国药监化妆品监管邮箱政务信息报送 请输入关键字 药品注册管理办法 2020年03月30日 发布 国家市场监督管理总局令 第27号 《药品注册管理办法》已于2020年1月15日经国家市场监督管理总局2020年第1次局务会议审议通过,现予公布,自2020 年7月1日起施行。 局长 肖亚庆 2020年1月22日 药品注册管理办法 (2020年1月22日国家市场监督管理总局令第27号公布) 第一章 总则 第一条为规范药品注册行为,保证药品的安全、有效和质量可控,根据《中华人民共和国药品管理法》(以下简称《药品 管理法》)、《中华人民共和国中医药法》、《中华人民共和国疫苗管理法》(以下简称《疫苗管理法》)、《中华人民共和国 行政许可法》、《中华人民共和国药品管理法实施条例》等法律、行政法规,制定本办法。 第二条 在中华人民共和国境内以药品上市为目的,从事药品研制、注册及监督管理活动,适用本办法。 第三条药品注册是指药品注册申请人(以下简称申请人)依照法定程序和相关要求提出药物临床试验、药品上市许可、再 注册等申请以及补充申请,药品监督管理部门基于法律法规和现有科学认知进行安全性、有效性和质量可控性等审查,决定是否 同意其申请的活动。 申请人取得药品注册证书后,为药品上市许可持有人(以下简称持有人)。 第四条药品注册按照中药、化学药和生物制品等进行分类注册管理。 中药注册按照中药创新药、中药改良型新药、古代经典名方中药复方制剂、同名同方药等进行分类。 化学药注册按照化学药创新药、化学药改良型新药、仿制药等进行分类。 生物制品注册按照生物制品创新药、生物制品改良型新药、已上市生物制品(含生物类似药)等进行分类。 中药、化学药和生物制品等药品的细化分类和相应的申报资料要求,由国家药品监督管理局根据注册药品的产品特性、创新 程度和审评管理需要组织制定,并向社会公布。 境外生产药品的注册申请,按照药品的细化分类和相应的申报资料要求执行。 第五条国家药品监督管理局主管全国药品注册管理工作,负责建立药品注册管理工作体系和制度,制定药品注册管理规 范,依法组织药品注册审评审批以及相关的监督管理工作。国家药品监督管理局药品审评中心(以下简称药品审评中心)负责药 物临床试验申请、药品上市许可申请、补充申请和境外生产药品再注册申请等的审评。中国食品药品检定研究院(以下简称中检院)、国家药典委员会(以下简称药典委)、国家药品监督管理局食品药品审核查验中心(以下简称药品核查中心)、国家药品 监督管理局药品评价中心(以下简称药品评价中心)、国家药品监督管理局行政事项受理服务和投诉举报中心、国家药品监督管
新药临床申请审批流程图
现在有三种申报方法 一.申报1.1类药进口(进口药) (1)方法:a.等药品在国外上市后,不在国内生产。申请分包进口(类似于代理)。 b.等药品在国外上市后,完善国内厂房技术,在国内生产。 (2)费用:临床实验37.6万,生产59.39万。 (3)难点:a.需要等药物在国外上市后才可以进行,延长周期。 b.如果在国内生产,需要完善国内厂房技术,延长周期、增加费用。 c.如果国外生产,分包进入国内时可以申请免除临床实验,但是不能确定可以申请成 功。 d.需要所有的临床资料,包括所有的实验数据。 二.申报1.1类新药临床试验(国产药) (1)方法:直接在国内申报1类新药,相关药理毒理研究资料由国外进行,提供相关证明,然后工艺研究、质量研究跟稳定性研究在国内进行,等国内药厂达到生产要求后,将药品在国内生产,按照国内药来报。 (2).费用:申请临床实验19.2万,申请生产43.2万。 (3)难点:a.需要等国内工厂具备相关生产条件后才可以进行申报。 b.无法申请避免临床。 三.申报国际多中心临床试验 (1)基本流程+费用+时间:主要临床基地伦理委员会审(费用...,时间...)+临床审批(费用37.6万,时间205天)+药品清关(费用...+时间...)(时间相对较短) (2)方法:申报程序与国内1.1类化学新药临床试验申报大致相同,但是需要更多的临床资料和
证明性文件。 (3)申请地点:国家药监局北京市西城区宣武门西大街28号大成广场3门一层(总局办公大楼 西侧),邮编100053 (4)流程图: 注:斜线前为一般审批时限,斜线后为特殊审批时限,均为工作日。 (5)申报资料列表: (一)概要部分
药品注册审评报告完整版
药品注册审评报告 Document serial number【NL89WT-NY98YT-NC8CB-NNUUT-NUT108】
国家食品药品监督管理局《2010年药品注册审批报告》 药品注册,是国家食品药品监督管理局依照《药品管理法》的规定,根据药品注册申请人的申请,对拟上市销售药品的安全性、有效性、质量可控性等进行审查,并决定是否同意其申请的审批过程。在药品研制、生产、流通、使用的全过程监管中,药品注册管理是从源头上对药品安全性和有效性实施监管的重要手段,其根本目的是通过科学评价,保证上市药品安全有效,保障和促进公众健康。 1 2010年药品注册管理的重要举措 2010年,药品注册管理工作继续践行科学监管理念,紧紧围绕“质量和效率”这个中心,以风险效益评估和风险管理为核心,坚持“规范审批、公开透明、鼓励创新”的原则,全面推进体制机制改革和法制建设,强化学药品物研究全程监管,进一步推进审评审批公开公平,提高审评审批效率,较好地履行了《药品管理法》赋予的职责。 完善药品注册管理法规体系 一是发布了《药物临床试验伦理审查工作指导原则》,规范和指导伦理委员会的药物临床试验伦理审查工作,加强药物临床试验的质量管理和对受试者的保护,提高药物临床试验伦理审查工作质量。