第二型糖原累积病的治疗
第九章-生化与分子遗传学(答案)

第九章生化与分子遗传学(答案)一、选择题(一)单项选择题1.基因突变对蛋白质所产生的影响不包括:A.影响活性蛋白质的生物合成B.影响蛋白质的一级结构C.改变蛋白质的空间结构D.改变蛋白质的活性中心E.影响蛋白质分子中肽键的形成2.原发性损害指:A.突变改变了protein的一级结构,使其失去正常功能B.突变改变了糖元的结构,使糖元利用障碍C.突变改变了脂肪的分子结构,使脂肪动员受阻D.突变改变了核酸的分子结构,使其不能传给下一代E.突变主要使蛋白质的亚基不能聚合*3.苯丙酮尿症的发病机理是苯丙氨酸羟化酶缺乏导致:A.代谢底物堆积B.代谢旁路产物堆积C.代谢中间产物堆积D.代谢终产物缺乏E.代谢终产物堆积4.半乳糖血症Ⅰ型的发病机理是由于基因突变导致酶遗传性缺乏使:A.代谢底物堆积B.代谢旁路产物堆积C.代谢中间产物堆积D.代谢终产物缺乏E.代谢终产物堆积5.色氨酸加氧酶缺乏症的发病机理是由于基因突变导致:A.5-羟色胺增多B.色氨酸不能被吸收C.色氨酸吸收过多D.烟酰胺生成过多E.代谢终产物堆积6.下列何种疾病不属于分子病?A. 肝豆状核变性B. 先天性睾丸发育不全综合征C. 血友病D. 镰形细胞贫血E. 家族性高胆固醇血症7.关于苯丙酮尿症(PKU),下列哪项说法是不正确的?A.可进行新生儿筛查B. 可进行产前检查C. 不通过DNA分析不能确定出携带者D. 是一种表现为智力低下的常染色体隐性遗传病E. 是由于遗传性缺乏苯丙氨酸羟化酶所致8.人类珠蛋白基因包括:A. 位于16p13上的类α珠蛋白基因簇,包括α和ζ基因B. 位于llpl5上的类β珠蛋白基因簇,包括α、β、γ、ε和ζ等基因C. 位于llpl5上的类α珠蛋白基因簇D. 位于16p13上的类β珠蛋白基因簇E. 位于Xp21的STR序列*9.镰状细胞贫血是由于血红蛋白β链上的第6位氨基酸被下列哪种氨基酸替代?A. 脯氨酸B. 色氨酸C. 苏氨酸D. 缬氨酸E. 亮氨酸*10.正常HbA的α链为141个氨基酸,有一种称为Hb Constant Spring的突变型,其α链为172个氨基酸,推测可能发生了:A. 无义突变B. 终止密码突变C. 移码突变D.错义突变E. 同义突变11.一个溶血性贫血病人,经检查Hb A2为30%,肽链裂解后见有α、γ、δ三种肽链,其最可能被诊断为:A.α—地中海贫血B.β-地中海贫血C. HbS病D. 高铁血红蛋白病E.遗传性非球形红细胞贫血12.由于代谢中间产物缺乏而引起的代谢缺陷是:A. 半乳糖血症B.白化病C. 糖原累积病D. 苯丙酮尿症E.家族性高胆固醇血症13.以下五种血液病中,哪一种不是遗传病?A. 红细胞G6PD缺乏症B. 地中海贫血C. 血友病D. 血小板无力症E. 特发性血小板减少性紫癜14.缺乏3个α基因引起:A. 静止型α-地中海贫血B. 轻性α—地中海贫血C. HbH病D. HbBart’s胎儿水肿综合征E.β—地中海贫血15.关于肌营养不良的遗传方式,下列哪项说法是错误的?A. Duchenne型肌营养不良为X连锁隐性遗传B. Becker型肌营养不良为常染色体隐性遗传C. 面肩肱型肌营养不良为常染色体显性遗传D. 肢带型肌营养不良为常染色体隐性遗传E. 强直型肌营养不良为常染色体隐性遗传*16.下列哪项是肝豆状核变性的典型症状?A. 视乳头水肿B. 视网膜粟粒样结节C. 黄斑区樱桃红色D. 角膜色素环(K-F环)E. 角膜弓*17.进行性肌营养不良的典型体征是:A. 躯干性共济失调B. 