分子动力学在材料科学中的应用

分子动力学在材料科学中的应用
摘要:本文综述了几种常见条件下的分子动力学模拟方法以及分子动力学模拟的最新发展趋势.介绍用分子动力学模拟方法研究固休的休相结构,表面问题,界面问题以及薄膜形成过程等方面的研究成果。
关键词:分子动力学; 计算机模拟; 材料科学
1引言
分子动力学(Molecular Dyanmica,简称MD)用于计算以固体、液体、气体为模型的单个分子运动,它是探索各种现象本质和某些新规律的一种强有力的计算机模拟方法,具有沟通宏观特性与微观结构的作用,对于许多在理论分析和实验观察上难以理解的现象可以做出一定的解释[1]。MD方法不要求模型过分简化,可以基于分子(原子、离子)的排列和运动的模拟结果直接计算求和以实现宏观现象中的数值估算。可以直接模拟许多宏观现象,取得和实验相符合或可以比较的结果,还可以提供微观结构、运动以及它们和体系宏观性质之间关系的极其明确的图象[2]。MD以其不带近似、跟踪粒子轨迹、模拟结果准确[3],而倍受研究者的关注,在物理、化学、材料、摩擦学等学科及纳米机械加工中得到广泛而成功的应用。本文主要评述MD方法在材料科学中的应用.
目前在材料微观结构的研究中,由于实验条件的限制,使得许多重要的微观结构的信息难以得到,如,对于由液态金属快速凝固的非晶转变过程,其微观结构的瞬时变化根本无法用实验仪器去测量。理论分析、实验测定及模拟计算已成为现代材料科学研究的3种主要方法[2]。20世纪90年代以来,由于计算机科学和技术的飞速发展,模拟计算的地位日渐突显。计算机模拟可以提供实验上尚无法获得或很难获得的信息。虽然计算机模拟不能完全取代实验,但可以用来指导实验的进行,从而促进理论和实践的发展,所以有必要对这一领域进行介绍。
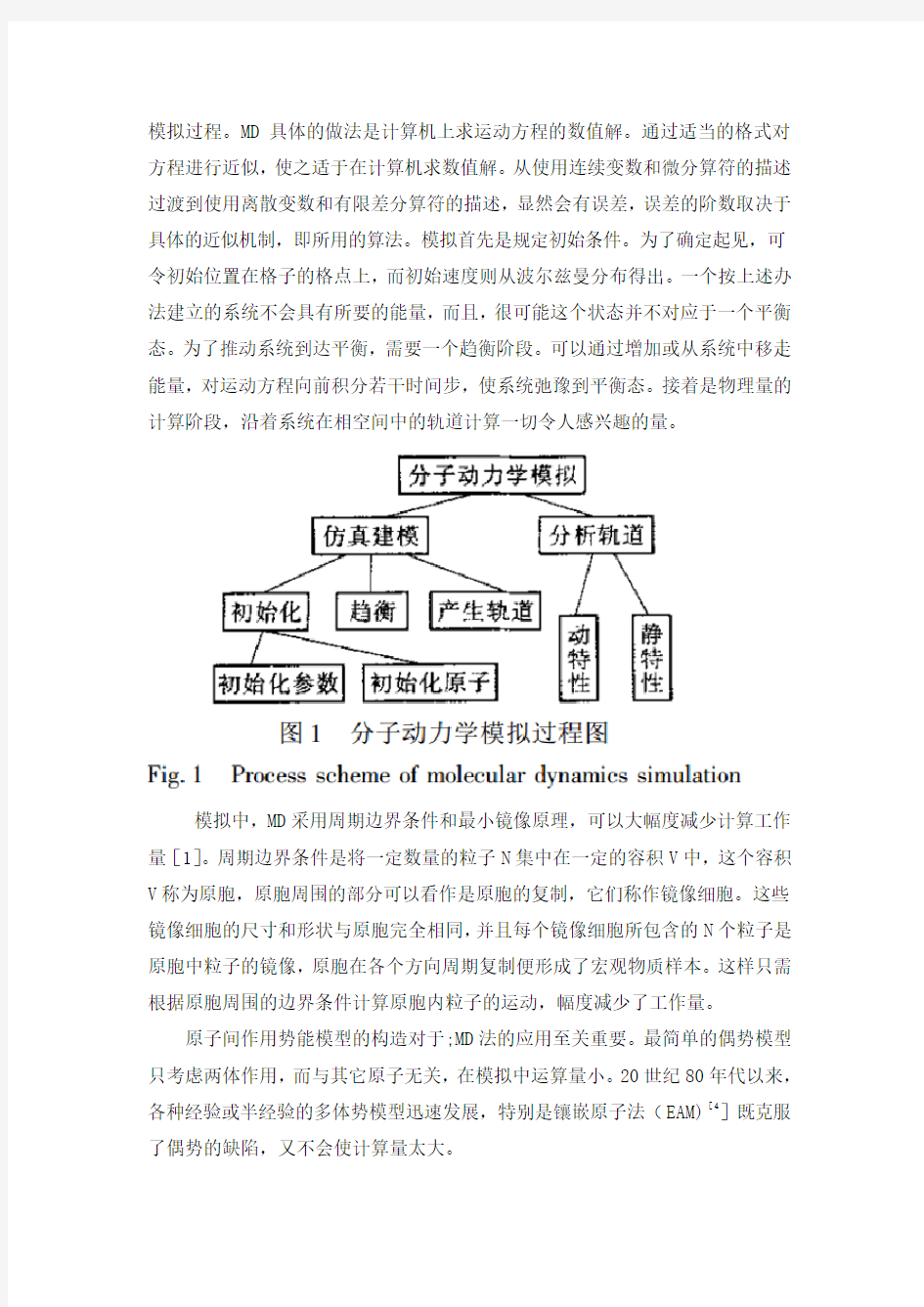
2 分子动力学基本原理
分子动力学将连续介质看成由N个原子或分子组成的粒子系统,各粒子之间的作用力可以通过量子力学势能函数求导得出,忽略量子效应后,运用经典牛顿力学建立系统粒子运动数学模型,通过数值求解得到粒子在相空间的运动轨迹,然后由统计物理学原理得出该系统相应的宏观动态、静态特性。图1所示是MD
模拟过程。MD具体的做法是计算机上求运动方程的数值解。通过适当的格式对方程进行近似,使之适于在计算机求数值解。从使用连续变数和微分算符的描述过渡到使用离散变数和有限差分算符的描述,显然会有误差,误差的阶数取决于具体的近似机制,即所用的算法。模拟首先是规定初始条件。为了确定起见,可令初始位置在格子的格点上,而初始速度则从波尔兹曼分布得出。一个按上述办法建立的系统不会具有所要的能量,而且,很可能这个状态并不对应于一个平衡态。为了推动系统到达平衡,需要一个趋衡阶段。可以通过增加或从系统中移走能量,对运动方程向前积分若干时间步,使系统弛豫到平衡态。接着是物理量的计算阶段,沿着系统在相空间中的轨道计算一切令人感兴趣的量。
模拟中,MD采用周期边界条件和最小镜像原理,可以大幅度减少计算工作量[1]。周期边界条件是将一定数量的粒子N集中在一定的容积V中,这个容积V称为原胞,原胞周围的部分可以看作是原胞的复制,它们称作镜像细胞。这些镜像细胞的尺寸和形状与原胞完全相同,并且每个镜像细胞所包含的N个粒子是原胞中粒子的镜像,原胞在各个方向周期复制便形成了宏观物质样本。这样只需根据原胞周围的边界条件计算原胞内粒子的运动,幅度减少了工作量。
原子间作用势能模型的构造对于;MD法的应用至关重要。最简单的偶势模型只考虑两体作用,而与其它原子无关,在模拟中运算量小。20世纪80年代以来,各种经验或半经验的多体势模型迅速发展,特别是镶嵌原子法(EAM)[4]既克服了偶势的缺陷,又不会使计算量太大。
3分子模拟主要技术细节
3.1 分子间作用势
分子动力学模拟的首要条件就是要知道分子间的相互作用势,分子间作用势函数确定后,通过势函数对“求导即可得出分子间的作用力。所以必须对分:子间的作用势进行研究,目前存在多种势能模型。分子间势函数的发展经历了对势一多体势的过程。对势认为原子之间的相互作用是两两之间的作用,与其他原子的位置无关;而实际上,在多原子体系中,一个原子的位置不同,将影响空间一定范围内的电子云分布,从而影响其他原子之间的有效相互作用,故多原子体系的势函数需更准确地用多体势表示。
在分子动力学模拟的初期,人们经常采用的是对势。常用的对势模型包括Lennard—Jones势、Morse势、Born—Lande势及Johnson势。在对势模型中,系统能量表示为原子对相互作用能量的加和。对于简单的、高对称性的晶体,对势能够描述原子间的相互作用。对势模型的主要缺点是忽略了多体原子间的相互作用,更具体地说,它是忽略了由于原子局域环境的变化引起的原子间相互作用的变化,因此需要建立新的模型。
基于EAM势的势函数还有很多种。这些多体势大都用于金属的微观模拟。为了将EAM势推广到共价键材料,需要考虑电子云的非球形对称。于是,Baskes 等提出了修正型嵌入原子核法(MEAM)。经过修正,Baskes理论已能解决上述问题,但其确定参数的过程相当复杂,应用上仍受到很大的限制。Pasianot等试图在总能量中加入一修正项,以修正原EAM模型中2条基本假设所引起的能量差。这一模型成功地描述了Nb、Fe、Cr等元素,但应用起来很繁杂。张邦维等综合考虑以上EAM模型的优缺点后,提出了分析型EAM理论,成为一个普适分析型EAM模型。胡望宇等在张邦维分析型EAM模型的基础上进行了改进,提出了改进的分析型EAM模型(MAEAM)。此外,还有许多形式的多体势函数形式,如Jacobsen 等在等效介质原理(EMT)的基础上提出的另一种函数形式,由于其简单、有效,因此也得到了广泛的应用。势函数确定后,通过对势函数求导即可得出分子间的作用力。