继续开展《药用原辅材料登记备案管理规定》、《药品标准管理办法》、《药物临床试验生物样本分析实验室管理规定》、《药物Ⅰ期临床试验管理指导原则》、《药物临床试验中严重不良事件报告与监测管理规定》及天然药物注册技术要求等法规和规范性文件的研究起草工作。 二是加快药品研究技术指导原则体系建设。成立了人用药物注册技术要求国际协调会(ICH)中国研究小组,对国际标准和技术规范进行深入研究。完成216个国外药品研究指导原则的翻译,其中150个拟转化实施的指导原则已对外征求意见,31个指导原则根据征求意见作了进一步修订,《药物致癌试验必要性的技术指导原则》正式对外发布执行。这些指导原则的建立,使得我国药品注册审评更加科学、规范,也将指导和促进我国新药研发向国际水平看齐。 确保药品注册审评科学、公正、公开 一是构建了已上市药品、药用辅料、溶出度测定方法等数据库,使药品技术审评建立在科学、量化的基础上。二是以仿制药为切入点,制定了化学药品仿制药电子申报资料格式,启动了电子申报,提高审评审批效率。三是通过重大专项专题会、创新品种沟通交流会、专家咨询会议,以及第三方验证、专家票决等方式,保证技术审评工作的科学性和公正性。四是进一步严格技术审评标准。对于高风险的疫苗、血液制品,坚持审评原则与国际接轨,严格技术审评,保证此类品种质量;对于需长期使用的新药,增加致癌性试验要求,确保用药安全;严格抗生素品种的立题审查及技术要求。五是继续加大信息公开力度。公开审评报告,使申报单位全面了解审评过程和审评决策依据;通过咨询日、开放日、主任信箱、网上信息反馈等多种形式不断加强与社会各界的沟通交流,促进审评工作的公开透明,逐步实现“阳光审评”。 加强中药、民族药的监管 一是会同卫生部、国家民委及国家中医药管理局共同发布了《全国民族医药近期重点工作实施方案(2010-2012年)》,强调要充分发挥民族医药在少数民族地区防病治病中的积极作用,加强对民族药的监督管理,保障民族地区广大人民群众用药安全。二是会同卫生部、国家中医药管理局等联合发布了《关于加强医疗机构中药制剂管理的意见》,要求发挥医疗机构中药制剂在中医临床诊疗中的积极作用,进一步促进医疗机构中药制剂科学、健康发展。三是贯彻落实国务院《关于扶持和促进中医药事业发展的若干意见》的要求,开展了中药民族药监管现状调研,进一步厘清促进中医药事业健康发展的思路、步骤和措施。 加强药品研究过程的监督管理 一是加强和完善药品注册现场核查。各省(区、市)全年累积派出几百个工作组、近3000人次,开展了药品注册现场检查,为药品技术审评工作提供了有力的保障。通过问卷调查和实地调研,对近几年药品注册现场核查工作进行了总结和分析,细化了现场检查的要点及相关格式要求。
药品注册现场核查常见问题(一)
药品注册现场核查常见问题(一) 《药品注册管理办法》要求“申请人应当提供充分可靠的研究数据,证明药品的安全性、有效性和质量可控性,并对全部资料的真实性负责”。2008年国 家食品药品监督管理局发布《药品注册现场核查管理规定》,规定应对所受理药品注册申请的研制情况进行实地确证,对原始记录进行审查,以确认申报资料的真实性、准确性和完整性。 原始记录是申请人或其委托人进行了相应的研制工作的证据性文件,也是药品研究机构撰写药品申报资料的依据。申报资料中申请人或其委托人完成实验工作所使用的物料、仪器设备,采用的实验条件、实验方法、操作步骤、实验过程,观察到的现象,测定的数据,得出的结果结论等均应在原始记录中有记载和体现,是对原始记录中记载的试验内容的总结与提炼。 真实、规范、完整的实验记录是保证药品研究结果真实可靠的基础。只有客观、准确、及时的记录整个药品研制的过程,真实地反映试验过程和结果,研究轨迹清楚、可追溯,研究过程可重复,才能证明申报资料的真实性,才能保证其申报资料的数据准确、可靠。 现将近年来,在药学研制现场核查时发现的原始记录中存在的问题以及产生的原因进行分析和总结。 一、存在真实性问题的记录 2008年《药品注册现场核查管理规定》发布前后,核查原始记录,发现许多原始记录基本是申报资料的“手抄本”,原始记录与申报资料高度一致,没有摸索和失败的记录,试验数据高度重复,如批生产记录,不同批次高度一致;溶出度、残留溶剂检查数据精密度极高。此类原始记录一般存在恶意造假的情况,一般是试验时只零散的记录了一些数据,甚至根本没有进行试验,申报资料参照模板整理完后,按照申报资料抄写原始记录,根据需要,随意的取舍,甚至编造试验及数据。 没有筛选、摸索过程和试验依据,没有失败的记录。常常出现申报资料的信息量大于原始记录,或者申报资料的内容是正确的,而原始记录是错误的情况。对于这类情况,在原始记录核查中会通过对试验可行性的审查、可疑数据的复查等,在现场进行确认,一经发现存在真实性问题,均按照不通过处理。 二、原始记录不规范问题解析 除发现存在真实性问题的品种外,核查时还发现记录不规范,不符合原始记录基本要求的情况,存在准确性和完整性的问题。 1 造成原始记录不规范的主要原因包括以下几方面 A. 事后誊写将物料量、测定数据、试验结果等随意记在一张纸上,或找一个临时的本子记录,试验完成后再誊写或整理实验记录。不仅有可能在誊写过程中出现笔误,增大了发生错误的几率,造成结果不准确,出现申报资料与原始记录不一致的情况;
最新药品注册管理办法