肢体性共济失调C. 剪刀式步态D. 毛细血管扩张E. 迈步呈鸭步态,上楼困难18.先天性肾上腺皮质增生症的最主要原因是:A. 肾上腺皮质功能减退B. 常染色体显性遗传C. 常染色体隐性遗传D. 肾上腺皮质激素合成过程中所需要的酶先天性缺乏E.下丘脑的病变引起19.先天性肾上腺皮质激素合成过程在先天性酶缺陷中下列哪点最多见?A. 21羟化酶缺陷B. 17羟化酶缺陷C. 18羟化酶缺陷D. 3β羟类固醇脱氢酶缺陷E. 20,22碳链酶缺陷20.治疗糖原累积病的最有效方法是?A. 纠正高血脂B. 纠正酸中毒C. 静脉维持稳定血糖D. 增加餐次E. 食用淀粉和果糖21.下列哪一型粘多糖累积病属于X-连锁隐性遗传?A. 粘多糖Ⅰ型(Hurler综合征和Scheie综合征)B. 粘多糖Ⅱ型(Hurter综合征)D.多糖Ⅲ型(Sanfilippo综合征) D. 粘多糖Ⅳ型(Morquio综合征)E. 粘多糖Ⅵ型(Maroleaux-Lamy综合征)22.苯丙酮尿症(PKU)引起的智力低下一般多在:A. 生后4个月左右被发现B. 新生儿期即被发现C. 生后2个月左右被发现D. 生后1年左右被发现E.生后2~3岁被发现*23.苯丙酮尿症在哪个年龄组开始治疗?一般应持续治疗几年?A. 出生后1个月开始治疗,应持续治疗1~2年B. 出生后2~3个月开始治疗.应持续治疗2~3年C. 出生后3~4个月开始治疗,应持续治疗3~4年D. 出生后1个月开始治疗,应持续治疗4~6年E. 出生后5~6个月开始治疗,应持续治疗4~6年24.患儿10个月,近一周来有抽搐发作3~4次。
2021年儿科单选题与答案解析(13)

2021年儿科单选题与答案解析13一、单选题(共60题)1.诊断急性肾小球肾炎者,下列哪项是正确的A:前驱无感染症状B:出现低白蛋白血症,高血压C:尿检查提示肾小球源性血尿、不同程度的尿蛋白D:血补体没有变化E:多伴有肝衰竭【答案】:C【解析】:诊断急性肾小球肾炎依据:在链球菌感染后1-3周出现血尿、水肿、血压高,尿常规提示肾小球源性血尿,不同程度蛋白尿,血清有链球菌感染的免疫学改变,血补体动态变化。
常见并发症有循环充血状态,甚至心力衰竭、肺水肿,高血压脑病,急性肾衰竭。
因此只有C选项表述正确。
2.先天性甲状腺功能低下按发病机制分类为A:原发性、继发性B:散发性、地方性C:碘缺乏性、非碘缺乏性D:遗传性、非遗传性E:碘缺乏性、非碘缺乏性、混合性【答案】:A【解析】:先天性甲状腺功能减低症是最常见的导致智力低下和发育落后的小儿内分泌疾病。
按发病机制分原发性和继发性。
3.麻疹易感者接触麻疹后为预防发病,注射免疫球蛋白时间是A:接触后14天以内B:接触后21天以内C:接触后10天以内D:接触后1天以内E:接触后5天以内【答案】:E【解析】:被动免疫:接触麻疹后5天内立即肌注免疫血清球蛋0.25ml/kg,可预防麻疹;若5天后注射者,仅能减轻症状。
被动免疫只能维持3~8周,以后应采取主动免疫。
4.先天性梅毒患儿的临床表现包括A:肝脾肿大、脑钙化B:骨损害、脑钙化C:贫血、血小板减少、脑钙化D:皮肤损害、肝脾肿大E:皮肤损害、脑钙化【答案】:D【解析】:临床表现大多数患儿出生时无症状,于2~3周后逐渐出现。
常见的症状有:①肝脾大;②皮肤黏膜损害:鼻炎为早期特征,于生后1周出现,可持续3个月之久,当鼻黏膜溃疡累及鼻软骨时形成“鞍鼻”,累及喉部引起声嘶。
皮疹常于生后2~3周出现,初为粉红、红色多行性斑丘疹,以后变为棕褐色,并有细小脱屑,掌、跖部还可见梅毒性天疱疮。