3.2周期性边界条件
原子和分子体系的分子模拟的目的是提供一个宏观样本的物性信息。在具有自
由边界的三维N个粒子的体系中处于界面的分子数正比于N1脂。在使用有限的原子数来模拟实际体系中原子的运动时,必须考虑表面对体系中原子运动的影响。为避免这种影响,可以通过周期性边界条件来实现。将含有N个粒子的体积当作具有与其相同单元无限周期点阵的原始单元(如图1是一个二维的示意图,在这个二维图象中每个单胞被其他的8个单胞所包围;在三维方向上每个单胞就会被26个单胞所包围),一给定粒子则与在此无限周期体系中的全部其他粒子相互作用。对有些模拟,在所有方向都用周期性边界条件是不合适的。比如在研究表面的分子吸附时,在与表面垂直方向上不能用周期性边界条件,而仅在平行于表面的2个方向需要应用周期性边界条件。
4 分子动力学在材料科学中的应用
4.1 分子动力学的适用范围
材料科学中计算模拟研究范围极为广泛,从埃量级的量子力学计算到连续介质层次的有限元或有限差分模型,可分为4个层次:电子、原子、显微组织和宏观层次(见图2)。;MD主要是原子尺度上研究体系中与时间和温度有关的性质的模拟方法。
最早将分子动力学方法用于材料研究中的是Vineyard 于1960年探讨材料辐射损伤的动力学规律。模拟结果给出了原子轨迹,这一工作使得过去对热力学性能的定性估计迈向对微观过程的定量研究.1964年Rahman用MD方法模拟液体氩,同时加进了周期性边界条件,结果他惊奇地发现可以用很少的粒子(864个)来反映真实系统的热力学性质。自此,凝聚态物质的分子动力学模拟成为可能,许多研究者纷纷投入这一研究工作。
最初应用是基于偶函数,如Lendard-Jones势函数和Morse势函数,模型简单,运算量小,而得以在材料科学中广泛应用。但由于其未考虑到体积相关项,在研究材料的弹性系数性质和预言金属的结合能及空位形成能时,难以获得准确的结果[5]。EAM多体势主要用于fcc型金属及其合金中,处理其结构、热力学、表面、缺陷及液态金属等问题,也应用于hcp及bcc型金属及合金,以及半导体Si。
一般,MD方法在中型机或微机上进行时,由于其内存和运算速度的限制,模拟研究只能限于500-1000个原子的小系统。因而模拟结果虽然也能揭示一些微观结构的特征和规律,但与实际的大系统情况有较大差异。在并行处理系统上对更大量的原子系统进行模拟研究[6],其结果必然会接近于实际,从而对生产实践将会更有实际指导意义。
4.2 分子动力学的应用
4.2.1 金属的液态结构
在目前实验条件下,液态金属的结构及其变化尚很难精确测定。王鲁红等人
[7]采用F-S型多体势描述了8种hcp型金属的液态微观结构并与实验相比较,模拟结果表明,Mg、Co和Zn的势函数可以较好的描述其液态结构,Ti和Zr的势函数则不能;由Be和Ru的势函数描述的液态结构较为合理,Hf 则与一般液态结构特点不一致。李辉等人[8]采用EAM多体势模型,很好地描述了液态过渡金属Ni的结构变化特性。
4.2.2 薄膜形成过程
薄膜研究是当今科学研究的热点之一。目前在很多薄膜制备方法中,都应用了低能离子轰击技术,在这些方法中,低能离子/表面相互作用在控制薄膜的微观结构方面起着重要作用。由于离子/表面相互作用发生在时间间隔小于10-12s 内,因而特别适合于用MD方法对这一过程进行描述。薄腊研究是当今科学研究的热点之一目前在很多薄摸制备方法中,都应用了低能离子轰击技术,如离子束增强沉积,等离子休辅助化学汽相沉积、溅射沉积、离子辅助分子束处延等。Garrison[9],Kitabtake[9]和郊正明四等人分别用低能粒子轰击Si(ool)一2xl表面,由此可用分子动力学方法研究低能粒子对表面原子行为的影响。郊等人的研究表明,10ev,100ev粒子的轰击,一方面增强了表面原子形成二聚体(dimer)的能力,使表面二聚体键数增加,另一方面也使表面原子的排列更趋无序。
Ethier和Lewis[10]模拟了纯Si、Si
0.5Ge
0.5
和纯Ge在si(loo)-2xl再构表面
上用分子束处延(MBE)法生长膜的过程,其结果给出了薄膜质量与衬底温度之间的关系,即衬底温度较低时,形成的结构有序性较差;在高的衬底温度下,发生外延生长。对于纯Ge的外延生长,只有最初的三层结晶,以后便出现岛状结构,这在定性上与实验和理论结果相一致。
4.2.3界面研究
文献中大量报道了近十儿年来关于晶界的一些分子动力学模拟结果.目前有关界面的分子动力学模拟研究开展的不多.金属一金属界面的分子动力学模拟研究还有一些报道.李明研究了Ag/Ni界面处裂纹扩展行为.Ag/Ni和Cu/Ni界面在弯曲状态下的力学行为作了分子动力学模拟研究,结果给出的力学性能曲线与宏观规律相符合,并给出了进行界面模拟时计算元胞的原子数为1000个左右.同时证明了界面的存在对复合材料性能影响很大,界面结构不同复合材料的性能也不相同。Yang等人研究了Ni(100)面涂弋层的结构和动力学行为,并对嵌人原子法
(EAM)和对势结果作了比较.Rahrnan等人对上述问题作了进一步研究,给出了单层Ag在Ni(loo)面上的结构与温度之间的关系。即室温下,Ag在Ni衬底上前后滑动,距Ni上表面平均距离为 2.15埃,温度为1200K时,Ag在Ni上形成孔泡;1300K时,Ag在Ni上形成单晶.这些工作足以说明用分子动力学方法研究界面问题是可行的.采用分子动力学模拟方法进行界面研究是十分有意义的工作,尤其是目前正急于解决的金属一陶瓷界面问题。