药品注册管理办法 (征求意见稿) 目录 第一章总则 第二章基本制度和要求 第三章药品上市注册 第一节药物临床试验 第二节药品上市许可 第三节关联审评审批 第四节药品注册核查 第五节药品注册检验 第四章药品加快上市注册 第一节突破性治疗药物程序 第二节附条件批准程序 第三节优先审评审批程序 第四节特别审批程序 第五章药品上市后变更和再注册 第一节药品上市后研究和变更 第二节药品再注册 第六章受理、补充资料和撤审 第七章争议解决 第八章工作时限 第九章监督管理 第十章法律责任 第十一章附则 —1 —
第一章总则 第一条【法律依据】为规范药品注册行为,保证药品的安全、有效和质量可控,根据《中华人民共和国药品管理法》(以下简称《药品管理法》)《中华人民共和国中医药法》《中华人民共和国疫苗管理法》(以下简称《疫苗管理法》)《中华人民共和国行政许可法》《中华人民共和国药品管理法实施条例》,制定本办法。 第二条【适用范围】在中华人民共和国境内以药品上市为目的,从事药品研制、注册及其监督管理活动,适用本办法。 第三条【药品注册定义】药品注册,是指药品注册申请人(以下简称申请人)依照法定程序和相关要求提出药品注册事项,药品监督管理部门基于法律法规和现有科学认知进行安全性、有效性和质量可控性等审查,作出是否同意其药品注册事项及其管理的过程。 申请人取得药品注册证书后,为药品上市许可持有人(以下简称持有人)。 第四条【药品注册事项】药品注册包括药物临床试验申请、药品上市许可申请、补充申请、再注册申请等许可事项,以及其他备案或者报告事项。 第五条【药品注册申请类别】药品注册申请类别,按照中药、化学药和生物制品等进行分类。 中药注册分类包括中药创新药、中药改良型新药、古代经典名方中药复方制剂、同名同方药等。 化学药注册分类包括化学药创新药、化学药改良型新药、仿制药等。
药品注册现场核查管理规定含附件的最全版
药品注册现场核查管理规定含附件的 最全版 1
2
关于印发药品注册现场核查管理规定的通知 国食药监注[ ]255号 05 月 23 日 发布 各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总 后卫生部药品监督管理局: 为规范药品研制秩序,保证药品注册现场核查工作质量,根据<药品 注册管理办法>的有关规定,国家局组织制定了<药品注册现场核查 管理规定>,现予印发,请遵照执行。 国家食品药品监督管理局 二○○八年五月二十三日 药品注册现场核查管理规定 3
第一章总则 第一条为加强药品注册现场核查管理,规范药品研制秩序,根据<中华人民共和国药品管理法>及其实施条例、<药品注册管理 办法>,制定本规定。 第二条药品注册现场核查分为研制现场核查和生产现场检查。 药品注册研制现场核查,是指药品监督管理部门对所受理药品注册申请的研制情况进行实地确证,对原始记录进行审查,确认申报资料真实性、准确性和完整性的过程。 药品注册生产现场检查,是指药品监督管理部门对所受理药品注册申请批准上市前的样品批量生产过程等进行实地检查,确认其是否与核定的或申报的生产工艺相符合的过程。 本规定所指的药品注册检验抽样,是指药品监督管理部门在药品注册现场核查过程中进行的取样、封样和通知检验。 第三条药品注册现场核查分为常规和有因。有因核查主要是指针对下列情形进行的现场核查: (一)药品审评过程中发现的问题; 4
(二)药品注册相关的举报问题; (三)药品监督管理部门认为需进行核查的其它情形。 第四条国家食品药品监督管理局负责全国药品注册现场核查的组织协调和监督管理。同时负责组织新药、生物制品批准上市前的生产现场检查;负责组织进口药品注册现场核查;负责组织对药品审评过程中发现的问题进行现场核查;负责组织涉及药品注册重大案件的有因核查。 第五条省、自治区、直辖市药品监督管理部门负责本行政区域内的下列药品注册现场核查: (一)负责所受理药品注册申请的研制现场核查; (二)负责所受理已上市药品改变剂型、改变给药途径注册申 请的生产现场检查; (三)负责所受理仿制药注册申请的生产现场检查; (四)负责所受理药品生产技术转让、变更处方和生产工艺可 能影响产品质量等补充申请的生产现场检查; (五)负责本行政区域内的有因核查。 研制工作跨省进行的药品注册申请,研制现场核查工作由受理该申请的省、自治区、直辖市药品监督管理部门负责,研制现场所在地省、自治区、直辖市药品监督管理部门应当予以协助。 5
药品注册审评报告
国家食品药品监督经管局《2010年药品注册审批报告》 药品注册,是国家食品药品监督经管局依照《药品经管法》的规定,根据药品注册申请人的申请,对拟上市销售药品的安全性、有效性、质量可控性等进行审查,并决定是否同意其申请的审批过程。在药品研制、生产、流通、使用的全过程监管中,药品注册经管是从源头上对药品安全性和有效性实施监管的重要手段,其根本目的是通过科学评价,保证上市药品安全有效,保障和促进公众健康。 1 2010年药品注册经管的重要举措 2010年,药品注册经管工作继续践行科学监经管念,紧紧围绕“质量和效率”这个中心,以风险效益评估和风险经管为核心,坚持“规范审批、公开透明、鼓励创新”的原则,全面推进体制机制改革和法制建设,强化学药品物研究全程监管,进一步推进审评审批公开公平,提高审评审批效率,较好地履行了《药品经管法》赋予的职责。 1.1 完善药品注册经管法规体系 一是发布了《药物临床实验伦理审查工作指导原则》,规范和指导伦理委员会的药物临床实验伦理审查工作,加强药物临床实验的质量经管和对受试者的保护,提高药物临床实验伦理审查工作质量。