皮疹常见于口周、鼻翼和肛周,皮损数月后呈放射状皲裂;5.家族性嗜血细胞综合征临床表现不正确的是A:发热B:肝脾大C:多与1岁以上发病D:皮疹E:出血【答案】:C【解析】:家族性嗜血细胞综合征多于1岁以内发病,少数可在生前发病或迟至8岁才开始。
低血糖的诊断与处理

低血糖的病理和临床表现(2) 低血糖病理:脑组织缺糖 充血、多发出血性瘀斑;脑水肿、点状坏死;神经细胞坏死、脑软化。 临床表现:1.大脑皮质受抑制:意识朦胧、定向力与识别力障碍、嗜睡、多汗、肌张力下降、震颤、精神失常等 皮质下中枢受累(基底节、下丘脑、自主神经):骚动不安、痛觉过敏、阵挛性或舞蹈样动作或幼稚动作(鬼脸)、瞳孔散大、强直性惊厥、锥体束阳性。 中脑受累:痉挛、惊厥、眼轴歪斜、病理征等 延脑受累:昏迷、去大脑强直、反射消失、瞳孔缩小等。
低血糖调节的生理(一)
单击此处添加正文,文字是您思想的提炼,为了演示发布的良好效果,请言简意赅地阐述您的观点。您的内容已经简明扼要,字字珠玑,但信息却千丝万缕、错综复杂,需要用更多的文字来表述;但请您尽可能提炼思想的精髓,否则容易造成观者的阅读压力,适得其反。正如我们都希望改变世界,希望给别人带去光明,但更多时候我们只需要播下一颗种子,自然有微风吹拂,雨露滋养。恰如其分地表达观点,往往事半功倍。当您的内容到达这个限度时,或许已经不纯粹作用于演示,极大可能运用于阅读领域;无论是传播观点、知识分享还是汇报工作,内容的详尽固然重要,但请一定注意信息框架的清晰,这样才能使内容层次分明,页面简洁易读。如果您的内容确实非常重要又难以精简,也请使用分段处理,对内容进行简单的梳理和提炼,这样会使逻辑框架相对清晰。
运动对糖代谢的影响运动
(5-10分钟):
肌肉利用葡萄糖增加(为空腹的数倍,胰岛素介导)
内源性葡萄糖生成增多(增加7-8倍;儿茶酚胺)
运动(10分钟以上):
肌糖原利用减少;
机体主要使用血糖和非酯化的脂肪酸提供能量
长时间剧烈运动(数小时):
肝糖原输出≤肌糖原消耗,血糖降低
长期规律运动:
胰岛素敏感性增强(肌肉血流增加,Glut4水平增加
糖尿病急症的治疗及争议问题

糖尿病酮症酸中毒
问题三:DKA是否需要补碱,补碱标准是 什么?
解答: 依据ADA2002指南 1、血气分析pH <7.2可考虑补碱 2、血气分析pH <7.0绝对补碱 3、常用的碱性药物为5%碳酸氢钠250~
500ml,以后根据病情,再决定是否需要补 充
糖尿病乳酸性酸中毒 (LA)
5
乳酸酸中毒
2
高渗性非酮症昏迷
处理原则: 1、积极补液 2、胰岛素的应用 3、补钾 4、并发症的治疗
高渗性非酮症昏迷
治疗: 1、积极补液
补液量的估计 • 按体重的12~15%为第一天补充的液体量,约69L/d • 头5h补充脱水的一半,第1h至少补充10002000ml ;8~12h内输入全天量1/2+尿量
问题二: DKA酮体无法消除的常见原因及处理对 策
解答: 1,输液量不足:这是纠正DKA最重要的措施 2,液体种类不合理:最常见的误区是大量输
盐,这样做一方面会在纠正DKA后发生体液潴 症所必需的。
3,血糖控制不理想,过高或过低都不利于酮 症的治疗
分类: 1、低血糖症 2、高渗性非酮症昏迷 3、糖尿病酮症酸中毒 4、乳酸酸中毒
糖尿病性低血糖症
低血糖症
定义:低血糖症是指人体内血糖低于正常
低限(50mg/dl [2.8mmol/L])引起相应症 状与体征的一种生理或病理状况。
概念区别:
低血糖症:血糖低,有症状 低血糖:血糖低,有或无症状 低血糖反应:有症状,有或无血糖低