4.2.4固体体相结构性能的研究
Parrinell和Rahman是最先采用分子动力学方法研究固体性能的,以此代替Milstein和Faber的静力学计算.其对象是Ni单晶在单轴压力下由面心立方结构向密排六方结构转变的过程,这是等温一等压分子动力学问题,所用势函数是Morse对势,其结果给出了应力一应变曲线,并与Milstein和Faber的汁算结果符合得相当好。Zhong[8]等人利用在嵌人原子法(Embedded一Atom一Method一EAM)的基础上发展起来的MBA势(Many一Body-Alloy)研究Pd一H系统的力学稳定性.所选取的势函数形式是Morse势.其中利用了Nose[15]和Rahman一Parrincllo分子动力学形式。结果表明氢脆的微观起因是氢饱和使某些区域的韧性和塑性增强,这与假定的氢增强局域塑性的机制相一致。
5 结论
从国内外分子动力学的模拟研究可以看出,随着计算机的普及及计算能力的提高,分子模拟方法的推广应用日益受到高度重视。模拟已逐渐从分子结构简单的体系扩展到分子结构复杂的体系,模拟粒子数已从一二百扩展到几千,并从单相扩展到多相、从均相扩展到非均相,位能函数也从简单扩展到复杂乃至要考虑量子效应。模拟方法也已经从Mc转至MD,以模拟真正的动力学过程。MD模拟方法也从平衡态模拟发展到外加场的非平衡态模拟。可以预料,分子模拟与材料的关系将更加紧密,它对材料科学工程的基础研究、工艺过程以及新产品的开发都会发挥更为明显的、不可替代的作用。
参考文献
[1]罗熙淳,梁迎春,董申. 分子动力学在纳米机械加工技术中的应用[J]中国机械工程,1999,10(60:692
[2]胡壮麒,王鲁红,刘轶电子和原子层次材料行为的计
算机模拟[J]材料研究学报,1099,12(1):1
[3] Nose S.J.Chem.Phys.1984.85:511
[4] Rice B D,Chcn J.Philos,Mag,Lctt.1990,61:139
[5]卢百平,张丰收,傅恒志材料科学中的分子动力学模拟
[6]编者西北工业大学2002年材料论坛论文集[C]+
西安:2002.1-4
[7]郭俊梅,邓德国,潘建生,等计算材料与材料设计[J]+
贵金属,1999,20(4):62
[8]董科军,刘让苏,郑采星,等液态金属凝固过程分子动力
学模拟的并行算法研究[J]+ 计算机科学与工程,2002,22(3):45
[9] Zhang W.Cai Y,Tomanck D. Phys.Rev.B,1992,46:8099
[10] Puska M J,Nicminen R M,Manninen M.Phys.Rev.B.1981,24:3037
分子动力学的模拟过程
分子动力学的模拟过程 分子动力学模拟作为一种应用广泛的模拟计算方法有其自身特定的模拟步骤,程序流程也相对固定。本节主要就分子动力学的模拟步骤和计算程序流程做一些简单介绍。 1. 分子动力学模拟步驟 分子动力学模拟是一种在微观尺度上进行的数值模拟方法。这种方法既可以得到一些使用传统方法,热力学分析法等无法获得的微观信息,又能够将实际实验研究中遇到的不利影响因素回避掉,从而达到实验研宄难以实现的控制条件。 分子动力学模拟的步骤为: (1)选取所要研究的系统并建立适当的模拟模型。 (2)设定模拟区域的边界条件,选取粒子间作用势模型。 (3)设定系统所有粒子的初始位置和初始速度。 (4)计算粒子间的相互作用力和势能,以及各个粒子的位置和速度。 (5)待体系达到平衡,统计获得体系的宏观特性。 分子动力学模拟的主要对象就是将实际物理模型抽象后的物理系统模型。因此,物理建模也是分子动力学模拟的一个重要的环节。而对于分子动力学模拟,主要还是势函数的选取,势函数是分子动力学模拟计算的核心。这是因为分子动力学模拟主要是计算分子间作用力,计算粒子的势能、位置及速度都离不开势函数的作用。系统中粒子初始位置的设定最好与实际模拟模型相符,这样可以使系统尽快达到平衡。另外,粒子的初始速度也最好与实际系统中分子的速度相当,这样可以减少计算机的模拟时间。 要想求解粒子的运动状态就必须把运动方程离散化,离散化的方法有经典Verlet算法、蛙跳算法(Leap-frog)、速度Veriet算法、Gear预估-校正法等。这些算法有其各自的优势,选取时可按照计算要求选择最合适的算法。 统计系统各物理量时,便又涉及到系统是选取了什么系综。只有知道了模拟系统采用的系综才能釆用相对应的统计方法更加准确,有效地进行统计计算,减少信息损失。 2. 分子动力学模拟程序流程 具体到分子动力学模拟程序的具体流程,主要包括: (1)设定和模拟相关的参数。 (2)模拟体系初始化。 (3)计算粒子间的作用力。 (4)求解运动方程。 (5)循环计算,待稳定后输出结果。 分子动力学模拟程序流程图如2.3所示。
分子动力学模拟方法概述(精)