继续开展《药用原辅材料登记备案经管规定》、《药品规范经管办法》、《药物临床实验生物样本分析实验室经管规定》、《药物Ⅰ期临床实验经管指导原则》、《药物临床实验中严重不良事件报告与监测经管规定》及天然药物注册技术要求等法规和规范性文件的研究起草工作。 二是加快药品研究技术指导原则体系建设。成立了人用药物注册技术要求国际协调会(ICH)中国研究小组,对国际规范和技术规范进行深入研究。完成216个国外药品研究指导原则的翻译,其中150个拟转化实施的指导原则已对外征求意见,31个指导原则根据征求意见作了进一步修订,《药物致癌实验必要性的技术指导原则》正式对外发布执行。这些指导原则的建立,使得我国药品注册审评更加科学、规范,也将指导和促进我国新药研发向国际水平看齐。 1.2 确保药品注册审评科学、公正、公开 一是构建了已上市药品、药用辅料、溶出度测定方法等数据库,使药品技术审评建立在科学、量化的基础上。二是以仿制药为切入点,制定了化学药品仿制药电子申报资料格式,启动了电子申报,提高审评审批效率。三是通过重大专项专题会、创新品种沟通交流会、专家咨询会议,以及第三方验证、专家票决等方式,保证技术审评工作的科学性和公正性。四是进一步严格技术审评规范。对于高风险的疫苗、血液制品,坚持审评原则与国际接轨,严格技术审评,保证此类品种质量;对于需长期使用的新药,增加致癌性实验要求,确保用药安全;严格抗生素品种的立题审查及技术要求。五是继续加大信息公开力度。公开审评报告,使申报单位全面了解审评过程和审评决策依据;通过咨询日、开放日、主任信箱、网上信息反馈等多种形式不断加强与社会各界的沟通交流,促进审评工作的公开透明,逐步实现“阳光审评”。 1.3 加强中药、民族药的监管 一是会同卫生部、国家民委及国家中医药经管局共同发布了《全国民族医药近期重点工作实施方案(2010-2012年)》,强调要充分发挥民族医药在少数民族地区防病治病中的积极作用,加强对民族药的监督经管,保障民族地区广大人民群众用药安全。二是会同卫生部、国家中医药经管局等联合发布了《关于加强医疗机构中药制剂经管的意见》,要求发挥医疗机构中药制剂在中医临床诊疗中的积极作用,进一步促进医疗机构中药制剂科学、健康发展。三是贯彻落实国务院《关于扶持和促进中医药事业发展的若干意见》的要求,开展了中药民族药监管现状调研,进一步厘清促进中医药事业健康发展的思路、步骤和措施。
新药申报与审批程序
按照《新药审批办法》第五章的规定:“新药的申报与审批分为临床研究和生产上市两个阶段。初审由省级药品监督管理部门负责,县市由国家药品监督管理局负责。” 现结合图1新药申报与审批基本程序示意图介绍该程 1.申报单位填写新药临床研究(或生产)申请表,连同申报的技术资料和样品报省、自治区、直辖市药品监督管理部门。省级药品监督管理部门进行初审,即对新药的各项原始资料是否齐全进行审查;同时,派员对试制条件进行实地考察,填写考察报告表。对已报齐所有应报资料的,正式通知申报单位收审;同时将样品和技术资料转省、自治区、直辖市药品检验所审核。对应报资料不全的,予以退审,将申请表和资料退回申报单位并提出退审理由。 2.省、自治区、直辖市药品检验所按新药审批各项技术要求完成对申报资料的审查和样品的检验。 药检所的审核系指对新药的药学(包括药理、毒理)研究资料进行审查和对样品进行实验检验;不包括为申报单位进行新的检测方法的研究。药检所审核完毕后,提出质量标准和对药学(包括药理、毒理)方面的综合审查意见,送省级药品监督管理部门。 3.省级药品监督管理部门初审通过同意上报的,在新药临床研究(或生产)申请表签署意见,连同申报的技术资料一式5份报国家药品监督管理局注册司进行形式审查。 3’.新生物制品和按《新药审批办法》第二十六条所列新药,由申报单位填写申请表,连同申报的技术资料一式五份直接报国家药品监督管理局注册司。样品检验和质量标准复核由中国药品生物制品检定所负责。4.国家药品监督管理局注册司经形式审查合格的,向申报单位发出收取审评费的通知。同时交药品审评中心安排技术审查、审评委员会审评及必要的复核等工作。 4.形式审查不合格的,予以退审。 5.技术审评通过后,将建议批准的或退审的审评报告及意见,报国家药品监督管理局药品注册司。
药品注册研制现场核查要点及判定原则
药品注册研制现场核查要点及判定原则 一、药品注册研制现场核查要点 (一)药学方面 1.工艺及处方研究 1.1研制人员是否从事过该项研制工作,并与申报资料的记载一致。 1.2工艺及处方研究是否具有与研究项目相适应的场所、设备和仪器。 1.3工艺及处方研究记录是否有筛选、摸索等试验过程的具体内容,工艺研究及其确定工艺的试验数据、时间是否与申报资料一致。 2.样品试制 2.1样品试制现场是否具有与试制该样品相适应的场所、设备,并能满足样品生产的要求,临床试验用样品和申报生产样品的生产条件是否符合《药品生产质量管理规范》的要求。申报生产所需样品的试制是否在本企业生产车间内进行。 2.2样品试制所需的原辅料、药材和提取物、直接接触药品的包装材料等是否具有合法来源(如供货协议、发票、药品批准证明性文件复印件等)。 2.3原辅料、药材和提取物、直接接触药品的包装材料等购入时间或供货时间与样品试制时间是否对应,购入量是否满足样品试制的需求。 2.4样品试制用的原辅料及直接接触药品的包装材料是否有检验报告书。 2.5样品试制是否具有制备记录或原始批生产记录,样品制备记录项目及其内容应齐全,如试制时间、试制过程及相关关键工艺参数、中间体检验记录等。 