输液注意事项 • 补液途径为静脉输注和/或胃肠道补液 • 心、肾功能 • 每日保持糖分在200g左右
高渗性非酮症昏迷
2、胰岛素的应用
z 首先静脉推注10U短效胰岛素 z 此后以2-5u/h的速度静脉点滴 z 当血糖降至13.9-16.7mmol/L后,开始输入5%
磷酸果糖激酶研究进展

磷酸果糖激酶研究进展杨益坤生命科学学院SA16008074摘要:磷酸果糖激酶掌握着糖酵解开始的钥匙,是糖酵解中最重要的控制点。
糖酵解速度主要取决于磷酸果糖激酶,它是个限速酶,其受多个途径调控:受ADP别构激活、ATP别构抑制;受柠檬酸、脂肪酸别构抑制;受果糖-2,6-二磷酸的调控;受H+抑制。
生物体对磷酸果糖激酶的调控十分精细。
对于磷酸果糖激酶的研究和应用主要在于对于糖尿病的治疗,磷酸果糖激酶的变构效应的研究,磷酸果糖激酶在糖酵解中的地位等等。
本文就磷酸果糖激酶的研究进展进行综述。
关键词:磷酸果糖激酶别构激活别构抑制柠檬酸结合位点乳酸糖尿病地位一.磷酸果糖激酶简介名称:磷酸果糖激酶(fructose-1,6-bisphosphate)磷酸果糖激酶1(Phosphofructokinase-1;PFK-1)是一种糖酵解作用里一种重要的酶,是一种由4个次单位组成的异位(allosteric)酵素,可受多种活化剂与抑制剂调控。
糖酵解(glycolysis)是指在细胞质中将葡萄糖分解成为丙酮酸的过程,期间每分解一分子葡萄糖产生两分子丙酮酸以及两分子ATP。
属于呼吸作用的第一个阶段,随后根据有氧与否将丙酮酸进行柠檬酸循环或发酵。
在糖解作用里,PFK-1是负责将果糖-6-磷酸与ATP 转变成为果糖-1,6-双磷酸与ADP。
糖酵解过程中,6-磷酸果糖生成1,6-二磷酸果糖( Fructosel,6bisphosphate,F-1,6-BP),催化此反应的酶是6-磷酸果糖激酶1(6-phosphofructokinase1,PFK1),这是糖酵解途径的第二次磷酸化反应,需要ATP与Mg2+参与,ΔG"0=-14.2KJ/mol,反应不可逆。
6-磷酸果糖激酶1是糖酵解过程的主要限速酶,是糖酵解过程中的主要调节点。
至此,糖酵解完成了代谢的第一个阶段。
催化反应:果糖-6-磷酸+ATP→果糖-1,6-二磷酸+ADP辅助因子:Mg2+催化机制:镁离子将ATP外侧的两个磷酸基团作用,使磷原子更容易接受孤对电子的亲和进攻,使两个磷原子之间的氧桥所共用的电子对向氧原子一方转移,ATP的磷酸集团有利于氧桥断裂,并与果糖-6-磷酸分子结合,生成果糖-1,6-二磷酸。
河北儿科模拟题2021年(26)_真题-无答案

河北儿科模拟题2021年(26) (总分90.31,考试时间120分钟)
多项选择题 1. 男孩3岁。生后4个月可见表情呆滞、易激惹,不能抬头,反复惊厥发作。2岁开始出现呕吐,喂养困难。现小儿智力明显落后,毛发棕黄,皮肤白嫩,尿为霉臭味,尿三氯化铁试验出现绿色。患儿发病的机制可能是 A. 肝细胞内苯丙氨酸羟化酶缺乏 B. 体内有过多的苯丙酮酸 C. 组氨酸羟化酶被抑制 D. 血酪氨酸浓度下降 E. 酪氨酸羟化酶被抑制 F. 二氢生物喋呤还原酶先天缺陷 G. 磷酸果糖激酶缺乏 H. 肌磷酸化酶缺乏 2. 男孩3岁。生后4个月可见表情呆滞、易激惹,不能抬头,反复惊厥发作。2岁开始出现呕吐,喂养困难。现小儿智力明显落后,毛发棕黄,皮肤白嫩,尿为霉臭味,尿三氯化铁试验出现绿色。该患儿诊断一旦明确,应尽早给予积极治疗,下列哪项正确 A. 主要是饮食疗法,开始治疗的年龄愈小,效果愈好。 B. 低苯丙氨酸饮食 C. 苯丙氨酸需要量,4岁以上约10~30mg/(kg·d) D. 维持血中苯丙氨酸浓度在2~10mg/dl为宜。 E. 饮食控制至少需持续到青春期以后 F. 限制淀粉类食物 G. 低铜饮食 H. L-DOPA主要用于BH4缺乏型PKU