《装备制造技术》 2007年第 10期 收稿日期 :2007-08-21 作者简介 :申海兰 , 24岁 , 女 , 河北人 , 在读研究生 , 研究方向为微机电系统。 分子动力学模拟方法概述 申海兰 , 赵靖松 (西安电子科技大学机电工程学院 , 陕西西安 710071 摘要 :介绍了分子动力学模拟的基本原理及常用的原子间相互作用势 , 如Lennard-Jones 势 ; 论述了几种常用的有限差分算法 , 如 Verlet 算法 ; 说明了分子动力学模拟的几种系综及感兴趣的宏观统计量的提取。关键词 :分子动力学模拟 ; 原子间相互作用势 ; 有限差分算法 ; 系综中图分类号 :O3 文献标识码 :A 文章编号 :1672-545X(200710-0029-02 从统计物理学中衍生出来的分子动力学模拟方法 (molec- ular dynamics simulation , M DS , 实践证明是一种描述纳米科技 研究对象的有效方法 , 得到越来越广泛的重视。所谓分子动力学模拟 , 是指对于原子核和电子所构成的多体系统 , 用计算机模拟原子核的运动过程 , 从而计算系统的结构和性质 , 其中每一个原子核被视为在全部其他原子核和电子所提供的经验势场作用下按牛顿定律运动 [1]。它被认为是本世纪以来除理论分析和实验观察之外的第三种科学研究手段 , 称之为“计算机实验” 手段 [2], 在物理学、化学、生物学和材料科学等许多领域中得到广泛地应用。
根据模拟对象的不同 , 将它分为平衡态分子动力学模拟 (EM DS (和非平衡态分子动力学模拟 (NEM DS 。其中 , EM DS 是分子动力学模拟的基础 ; NEM DS 适用于非线性响应系统的模拟 [3]。下面主要介绍 EM DS 。 1分子动力学方法的基本原理 计算中根据以下基本假设 [4]: (1 所有粒子的运动都遵循经典牛顿力学规律。 (2 粒子之间的相互作用满足叠加原理。 显然这两条忽略了量子效应和多体作用 , 与真实物理系统存在一定差别 , 仍然属于近似计算。 假设 N 为模拟系统的原子数 , 第 i 个原子的质量为 m i , 位置坐标向量为 r i , 速度为 v i =r ? i , 加速度为 a i =r ?? i , 受到的作用力为 F i , 原子 i 与原子 j 之间距离为 r ij =r i -r j , 原子 j 对原子 i 的作用力为 f ij , 原子 i 和原子 j 相互作用势能为 ! (r ij , 系统总的势能为 U (r 1, r 2, K r N = N i =1! j ≠ i ! " (r ij , 所有的物理量都是随时 间变化的 , 即 A=A (t , 控制方程如下 : m i r ?? i =F i =j ≠ i
分子动力学在材料科学中的应用
分子动力学在材料科学中的应用 摘要:本文综述了几种常见条件下的分子动力学模拟方法以及分子动力学模拟的最新发展趋势.介绍用分子动力学模拟方法研究固休的休相结构,表面问题,界面问题以及薄膜形成过程等方面的研究成果。 关键词:分子动力学; 计算机模拟; 材料科学 1引言 分子动力学(Molecular Dyanmica,简称MD)用于计算以固体、液体、气体为模型的单个分子运动,它是探索各种现象本质和某些新规律的一种强有力的计算机模拟方法,具有沟通宏观特性与微观结构的作用,对于许多在理论分析和实验观察上难以理解的现象可以做出一定的解释[1]。MD方法不要求模型过分简化,可以基于分子(原子、离子)的排列和运动的模拟结果直接计算求和以实现宏观现象中的数值估算。可以直接模拟许多宏观现象,取得和实验相符合或可以比较的结果,还可以提供微观结构、运动以及它们和体系宏观性质之间关系的极其明确的图象[2]。MD以其不带近似、跟踪粒子轨迹、模拟结果准确[3],而倍受研究者的关注,在物理、化学、材料、摩擦学等学科及纳米机械加工中得到广泛而成功的应用。本文主要评述MD方法在材料科学中的应用. 目前在材料微观结构的研究中,由于实验条件的限制,使得许多重要的微观结构的信息难以得到,如,对于由液态金属快速凝固的非晶转变过程,其微观结构的瞬时变化根本无法用实验仪器去测量。理论分析、实验测定及模拟计算已成为现代材料科学研究的3种主要方法[2]。20世纪90年代以来,由于计算机科学和技术的飞速发展,模拟计算的地位日渐突显。计算机模拟可以提供实验上尚无法获得或很难获得的信息。虽然计算机模拟不能完全取代实验,但可以用来指导实验的进行,从而促进理论和实践的发展,所以有必要对这一领域进行介绍。 2 分子动力学基本原理 分子动力学将连续介质看成由N个原子或分子组成的粒子系统,各粒子之间的作用力可以通过量子力学势能函数求导得出,忽略量子效应后,运用经典牛顿力学建立系统粒子运动数学模型,通过数值求解得到粒子在相空间的运动轨迹,然后由统计物理学原理得出该系统相应的宏观动态、静态特性。图1所示是MD
分子动力学方法模拟基本步骤
分子动力学方法模拟基本步骤 1.第一步 即模型的设定,也就是势函数的选取。势函数的研究和物理系统上对物质的描述研究息息相关。最早是硬球势,即小于临界值时无穷大,大于等于临界值时为零。常用的是LJ势函数,还有EAM势函数,不同的物质状态描述用不同的势函数。 模型势函数一旦确定,就可以根据物理学规律求得模拟中的守恒量。 2 第二步 给定初始条件。运动方程的求解需要知道粒子的初始位置和速度,不同的算法要求不同的初始条件。如:verlet算法需要两组坐标来启动计算,一组零时刻的坐标,一组是前进一个时间步的坐标或者一组零时刻的速度值。 一般意思上讲系统的初始条件不可能知道,实际上也不需要精确选择代求系统的初始条件,因为模拟实践足够长时,系统就会忘掉初始条件。当然,合理的初始条件可以加快系统趋于平衡的时间和步伐,获得好的精度。 常用的初始条件可以选择为:令初始位置在差分划分网格的格子上,初始速度则从玻尔兹曼分布随机抽样得到;令初始位置随机的偏离差分划分网格的格子上,初始速度为零;令初始位置随机的偏离差分划分网格的格子上,初始速度也是从玻尔兹曼分布随机抽样得到。 第三步 趋于平衡计算。在边界条件和初始条件给定后就可以解运动方程,进行分子动力学模拟。但这样计算出的系统是不会具有所要求的系统的能量,并且这个状态本身也还不是一个平衡态。 为使得系统平衡,模拟中设计一个趋衡过程,即在这个过程中,我们增加或者从系统中移出能量,直到持续给出确定的能量值。我们称这时的系统已经达到平衡。这段达到平衡的时间成为驰豫时间。 分子动力学中,时间步长的大小选择十分重要,决定了模拟所需要的时间。为了减小误差,步长要小,但小了系统模拟的驰豫时间就长了。因此根据经验选择适当的步长。如,对一个具有几百个氩气Ar分子的体系,lj势函数,发现取h为0.01量级,可以得到很好的相图。这里选择的h是没有量纲的,实际上这样选择的h对应的时间在10-14s的量级呢。如果模拟1000步,系统达到平衡,驰豫时间只有10-11s。 第四步 宏观物理量的计算。实际计算宏观的物理量往往是在模拟的最后揭短进行的。它是沿相空间轨迹求平均来计算得到的(时间平均代替系综平均)