2.6样品试制量、剩余量与使用量之间的关系是否对应一致。 2.7尚在进行的长期稳定性研究是否有留样,该样品所用直接接触药品的包装材料是否与申报资料一致。 2.8申报生产所需样品的原始批生产记录是否与申报工艺对应。 3.质量、稳定性研究及样品检验 3.1研究人员是否从事过该项研究工作,并与申报资料的记载一致。 3.2质量、稳定性研究及检验现场是否具有与研究项目相适应的场所、设备和仪器。 3.3研究期间的仪器设备是否校验合格,是否具有使用记录,记录时间与研究时间是否对应一致,记录内容是否与申报资料一致。 3.4用于质量、稳定性研究的样品批号、研究时间与样品试制时间的关系是否相对应。 3.5对照研究所用对照药品是否具有来源证明。 3.6所用的对照品/标准品是否具有合法来源,如为工作对照品,是否有完整的标化记录。 3.7质量研究各项目以及方法学考察内容是否完整,各检验项目中是否记录了所有的原始数据,数据格式是否与所用的仪器设备匹配,质量研究各项目(鉴别、检查、含量测定等)是否有实验记录、实验图谱及实验方法学考察内容。 3.8质量研究及稳定性研究实验图谱是否可溯源,IR、UV、HPLC、GC等具数字信号处理系统打印的图谱是否具有可追溯的关键信息(如带有存盘路径的图谱原始数据文件名和数据采集时间),各图谱的电子版是否保存完好;需目视检查的项目(如薄层色谱、纸色谱、电泳等)是否有照片或数码照相所得的电子文件。 3.9质量研究及稳定性研究原始实验图谱是否真实可信,是否存在篡改图谱信息(如采集时间)、一图多用等现象。 3.10稳定性研究过程中各时间点的实验数据是否合乎常规,原始记录数据与申报资料是否一致。 4.委托研究 其他部门或单位进行的研究、试制、检测等工作,是否有委托证明材料。委托证明材料
药品注册审评报告
国家食品药品监督管理局《2010年药品注册审批报告》 ????药品注册,是国家食品药品监督管理局依照《药品管理法》的规定,根据药品注册申请人的申请,对拟上市销售药品的安全性、有效性、质量可控性等进行审查,并决定是否同意其申请的审批过程。在药品研制、生产、流通、使用的全过程监管中,药品注册管理是从源头上对药品安全性和有效性实施监管的重要手段,其根本目的是通过科学评价,保证上市药品安全有效,保障和促进公众健康。 ??? ?1 2010年药品注册管理的重要举措 ????2010年,药品注册管理工作继续践行科学监管理念,紧紧围绕“质量和效率”这个中心,以风险效益评估和风险管理为核心,坚持“规范审批、公开透明、鼓励创新”的原则,全面推进体制机制改革和法制建设,强化学药品物研究全程监管,进一步推进审评审批公开公平,提高审评审批效率,较好地履行了《药品管理法》赋予的职责。 ??? ?1.1 完善药品注册管理法规体系 ??? ?一是发布了《药物临床试验伦理审查工作指导原则》,规范和指导伦理委员会的药物临床试验伦理审查工作,加强药物临床试验的质量管理和对受试者的保护,提高药物临床试验伦理审查工作质量。继续开展《药用原辅材料登记备案管理规定》、《药品标准管理办法》、《药物临床试验生物样本分析实验室管理规定》、《药物Ⅰ期临床试验管理指导原则》、《药物临床试验中严重不良事件报告与监测管理规定》及天然药物注册技术要求等法规和规范性文件的研究起草工作。 ???? 二是加快药品研究技术指导原则体系建设。成立了人用药物注册技术要求国际协调会(ICH)中国研究小组,对国际标准和技术规范进行深入研究。完成216个国外药品研究指导原则的翻译,其中150个拟转化实施的指导原则已对外征求意见,31个指导原则根据征求意见作了进一步修订,《药物致癌试验必要性的技术指导原则》正式对外发布执行。这些指导原则的建立,使得我国药品注册审评更加科学、规范,也将指导和促进我国新药研发向国际水平看齐。 ??? ?1.2 确保药品注册审评科学、公正、公开 ????一是构建了已上市药品、药用辅料、溶出度测定方法等数据库,使药品技术审评建立在科学、量化的基础上。二是以仿制药为切入点,制定了化学药品仿制药电子申报资料格式,启动了电子申报,提高审评审批效率。三是通过重大专项专题会、创新品种沟通交流会、专家咨询会议,以及第三方验证、专家票决等方式,保证技术审评工作的科学性和公正性。四是进一步严格技术审评标准。对于高风险的疫苗、血液制品,坚持审评原则与国际接轨,严格技术审评,保证此类品种质量;对于需长期使用的新药,增加致癌性试验要求,确保用药安全;严格抗生素品种的立题审查及技术要求。五是继续加大信息公开力度。公开审评报告,使申报单位全面了解审评过程和审评决策依据;通过咨询日、开放日、主任信箱、网上信息反馈等多种形式不断加强与社会各界的沟通交流,促进审评工作的公开透明,逐步实现“阳光审评”。 ???? 1.3 加强中药、民族药的监管 ????一是会同卫生部、国家民委及国家中医药管理局共同发布了《全国民族医药近期重点工作实施方案(2010-2012年)》,强调要充分发挥民族医药在少数民族地区防病治病中的积极作用,加强对民族药的监督管理,保障民族地区广大人民群众用药安全。二是会同卫生部、国家中医药管理局等联合发布了《关于加强医疗机构中药制剂管理的意见》,要求发挥医疗机构中药制剂在中医临床诊疗中的积极作用,进一步促进医疗机构中药制剂科学、健康发展。三是贯彻落实国务院《关于扶持和促进中医药事业发展的若干意见》的要求,开展了中药民族药监管现状调研,进一步厘清促进中医药事业健康发展的思路、步骤和措施。 ????1.4 加强药品研究过程的监督管理 ????一是加强和完善药品注册现场核查。各省(区、市)全年累积派出几百个工作组、近3000人次,开展了药品注册现场检查,为药品技术审评工作提供了有力的保障。通过问卷调查和实地调研,对近几年药品注册现场核查工作进行了总结和分析,细化了现场检查的要点及相关格式要求。