A1/A2题型 1. 患儿2岁,智力低下,体格发育迟缓,眼距宽,鼻梁低平,眼裂小,有内眦赘皮,外耳小,舌常伸出口外,头围小于正常,出牙延迟,四肢短,关节可过度弯曲,atd角增大。该病可有下列情况,除外 A. 约30%的患儿伴有先天性心脏病 B. 免疫功能正常 C. 白血病的发生率较正常儿增高10~30倍 D. 性发育延迟 E. 30岁以后常出现老年性痴呆症状 2. 9岁女孩,因进行性面色苍白,肝、脾、淋巴结肿大3年入院。查体:肝肋下5cm,脾肋下6cm,腹壁静脉显露,血象示中度贫血和血小板下降,骨髓检查发现有富含胞浆,内充满洋葱皮样条纹结构,有数个偏心核、直径约20~80μm的细胞。最可能的诊断是 A. 慢性粒细胞性白血病 B. 尼曼-匹克病 C. 肝硬化伴脾亢 D. 戈谢病 E. 肝豆状核变性 3. 2岁女孩,生后体温低,少哭、便秘,面貌特殊,眼距宽,鼻梁平,舌厚肥大。现眼睑水肿,皮肤粗糙、苍黄,智力低下,身高70cm,腕部X线片可见一个骨化中心。该患儿应如何治疗( ) A. 护肝治疗 B. 补充维生素D和钙剂 C. 低苯丙氨酸饮食 D. 终生给予甲状腺激素制剂 E. 不需用药,继续观察 4. 一2岁女孩。出生时正常,母乳喂养,3个月后逐渐智能落后,时有抽搐发作。头发呈棕黄色,肤色白,尿有鼠尿臭味,临床疑为苯丙酮尿症。假如该患儿为刚出生不久,为早期诊断,应选择下列哪项检查( ) A. Gutheie细菌生长抑制试验 B. 尿三氯化铁试验 C. 尿有机酸分析 D. 尿蝶呤分析 E. 血氨基酸分析 5. 1岁小孩,矮小,发育落后,面色苍白,多汗,气促,软弱,无发绀,肝大达肋缘下6cm,质中缘钝,空腹血糖值为2.1mmol/L,轻度血乳酸增高,血气分析示酸中毒。胸片示心脏明显增大。最可能的诊断 A. 先天性心脏病伴心功能不全 B. 糖原累积症 C. 黏多糖病 D. 肝豆状核变性 E. 心肌病 6. 9岁女孩,因进行性面色苍白,肝、脾、淋巴结肿大3年入院。查体:肝肋下5cm,脾肋下6cm,腹壁静脉显露,血象示中度贫血和血小板下降,骨髓检查发现有富含胞浆,内充满洋葱皮样条纹结构,有数个偏心核、直径约20~80μm的细胞。进一步检查 A. 骨髓检查找幼稚细胞 B. 找尼曼-匹克细胞 C. 肝活检 D. 血清铜蓝蛋白测定 E. 白细胞或皮肤成纤维细胞的葡糖脑苷酶酶活性测定 7. 一2岁女孩。出生时正常,母乳喂养,3个月后逐渐智能落后,时有抽搐发作。头发呈棕黄色,肤色白,尿有鼠尿臭味,临床疑为苯丙酮尿症。本症患者中哪一种氨基酸血浓度升高( ) A. 酪氨酸 B. 色氨酸 C. 苯丙氨酸 D. 丙氨酸 E. 丝氨酸 8. 1岁小孩,矮小,发育落后,面色苍白,多汗,气促,软弱,无发绀,肝大达肋缘下6cm,质中缘钝,空腹血糖值为2.1mmol/L,轻度血乳酸增高,血气分析示酸中毒。胸片示心脏明显增大。最可能是疾病的哪种类型 A. 先天性心脏病左向右分流型 B. 糖原累积症Ⅰ型 C. 黏多糖病Ⅰ型 D. 糖原累积症Ⅱ型 E. 心内膜弹力纤维增生症 9. 9岁女孩,因进行性面色苍白,肝、脾、淋巴结肿大3年入院。查体:肝肋下5cm,脾肋下6cm,腹壁静脉显露,血象示中度贫血和血小板下降,骨髓检查发现有富含胞浆,内充满洋葱皮样条纹结构,有数个偏心核、直径约20~80μm的细胞。