分子动力学模拟
分子动力学模拟 分子动力学就是一门结合物理,数学与化学的综合技术。分子动力学就是一套分子模拟方法,该方法主要就是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量与其她宏观性质。 这门技术的发展进程就是: 1980年:恒压条件下的动力学方法(Andersenの方法、Parrinello-Rahman法) 1983年:非平衡态动力学方法(Gillan and Dixon) 1984年:恒温条件下的动力学方法(能势‐フーバーの方法) 1985年:第一原理分子动力学法(→カー?パリネロ法) 1991年:巨正则系综的分子动力学方法(Cagin and Pettit)、 最新的巨正则系综,即为组成系综的系统与一温度为T、化学势为μ的很大的热源、粒子源相接触,此时系统不仅同热源有能量交换,而且可以同粒子源有粒子的交换,最后达到平衡,这种系综称巨正则系综。 进行分子动力学模拟的第一步就是确定起始构型,一个能量较低的起始构型就是进行分子模拟的基础,一般分子的其实构型主要就是来自实验数据或量子化学计算。在确定起始构型之后要赋予构成分子的各个原子速度,这一速度就是根据玻尔兹曼分布随机生成,由于速度的分布符合玻尔兹曼统计,因此在这个阶段,体系的温度就是恒定的。另外,在随机生成各个原子的运动速度之后须进行调整,使得体系总体在各个方向上的动量之与为零,即保证体系没有平动位移。 由上一步确定的分子组建平衡相,在构建平衡相的时候会对构型、温度等参数加以监控。 进入生产相之后体系中的分子与分子中的原子开始根据初始速度运动,可以想象其间会发生吸引、排斥乃至碰撞,这时就根据牛顿力学与预先给定的粒子间相互作用势来对各个例子的运动轨迹进行计算,在这个过程中,体系总能量不变,但分子内部势能与动能不断相互转化,从而体系的温度也不断变化,在整个过程中,体系会遍历势能面上的各个点,计算的样本正就是在这个过程中抽取的。 用抽样所得体系的各个状态计算当时体系的势能,进而计算构型积分。 作用势的选择与动力学计算的关系极为密切,选择不同的作用势,体系的势能面会有不同的形状,动力学计算所得的分子运动与分子内部运动的轨迹也会不同,进而影响到抽样的结果与抽样结果的势能计算,在计算宏观体积与微观成分关系的时候主要采用刚球模型的二体势,计算系统能量,熵等关系时早期多采用Lennard-Jones、morse势等双体势模型,对于金属计算,主要采用morse势,但就是由于通过实验拟合的对势容易导致柯西关系,与实验不符,因此在后来的模拟中有人提出采用EAM等多体势模型,或者采用第一性原理计算结果通过一定的物理方法来拟合二体势函数。但就是对于二体势模型,多体势往往缺乏明确的表达式,参量很多,模拟收敛速度很慢,给应用带来很大困难,因此在一般应用中,通过第一性原理计算结果拟合势函数的L-J,morse等势模型的应用仍非常广泛。 分子动力学计算的基本思想就是赋予分子体系初始运动状态之后,利用分子的自然运动在相空间中抽取样本进行统计计算,时间步长就就是抽样的间隔,因而时间步长的选取对动力学模拟非常重要。太长的时间步长会造成分子间的激烈碰撞,体系数据溢出;太短的时间步长会降低模拟过程搜索相空间的能力,因此一般选取的时间步长为体系各个自由度中最短运动周期的十分之一。但就是通常情况下,体系各自由度中运动周期最短的就是各个化学键的振动,而这种运动对计算某些宏观性质并不产生影响,因此就产生了屏蔽分子内部振动或其她无关运动的约束动力学,约束动力学可以有效地增长分子动力学模拟时间步长,提高搜索相空间的能
分子动力学软件选择
分子动力学软件选择 There are widely used packages like AMBER, CHARMm and X-PLOR https://www.360docs.net/doc/0113737966.html,/amber/amber.html https://www.360docs.net/doc/0113737966.html,/ https://www.360docs.net/doc/0113737966.html,/ CHARMm and X-PLOR both use the same forcefield. Amber's is different. If you're Wintel-bound, you could try Hyperchem, which has a free downloadable demo: https://www.360docs.net/doc/0113737966.html,/products/hc5_features.html It has a nice structure build capability (the other packages have powerful languages, but can be intimidating to new users). OpenSource adherents can find a wealth of free packages at SAL, an excellent site: https://www.360docs.net/doc/0113737966.html,/Z/2/index.shtml My personal favourites are MMTK, EGO and VMD/NAMD. I compiled a list of free and commerical programs at https://www.360docs.net/doc/0113737966.html,/chemistry/soft_mod_en.html modeling in solution is possible e.g. with these programs (to the best of my knowledge): commercial: AMSOL, GROMOS, Titan free: GAMESOL, GROMACS, MOIL, OMNISOL, Tinker You find links to all of these programs at https://www.360docs.net/doc/0113737966.html,/chemistry/soft_mod_en.html PAPA (计算粒状物料的三维并行分子动力学计算程序) 【URL】http://www.ica1.uni-stuttgart.de/Research/Software_P3T/papa.html 【作者】 ICA 1 Group, Institute of Computer Applications (ICA) of the University of Stuttgart 【语言版本】 English 【收费情况】免费
MS分子动力学模拟具体实施步骤
第3章 铁基块体非晶合金‐纳米晶转变的动力学模拟过程 3.1 Discover模块 3.1.1 原子力场的分配 在使用Discover模块建立基于力场的计算中,涉及几个步骤。主要有:选择力场、指定原子类型、计算或指定电荷、选择non‐bond cutoffs。 在这些步骤中,指定原子类型和计算电荷一般是自动执行的。然而,在某些情形下需要手动指定原子类型。原子定型使用预定义的规则对结构中的每个原子指定原子类型。在为特定的系统确定能量和力时,定型原子使工作者能使用正确的力场参数。通常,原子定型由Discover使用定型引擎的基本规则来自动执行,所以不需要手动原子定型。然而,在特殊情形下,人们不得不手动的定型原子,以确保它们被正确地设置。 图 3-1 1)计算并显示原子类型:点击Edit→Atom Selection,如图3‐1所示 图3-2 弹出对话框,如图3‐2所示 从右边的…的元素周期表中选择Fe,再点Select,此时所建晶胞中所有Fe
原子都将被选中,原子被红色线圈住即表示原子被选中。再编辑集合,点击Edit →Edit Sets,如图3‐3、3‐4所示。 图3-3 图3-4 弹出对话框见图3‐4,点击New...,给原子集合设定一个名字。这里设置为Fe,则3D视图中会显示“Fe”字样,再分配力场:在工具栏上点击Discover按 钮,从下拉列表中选择Setup,显示Discover Setup对话框,选择Typing选项卡,见图3‐5。 图3-5 在Forcefield types里选择相应原子力场,再点Assign(分配)按钮进行原子力场分配。注意原子力场中的价态要与Properties Project里的原子价态(Formalcharge)一致。
vasp做分子动力学
vasp做分子动力学的好处,由于vasp是近些年开发的比较成熟的软件,在做电子scf速度方面有较好的优势。 缺点:可选系综太少。 尽管如此,对于大多数有关分子动力学的任务还是可以胜任的。 主要使用的系综是NVT和NVE。 下面我将对主要参数进行介绍! 一般做分子动力学的时候都需要较多原子,一般都超过100个。 当原子数多的时候,k点实际就需要较少了。有的时候用一个k点就行,不过这都需要严格的测试。通常超过200个原子的时候,用一个k点,即Gamma点就可以了。 INCAR: EDIFF 一般来说,用1E-4或者1E-5都可以,这个参数只是对第一个离子步的自洽影响大一些,对于长时间的分子动力学的模拟,精度小一点也无所谓,但不能太小。 IBRION=0 分子动力学模拟 IALGO=48 一般用48,对于原子数较多,这个优化方式较好。 NSW=1000 多少个时间步长。 POTIM=3 时间步长,单位fs,通常1到3. ISIF=2 计算外界的压力. NBLOCK= 1 多少个时间步长,写一次CONTCAR,CHG和CHGCAR,PCDAT. KBLOCK=50 NBLOCK*KBLOCK个步长写一次XDATCAR. ISMEAR=-1 费米迪拉克分布. SIGMA =0.05 单位:电子伏 NELMIN=8 一般用6到8,最小的电子scf数.太少的话,收敛的不好. LREAL=A APACO=10 径向分布函数距离,单位是埃. NPACO=200 径向分布函数插的点数. LCHARG=F 尽量不写电荷密度,否则CHG文件太大. TEBEG=300 初始温度. TEEND=300 终态温度。