国家食品药品监管总局关于解决药品注册申请积压实行优先审评审批的意见
国家食品药品监管总局关于解决药品注册申请积压实行优先 审评审批的意见 【法规类别】药品管理 【发文字号】食药监药化管[2016]19号 【失效依据】食品药品监管总局关于鼓励药品创新实行优先审评审批的意见 【发布部门】国家食品药品监督管理总局 【发布日期】2016.02.24 【实施日期】2016.02.24 【时效性】失效 【效力级别】XE0303 国家食品药品监管总局关于解决药品注册申请积压实行优先审评审批的意见 (食药监药化管〔2016〕19号) 各省、自治区、直辖市食品药品监督管理局,新疆生产建设兵团食品药品监督管理局:为加强药品注册管理,加快具有临床价值的新药和临床急需仿制药的研发上市,解决药品注册申请积压的矛盾,现提出以下意见。 一、优先审评审批的范围 (一)具有明显临床价值,符合下列情形之一的药品注册申请: 1.未在中国境内外上市销售的创新药注册申请。 2.转移到中国境内生产的创新药注册申请。
3.使用先进制剂技术、创新治疗手段、具有明显治疗优势的药品注册申请。 4.专利到期前3年的药品临床试验申请和专利到期前1年的药品生产申请。 5.申请人在美国、欧盟同步申请并获准开展药物临床试验的新药临床试验申请;在中国境内用同一生产线生产并在美国、欧盟药品审批机构同步申请上市且通过了其现场检查的药品注册申请。 6.在重大疾病防治中具有清晰的临床定位的中药(含民族药)注册申请。 7.列入国家科技重大专项或国家重点研发计划的新药注册申请。 (二)防治下列疾病且具有明显临床优势的药品注册申请: 1.艾滋病; 2.肺结核; 3.病毒性肝炎; 4.罕见病; 5.恶性肿瘤; 6.儿童用药品; 7.老年人特有和多发的疾病。 (三)其他 1.在仿制药质量一致性评价中,需改变已批准工艺重新申报的补充申请; 2.列入《关于开展药物临床试验数据自查核查工作的公告》(食品药品监管总局2015年第117号)的自查核查项目,申请人主动撤回并改为按与原研药质量和疗效一致的标准完善后重新申报的仿制药注册申请; 3.临床急需、市场短缺的药品注册申请。具体品种名单由国家卫生计生委和工业和信息化部提出,食品药品监管总局药品审评中心(以下简称药审中心)组织相关部门和专家论证后确定。
药品审批
某药业研制一个已在国内上市销售的进口化学药制剂,其原料药未进口,注册时需要提交哪些研究资料?试提出加快药品审批进度的建议。 从题目“某药业研制一个已在国内上市销售的进口化学药制剂,其原料药未进口,注册时需要提交哪些研究资料?”可知该药品在注册分类里属于第6类——已有国家药品标准的原料药或制剂。在注册时需要申报的资料项目包括综述资料、药学研究资料、药理毒理研究资料以及临床试验资料。 首先,每个注册药品都要提交综述资料,其中包括:药品名称、证明性文件、立题目的与依据、对主要研究结果的总结及评价、药品说明书样稿、起草说明及最新参考文献包装和标签设计样稿。 制剂处方及工艺的研究资料及文献资料(单独申请注册药物制剂,必须提供原料药的合法来源证明文件,一式2 份,分别放入资料项目2 的资料和资料项目13 号的资料中。使用国产原料药的申请人,应当提供该原料药的药品批准证明文件、检验报告书、药品标准、原料药生产企业的营业执照、《药品生产许可证》、《药品生产质量管理规范》认证证书、与该原料药生产企业签订的供货协议、销售发票等的复印件。使用进口原料药的,应当提供与该原料药生产企业或国内合法的销售代理商签订的供货协议、《进口药品注册证》或者《医药产品注册证》、口岸药品检验所检验报告书、药品标准复印件等。药品注册过程中,研制制剂所用的进口原料药未取得《进口药品注册证》或者《医药产品注册证》的,必须经国家食品药品监督管理局批准。)提供资料包括确证化学结构或者或组份的试验资料及文献资料、质量研究工作的试验资料及文献资料、药品标准草案及起草说明、样品的检验报告书、辅料的来源及质量标准、药物稳定性研究的试验资料及文献资料和直接接触药品的包装材料和容器的选择依据及质量标准。 药理毒理资料包括药理毒理研究资料项下的16项药理毒理研究资料综述,以及说明项下第17项——局部用药除按所属注册分类及项目报送相应资料外,应当报送资料项目21,必要时应当进行局部吸收试验。资料项目21为过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等特殊安全性试验资料和文献资料。 关于临床试验资料部分,除国内外相关的临床试验资料综述以外,对于注册
药品注册现场核查管理规定
药品注册现场核查管理规定 第一章总则 (2) 第二章药品注册研制现场核查 (4) 第一节药物临床前研究现场核查 (4) 第二节药物临床试验现场核查 (5) 第三节申报生产研制现场核查 (5) 第三章药品注册生产现场检查 (6) 第一节新药、生物制品生产现场检查 (6) 第二节已上市药品改变剂型、改变给药途径生产现场检查 (7) 第三节仿制药生产现场检查 (7) 第四节补充申请生产现场检查 (8) 第五章药品注册检验抽样要求 (9) 第六章核查人员管理 (10) 第七章附则 (10)