诊断明确后采用哪种治疗 A. Ceredase注射 B. 化疗 C. 脾切除 D. 肝移植 E. 青霉胺口服 10. 12岁女孩身高118cm,小学4年级,学习成绩欠佳,出生时手足背肿。体检时发现乳房未发育,乳头间距宽,外生殖器呈婴儿型,未有过月经,无其他第二性征。初步诊断为先天性卵巢发育不全综合征。如确诊为先天性卵巢发育不全综合征应如何治疗( ) A. 无需治疗 B. 补充雌激素 C. 应用重组人生长激素+雌激素 D. 补充甲状腺素+雌激素 E. 单纯应用人生长激素 11. 一2岁女孩。出生时正常,母乳喂养,3个月后逐渐智能落后,时有抽搐发作。头发呈棕黄色,肤色白,尿有鼠尿臭味,临床疑为苯丙酮尿症。该患儿出生后最合适的食物是( ) A. 母乳 B. 牛乳 C. 米粉 D. 低苯丙氨酸奶粉 E. 无苯丙氨酸奶粉 12. 男孩,5岁,矮小,轻度驼背,智力发育落后就诊。查体:头大,颈短,鼻梁低,唇厚舌大,表情呆板,皮肤粗糙,腹大,脐疝,肝肋下5cm,脾肋下4cm,四肢不能伸直。最可能的诊断是 A. 甲状腺功能减低症 B. 糖原累积病 C. 肝豆状核变性 D. 黏多糖病 E. 苯丙酮尿症 13. 男,11岁,肝大、肢体震颤、步态不稳6个月,口齿不清,动作僵硬。肝肋下4cm,质 硬,裂隙灯下可见角膜边缘有棕黄色环。最可能的诊断 A. 肝糖原累积症 B. 病毒性肝炎 C. 溶血性贫血 D. 肝豆状核变性 E. 尼曼-匹克病 14. 12岁女孩身高118cm,小学4年级,学习成绩欠佳,出生时手足背肿。体检时发现乳房未发育,乳头间距宽,外生殖器呈婴儿型,未有过月经,无其他第二性征。初步诊断为先天性卵巢发育不全综合征。治疗的目的为( ) A. 使患儿成年后获得生育能力 B. 达到治愈 C. 改善最终成人期身高和性征发育,以获得患儿的心理健康 D. 防止下一代发生类似情况 E. 安慰父母 15. 男孩,5岁,矮小,轻度驼背,智力发育落后就诊。查体:头大,颈短,鼻梁低,唇厚舌大,表情呆板,皮肤粗糙,腹大,脐疝,肝肋下5cm,脾肋下4cm,四肢不能伸直。进一步检查 A. 甲状腺功能 B. 糖代谢功能试验 C. 肝功能+铜蓝蛋白 D. 骨骼X片+尿黏多糖 E. 血苯丙氨酸 16. 男,11岁,肝大、肢体震颤、步态不稳6个月,口齿不清,动作僵硬。肝肋下4cm,质硬,裂隙灯下可见角膜边缘有棕黄色环。进一步检查明确诊断 A. 脑电图 B. 肝脏B超 C. 头颅MRI D. 血清铜蓝蛋白 E. 肌电图 17. 男孩4岁,自幼生长缓慢,多次晨起发生惊厥,来诊时见患儿矮胖,肝肿大达肋缘下6cm,质地中等;空腹血糖值为2.3mmol/L,注射肾上腺素后血糖升高甚微。肝活检切片见肝细胞内大量PAS阳性物质积聚伴多量脂肪滴。诊断首先考虑( ) A. 脑苷脂沉积症 B. 肝豆状核变性 C. 半乳糖血症 D. 糖原累积症 E. 脂肪肝 18. 男,11岁,肝大、肢体震颤、步态不稳6个月,口齿不清,动作僵硬。肝肋下4cm,质硬,裂隙灯下可见角膜边缘有棕黄色环。治疗采用 A. 酶替代治疗 B. 保肝治疗 C. 脾切除 D. 肝移植 E. 青霉胺口服 19. 男孩4岁,自幼生长缓慢,多次晨起发生惊厥,来诊时见患儿矮胖,肝肿大达肋缘下6cm,质地中等;空腹血糖值为2.3mmol/L,注射肾上腺素后血糖升高甚微。肝活检切片见肝细胞内大量PAS阳性物质积聚伴多量脂肪滴。经肝细胞酶检测发现葡萄糖-6-磷酸酶缺陷,确诊为1型糖原累积症,当前的治疗原则为( ) A. 