不设的话,等于TEBEG. SMASS -3 NVE ensemble;-1 用来做模拟退火;大于0 NVT 系综。 ///////////////////////////////////////////////////////////////////// ///////////////////////////////////////////////////////////////////// 1)收敛判据的选择 结构弛豫的判据一般有两种选择:能量和力。这两者是相关的,理想情况下,能量收敛到基态,力也应该是收敛到平衡态的。但是数值计算过程上的差异导致以二者为判据的收敛速度差异很大,力收敛速度绝大部分情况下都慢于能量收敛速度。这是因为力的计算是在能量的基础上进行的,能量对坐标的一阶导数得到力。计算量的增大和误差的传递导致力收敛慢。 到底是以能量为收敛判据,还是以力为收敛判据呢?关心能量的人,觉得以能量
分子动力学模拟基础知识
分子动力学模拟基础知识 ? Molecular Dynamics Simulation o MD: Theoretical Background Newtonian Mechanics and Numerical Integration The Liouville Operator Formalism to Generating MD Integration Schemes o Case Study 1: An MD Code for the Lennard-Jones Fluid Introduction The Code, mdlj.c o Case Study 2: Static Properties of the Lennard-Jones Fluid (Case Study 4 in F&S) o Case Study 3: Dynamical Properties: The Self-Diffusion Coefficient ? Ensembles o Molecular Dynamics at Constant Temperature Velocity Scaling: Isokinetics and the Berendsen Thermostat Stochastic NVT Thermostats: Andersen, Langevin, and Dissipative Particle Dynamics The Nosé-Hoover Chain Molecular Dynamics at Constant Pressure: The Berendsen Barostat Molecular Dynamics Simulation We saw that the Metropolis Monte Carlo simulation technique generates a sequence of states with appropriate probabilities for computing ensemble averages (Eq. 1). Generating states probabilitistically is not the only way to explore phase space. The idea behind the Molecular Dynamics (MD) technique is that we can observe our dynamical system explore phase space by solving all particle equations of motion . We treat the particles as classical objects that, at least at this stage of the course, obey Newtonian mechanics. Not only does this in principle provide us with a properly weighted sequence of states over which we can compute ensemble averages, it additionally gives us time-resolved information, something that Metropolis Monte Carlo cannot provide. The ``ensemble averages'' computed in traditional MD simulations are in practice time averages : (99) The ergodic hypothesis partially requires that the measurement time, , i , in the system. The price we pay for this extra information is that we must at least access if not store particle velocities in addition to positions, and we must compute interparticle forces in addition to potential energy. We will introduce and explore MD in this section.