第一章总则 第一条为加强药品注册现场核查管理,规范药品研制秩序,根据《中华人民共和国药品管理法》及其实施条例、《药品注册管理办法》,制定本规定。 第二条药品注册现场核查分为研制现场核查和生产现场检查。 药品注册研制现场核查,是指药品监督管理部门对所受理药品注册申请的研制情况进行实地确证,对原始记录进行审查,确认申报资料真实性、准确性和完整性的过程。 药品注册生产现场检查,是指药品监督管理部门对所受理药品注册申请批准上市前的样品批量生产过程等进行实地检查,确认其是否与核定的或申报的生产工艺相符合的过程。 本规定所指的药品注册检验抽样,是指药品监督管理部门在药品注册现场核查过程中进行的取样、封样和通知检验。 第三条药品注册现场核查分为常规和有因。有因核查主要是指针对下列情形进行的现场核查: (一)药品审评过程中发现的问题; (二)药品注册相关的举报问题; (三)药品监督管理部门认为需进行核查的其他情形。 第四条国家食品药品监督管理局负责全国药品注册现场核查的组织协调和监督管理。同时负责组织新药、生物制品批准上市前的生产现场检查;负责组织进口药品注册现场核查;负责组织对药品审评过程中发现的问题进行现场核查;负责组织涉及药品注册重大案件的有因核查。 第五条省、自治区、直辖市药品监督管理部门负责本行政区域内的下列药品注册现场核查: (一)负责所受理药品注册申请的研制现场核查; (二)负责所受理已上市药品改变剂型、改变给药途径注册申请的生产现场
关于药品注册审评审批若干政策的公告(2015年第230号)的解读和考量
关于药品注册审评审批若干政策的公告(2015年第230号)的解读和考量 2015-11-13kikidoa临床试验前沿信息 临床试验前沿信息 关于药品注册审评审批若干政策的公告(2015年第230 号)的解读和考量 CFDA这效率现在简直可以登峰造极,也反映出国家治理拥堵,改革现有审批制度的决心。在昨晚重磅炸弹丢出了,业界非常关心的注册册审评审批若干政策落地,这一纸通告直接越过了《药品注册管理办法》,让我们瞠目结舌。 以下我对“国家食品药品监督管理总局关于药品注册审评审批若干政策的公告(2015年第230号)”文件的解读和考量。 关于药品注册审评审批若干政策的公告Kikidoa's Comments 根据《中华人民共和国药品管理法》、《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)等有关规定,为解决药品注册申请积压问题,提高药品审评审批质量和效率,经国务院同意,实行如下药品注册审评审批政策。目的很直接:就是为了解决审批积压,提高审批质量和效率。 但附带的目的有些多:1)颠覆了临床试验这个行业规则,又会塑造一个游戏规则。2)基本一刀切掉了多年的3类品种,自我否定了这么多年劳民伤财的3类新药临床试验;但是很可能把一些3类药推向了创新药的节奏,在抢歪果仁还没进来之前,想做3类以后难啊。…… 一、提高仿制药审批标准 仿制药按与原研药质量和疗效一致的原则受理和审评审批。其中,对已在中国境外上市但尚未在境内上市药品的仿制药注册申请,应与原研药进行生物等效性研究并按国际通行技术要求开展临床试验,所使用的原研药由企业自行采购,向国家食品药品监督管理总局申请一次性进口;未能与原研药进行对比研究的,应按照创新药的技术要求开展研究。 已经受理的仿制药注册申请,实行分类处理: (一)中国境内已有批准上市原研药,申请注册的仿制药没有达到与原研药质量和疗效一致的,不予批准。 (二)中国境外已上市但境内没有批准上市原研药,申请仿制药注册的企业可以选择按原规定进行审评审批,但在药品批准上市3年内需按照国发首先按照《国发〔2015〕44号》文件定义的仿制药:由现行的“仿已有国家标准的药品”调整为“仿与原研药品质量和疗效一致的药品”。但是这一定义标准还略显笼统,详细的定义估计还应该以定稿的《化学药品注册分类改革工作方案》中为主,目前还在征求意见(要求同的活性成份、剂型、规格、适应症、给药途径和用法用量与原研完全相同)。 仿制药的临床试验的要求也基本确定:与原研药进行生物等效性研究并按国际通行技术要求开展临床试验。但这个仍很难理解。 1)无论原研产品是否在国内上市,必须进行生物等效性的临床试验;(未上市可以申请一次性进口,这次应该会好批些了);未与原研药进行对比研究的,按照创新药开展临床试验。[创新药的定义:创是指含有新的结构明确的具有生理或药理作用的分子或离子,包括用拆分或者合成等方法制得的已知活性成份的光学异构体及其制剂。] 2)国际通行的技术要求开展临床试验——这个太含糊了,真的有能通用的吗? 通行的技术:品牌药未有在本国上市之前就没有
药品注册现场核查管理制度
《药品注册现场核查管理规定》 (国食药监注[2008]255号) 各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总后卫生部药品监督管理局:为规范药品研制秩序,保证药品注册现场核查工作质量,根据《药品注册管理办法》的有关规定,国家局组织制定了《药品注册现场核查管理规定》,现予印发,请遵照执行。 ?国家食品药品监督管理局?二00八年五月二十三日 药品注册现场核查管理规定 第一章总则 第一条为加强药品注册现场核查管理,规范药品研制秩序,根据《中华人民共和国药品管理法》及其实施条例、《药品注册管理办法》,制定本规定。 第二条药品注册现场核查分为研制现场核查和生产现场检查。?