首先保持血糖正常并预防和积极治疗感染及控制酸中毒 B. 进行肝移植 C. 酶替代治疗 D. 保肝+控制感染和酸中毒 E. 控制酸中毒 20. 患儿2岁,智力低下,体格发育迟缓,眼距宽,鼻梁低平,眼裂小,有内眦赘皮,外耳小,舌常伸出口外,头围小于正常,出牙延迟,四肢短,关节可过度弯曲,atd角增大。最可能的诊断为 A. 21-三体综合征 B. 高精氨酸血症 C. 先天性甲状腺功能低下 D. 苯丙酮尿症 E. 半乳糖血症 21. 患儿9个月,生后6个月起进食后经常呕吐,皮肤湿疹,智力及体格发育渐落后于同龄儿,喜睡,有癫痫小发作,尿有鼠尿臭味,毛发及虹膜颜色浅,皮肤白,尿三氯化铁试验阳性。最可能的诊断为 A. 苯丙酮尿症 B. 呆小病 C. 癫痫 D. 先天愚型 E. 组氨酸血症 22. 2岁女孩,生后体温低,少哭、便秘,面貌特殊,眼距宽,鼻梁平,舌厚肥大。现眼睑水肿,皮肤粗糙、苍黄,智力低下,身高70cm,腕部X线片可见一个骨化中心。最可能的诊断为( ) A. 先天愚型 B. 先天性甲状腺功能减低症 C. 黏多糖病 D. 苯丙酮尿症 E. 软骨发育不良 23. 男孩4岁,自幼生长缓慢,多次晨起发生惊厥,来诊时见患儿矮胖,肝肿大达肋缘下6cm,质地中等;空腹血糖值为2.3mmol/L,注射肾上腺素后血糖升高甚微。肝活检切片见肝细胞内大量PAS阳性物质积聚伴多量脂肪滴。肝糖原累积症保持血糖正常的最合理方法为( ) A. 应用升血糖药物 B. 静脉输葡萄糖液 C. 升血糖药物+静脉输葡萄糖液 D. 频频摄入碳水化物或频频口服生玉米淀粉 E. 手术治疗 24. 患儿2岁,智力低下,体格发育迟缓,眼距宽,鼻梁低平,眼裂小,有内眦赘皮,外耳小,舌常伸出口外,头围小于正常,出牙延迟,四肢短,关节可过度弯曲,atd角增大。下
03糖代谢-4糖原的合成与分解

腺苷酸环化酶(活性)
肾上腺素或 胰高血糖素
1、腺苷酸环化酶
的共价修饰反 应是酶促反应, 只要有少量信 号分子(如激 素)存在,即 可通过加速这 种酶促反应, 而使大量的另 一种酶发生化 学修饰,从而 获得放大效应。 这种调节方式 快速、效率极 高。
30
2、ATP
cAMP
2
102
3
3、蛋白激酶
(无活性)
-
磷酸化酶b激酶 磷酸化酶b激酶-P 糖原合酶 (有活性) Pi 糖原合酶-P 磷酸化酶b (无活性)
磷蛋白磷酸酶-1
Pi
+
–
磷蛋白磷酸酶-1
磷酸化酶a-P
磷蛋白磷酸酶-1
Pi
–
PKA(有活性)
28
–
磷蛋白磷酸酶抑制剂-P 磷蛋白磷酸酶抑制剂
激素通过cAMP-蛋白激酶调节代谢示意图
激素 受体 G蛋白 环化酶
糖原累积症
糖原累积症(glycogen storage diseases)是一 类遗传性代谢病,其特点为体内某些器官组织 中有大量糖原堆积。会导致肝硬化和肝功能衰 竭。除肝病变外,大部分患者有肌无力,尤其 疾走和爬山时,部分患者有肌肉萎缩。糖原可累
积在心脏,出现心脏增大。
引起糖原累积症的原因是:
糖原合成与分解均在细胞质中进行……
23
代谢调节与关键酶
酶定位的区域化:“合成”、“分解”割据 一方。 酶活性的调节:“增加一个,降低一个”。防 止无效循环。 共价修饰调节:难点、重点 磷酸化酶 酶活性的调节 Gn
糖原合酶 别构调节
G
24
1、糖原代谢的共价修饰调节
酶分子中的某些基团,在其它酶的催化下,可以 共价结合或脱去,引起酶分子构象的改变,使其活性 得到调节,这种方式称为酶的共价修饰(Covalent moldification )。磷酸化/去磷酸化是主要形式。
血脂异常的诊断与治疗
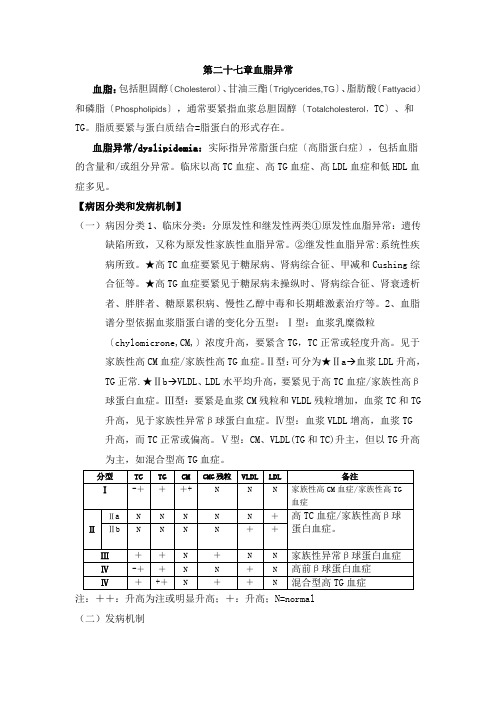
第二十七章血脂异常血脂:包括胆固醇〔Cholesterol〕、甘油三酯〔Triglycerides,TG〕、脂肪酸〔Fattyacid〕和磷脂〔Phospholipids〕,通常要紧指血浆总胆固醇〔Totalcholesterol,TC〕、和TG。
脂质要紧与蛋白质结合=脂蛋白的形式存在。
血脂异常/dyslipidemia:实际指异常脂蛋白症〔高脂蛋白症〕,包括血脂的含量和/或组分异常。
临床以高TC血症、高TG血症、高LDL血症和低HDL血症多见。
【病因分类和发病机制】(一)病因分类1、临床分类:分原发性和继发性两类①原发性血脂异常:遗传缺陷所致,又称为原发性家族性血脂异常。
②继发性血脂异常:系统性疾病所致。
★高TC血症要紧见于糖尿病、肾病综合征、甲减和Cushing综合征等。
★高TG血症要紧见于糖尿病未操纵时、肾病综合征、肾衰透析者、胖胖者、糖原累积病、慢性乙醇中毒和长期雌激素治疗等。
2、血脂谱分型依据血浆脂蛋白谱的变化分五型:Ⅰ型:血浆乳糜微粒〔chylomicrone,CM,〕浓度升高,要紧含TG,TC正常或轻度升高。
见于家族性高CM血症/家族性高TG血症。
Ⅱ型:可分为★Ⅱa→血浆LDL升高,TG正常.★Ⅱb→VLDL、LDL水平均升高,要紧见于高TC血症/家族性高β球蛋白血症。
Ⅲ型:要紧是血浆CM残粒和VLDL残粒增加,血浆TC和TG 升高,见于家族性异常β球蛋白血症。
Ⅳ型:血浆VLDL增高,血浆TG升高,而TC正常或偏高。
Ⅴ型:CM、VLDL(TG和TC)升主,但以TG升高为主,如混合型高TG血症。
注:++:升高为注或明显升高;+:升高;N=normal(二)发病机制1、获得性因素★高脂饮食:常见★体重增加:原发性胖胖及胰岛素抵抗为特征的代谢综合征是血浆TC、TG升高的常见缘故。
★增龄:血浆TC随年龄而升高〔缘故:机体分解代谢下落,LDL受体活性落低〕。
★雌激素缺乏:雌激素增加LDL分解。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第二型糖原累积病的治疗
*导读:于1963年由Hers发现,其病因为溶酶体中α1,4-葡萄糖苷酶(αl,4glucosidase)即酸性麦芽糖酶缺乏,以致糖原及麦芽糖不能转化为葡萄糖而被利用,全身组织均有糖原沉着。
……
此型亦称Pompe"s病,于1963年由Hers发现,其病因为溶酶体中α1,4-葡萄糖苷酶(αl,4glucosidase)即酸性麦芽糖酶缺乏,以致糖原及麦芽糖不能转化为葡萄糖而被利用,全身组织均有糖原沉着,包括脑及视网膜毛细血管等。
本病不伴有低血糖,酮症,高脂血症或其他中间代谢异常。
本病可分为婴儿型,青少年型及成年型。
婴儿型多在3~6个月内出现症状,有的在出生时即有表现。
临床表现为骨骼肌张力低,心脏明显扩大,舌体增大及不同程度的肝肿大,脸容可似克汀病婴儿,常有呛咳,呼吸困难,EKG可表现为QRS波增宽,PR间期缩短,多数患儿在2~3岁时因心脏病变及呼吸肌衰竭而死亡。
青少年型表现为进行性肌营养不良,病人有步态异常,但无心脏表现。
成年发病者仅表现为骨骼肌无力,症状有时非常轻微可无症状。
治疗
至今防治尚缺乏有效措施。
有人采用黑色曲菌(Aspergillus niger)的a-糖甙酶由腹腔注入大白鼠后,发现肝,心及肌肉糖
原减少,也有试用于患者的,仅能使肝糖原暂时下降,但临床上尚属研究阶段。