药品注册研制现场核查,是指药品监督管理部门对所受理药品注册申请的研制情况进行实地确证,对原始记录进行审查,确认申报资料真实性、准确性和完整性的过程。 药品注册生产现场检查,是指药品监督管理部门对所受理药品注册申请批准上市前的样品批量生产过程等进行实地检查,确认其是否与核定的或申报的生产工艺相符合的过程。 本规定所指的药品注册检验抽样,是指药品监督管理部门在药品注册现场核查过程中进行的取样、封样和通知检验。 第三条药品注册现场核查分为常规和有因。有因核查主要是指针对下列情形进行的现场核查:?(一)药品审评过程中发现的问题;? (二)药品注册相关的举报问题;? (三)药品监督管理部门认为需进行核查的其他情形。 第四条国家食品药品监督管理局负责全国药品注册现场核查的组织协调和监督管理。同时负责组织新药、生物制品批准上市前的生产现场检查;负责组织进口药品注册现场核查;负责组织对药品审评过程中发现的问题进行现场核查;负责组织涉及药品注册重大案件的有因核查。 第五条省、自治区、直辖市药品监督管理部门负责本行政区域内的下列药品注册现场核查:? (一)负责所受理药品注册申请的研制现场核查; (二)负责所受理已上市药品改变剂型、改变给药途径注册申请的生产现场检查;?(三)负责所受理仿制药注册申请的生产现场检查; (四)负责所受理药品生产技术转让、变更处方和生产工艺可能影响产品质量等补充申请的生产现场检查; (五)负责本行政区域内的有因核查。
药品审批流程+受理号含义+评审时间
药品审批流程
2005年以后受理号分为四部分,第一部分是申请的基本信息(四位字母),第二部份是年份(第五、六位),第三部分是流水号(第七至十一位),第四部分(十二、十三位)是受理单位标识。药品、辅料注册申请受理号共十三位,药包材注册申请受理号共十二位。 前面的四位字母意思分别是 第一位:C表示国产,J表示进口 第二位:X表示新药,Y表示已有国家标准(即仿制药) 第三位:H表示化学药品,Z表示中药,S表示生物制品,F表示辅料 第四位:L表示申请临床,S表示申请上市(即生产),B表示补充申请,Z表示再注册,F 表示分包装 如“CXHL0600001甘”表示国产新药化学药品临床申请,是2006年受理的该类型第一个注册申请,甘肃省局上报;“JYHF0600001桂”表示进口已有国家标准化学药品分包装申请,是2006年受理的该类型第一个注册申请,广西区局上报。 2005年以前受理号的大体意思是: X:表示申请国产注册或补充(新药) Y:表示申请国产注册或补充(已有国家标准,即仿制药) FX:申请仿制(西药) FZ:申请仿制(中药) FX、FZ前面加B,即BFX或BFZ即为补充申请的意思 BFX/BFZ:申请仿制补充(西药/中药) :申请新药证书及生产(化药) CXL:申请新药临床研究(化药) CXZ:申请新药试生产转正式生产(化药) CXS、CXL、CXZ或CX后面加B即为补充的意思CZS:申请新药证书及生产(中药)CZL:申请新药临床研究(中药) CZZ:申请新药试生产转正式生产(中药) CZS、CZL、CZZ或CZ后面加B即为补充的意思 J:为申请进口注册或补充 B:申请进口药品补充 A:申请进口药品注册证 AS:申请进口药品注册证(生物制品) AZ:申请进口药品注册证(中药) H:申请进口药品注册证换发 HS:申请进口药品注册证换发(生物制品) HZ:申请进口药品注册证换发(中药) CSS:申请生物制品试生产转正式生产 CSL:申请生物制品临床研究 CSZ:申请生物制品试生产转正式生产 CSS、CSL、CSZ、CS后面加B即为补充的意思
《药品注册管理办法》2020年版整理
国家市场监督管理总局令 第27号 《药品注册管理办法》已于2020年1月15日经国家市场监督管理总局2020年第1次局务会议审议通过,现予公布,自2020年7月1日起施行。 局长肖亚庆 2020年1月22日 药品注册管理办法 (2020年1月22日国家市场监督管理总局令第27号公布) 第一章总则 第一条为规药品注册行为,保证药品的安全、有效和质量可控,根据《中华人民国药品管理法》(以下简称《药品管理法》)、《中华人民国中医药法》、《中华人民国疫苗管理法》(以下简称《疫苗管理法》)、《中华人民国行政许可法》、《中华人民国药品管理法实施条例》等法律、行政法规,制定本办法。 第二条在中华人民国境以药品上市为目的,从事药品研制、注册及监督管理活动,适用本办法。
第三条药品注册是指药品注册申请人(以下简称申请人)依照法定程序和相关要求提出药物临床试验、药品上市许可、再注册等申请以及补充申请,药品监督管理部门基于法律法规和现有科学认知进行安全性、有效性和质量可控性等审查,决定是否同意其申请的活动。 申请人取得药品注册证书后,为药品上市许可持有人(以下简称持有人)。 第四条药品注册按照中药、化学药和生物制品等进行分类注册管理。 中药注册按照中药创新药、中药改良型新药、古代经典名方中药复方制剂、同名同方药等进行分类。 化学药注册按照化学药创新药、化学药改良型新药、仿制药等进行分类。 生物制品注册按照生物制品创新药、生物制品改良型新药、已上市生物制品(含生物类似药)等进行分类。 中药、化学药和生物制品等药品的细化分类和相应的申报资料要求,由国家药品监督管理局根据注册药品的产品特性、创新程度和审评管理需要组织制定,并向社会公布。 境外生产药品的注册申请,按照药品的细化分类和相应的申报资料要求执行。 第五条国家药品监督管理局主管全国药品注册管理工作,负责建立药品注册管理工作体系和制度,制定药品注册管理规,依法组织药品注册审评审批以及