分子动力学模拟及其在材料中的研究进展汇总
《材料计算设计基础》 学号: 流水号: 姓名: 完成日期:
分子动力学模拟及其在材料中的研究进展 摘要:本文综述了分子动力学模拟技术的发展,介绍了分子动力学的分类、运动方程的求解、初始条件和边界条件的选取、平衡系综及其控制、感兴趣量的提取以及分子动力学模拟在材料中的研究进展。 关键词:分子动力学模拟平衡态系综金属材料感兴趣量径向分布函数 引言 科学工作者在长期的科学研究实践中发现,当实验研究方法不能满足研究工作的需求时,用计算机模拟却可以提供实验上尚无法获得或很难获得的重要信息;尽管计算机模拟不能完全取代实验,但可以用来指导实验,并验证某些理论假设,从而促进理论和实验的发展。特别是在材料形成过程中许多与原子有关的微观细节,在实验中基本上是无法获得的,而在计算机模拟中即可以方便地得到。这种优点使分子动力学模拟在金属材料研究中显得非常有吸引力。 分子动力学MD (Molecular Dynamics)模拟就是用计算机方法来表示统计力学,作为实验的一个辅助手段。MD模拟就是对于原子核和电子所构成的多体系统,求解运动方程(如牛顿方程、哈密顿方程或拉格朗日方程),其中每一个原子核被视为在全部其它原子核和电子作用下运动,通过分析系统中各粒子的受力情况,用经典或量子的方法求解系统中各粒子在某时刻的位置和速度,以确定粒子的运动状态,进而计算系统的结构和性质。该模拟技术主要涉及粒子运动的动力学问题,与蒙特卡罗模拟方法(简称MC)相比,分子动力学是一种“确定性方法”, 它所计算的是时间平均,而MC进行的是系综平均。然而按照统计力学各态历经假设,时间平均等价于系综平均。因此,两种方法严格的比较计算能给出几乎相同的结果。 经典的分子动力学方法是Alder等于1957年提出并首先在“硬球”液体模型下应用,发现了由Kirkwood在1939年根据统计力学预言的“刚性球组成的集合系统会发生有液相到结晶相的转变”。后来人们称这种相变为Alder相变。Rahman
分子动力学方法
分子动力学方法 一、引言 计算机模拟中的另一类确定性模拟方法,即统计物理中的所谓合于动力学方法(Molecular Dynamics Method)。这种方法是按该体系内部的内禀动力学规律来计算并确定位形的转变。它首先需要建立一组分子的运动方程,并通过直接对系统中的一个个分子运动方程进行数值求解,得到每个时刻各个分子的坐标与动量,即在相空间的运动轨迹,再利用统计计算方法得到多体系统的静态和动态特性,从而得到系统的宏观性质。在这样的处理过程中我们可以看出:MD方法中不存在任何随机因素。在MD方法处理过程中方程组的建立是通过对物理体系的微观数学描述给出的。在这个微观的物理体系中,每个分子都各自服从经典的牛顿力学。每个分子运动的内禀动力学是用理论力学上的哈密顿量或者拉格朗日量来描述,也可以直接用牛顿运动方程来描述。确定性方法是实现Boltzman的统计力学途径。这种方法可以处理与时间有关的过程,因而可以处理非平衡态问题。但是使用该方法的程序较复杂,讨算量大,占内存也多、本节将介绍分子动力学方法及其应用。 原则上,MD方法所适用的微观物理体系并无什么限制。这个方法适用的体系既可以是少体系统,也可以是多体系统;既可以是点粒子体系,也可以是具有内部结构的体系;处理的微观客体既可以是分子,也可以是其他的微观粒子。 实际上,MD模拟方法和随机模拟方法一样都面临着两个基本限制:一个是有限观测时间的限制;另一个是有限系统大小的限制。通常人们感兴趣的是体系在热力学极限下(即粒子数日趋于无穷时)的性质。但是计算机模拟允许的体系大小要比热力学极限小得多,因此可能会出现有限尺寸效应。为了减小有限尺寸效应,人们往往引入周期性、全反射、漫反射等边界条件。当然边界条件的引入显然会影响体系的某些性质。 对于MD方法,向然的系综是微正则系综,这时能量是运动常量。然而,当我们想要研究温度和(或)压力是运动常量的系统时,系统不再是封闭的。例如当温度为常量的系统可以认为系统是放置在一个热俗中。当然,在MD方法中我们只是在想像中将系统放入热浴中。实际上,在模拟计算中具体所采取的做法是对一些自由度加以约束。例如在恒温体系的情况下,体系的平均动能是一个不变量。这时我们可以设计一个算法,使平均动能被约束在一个给定值上。由于这个约束,我们并不是在真正处理一个正则系综,而实际上仅仅是复制了这个系综的位形部分。只要这一约束不破坏从一个状态到另一个状态的马尔科夫特性,这种做法就是正确的。不过其动力学性质可能会受到这一约束的影响。 自五十年代中期开始,MD方法得到了广泛的应用。它与蒙特卡洛方法一起已经成为计算机模拟的重要方法。应用MD方法取得了许多重要成果,例如气体或液体的状态方程、相变问题、吸附问题等,以及非平衡过程的研究。其应用已从化学反应、生物学的蛋白质到重离子碰撞等广泛的学科研究领域。 二、分子运动方程的数值求解 采用MD方法时,必须对一组分于运动微分方程做数值求解。从计算数学的角度来看,这个求解是一个初值问题。实际上计算数学为了求解这种问题己经发展了许多的算法,但并不是所有的这些算法都可以用来解决物理问题。下面我们先以一个一维谐振子为例,来看一下如何用计算机数值计算方法求解初值问题。一维谐振子的经典哈密顿量为 (2.1) 这里的哈密顿量(即能量)为守恒量。假定初始条件为x(p)、p(0),则它的哈密顿方程是对时间的一阶微分方程 (2.2) 现在我们要用数值积分方法计算在相空间中的运动轨迹(X(t)、p(t)) 。我们采用有限差分法,将微分方程变为有限差分方程,以便在计算机上做数值求解,并得到空间坐标和动量随时间的演化关系。首先,
分子动力学模拟
分子动力学模拟 The Standardization Office was revised on the afternoon of December 13, 2020
分子动力学模拟 分子动力学是一门结合物理,数学和化学的综合技术。分子动力学是一套分子模拟方法,该方法主要是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。 这门技术的发展进程是: 1980年:恒压条件下的动力学方法(Andersenの方法、Parrinello-Rahman法)1983年:非平衡态动力学方法(Gillan and Dixon) 1984年:恒温条件下的动力学方法(能势‐フーバーの方法) 1985年:第一原理分子动力学法(→カー?パリネロ法) 1991年:巨正则系综的分子动力学方法(Cagin and Pettit). 最新的巨正则系综,即为组成系综的系统与一温度为T、化学势为μ的很大的热源、粒子源相接触,此时系统不仅同热源有能量交换,而且可以同粒子源有粒子的交换,最后达到平衡,这种系综称巨正则系综。 进行分子动力学模拟的第一步是确定起始构型,一个能量较低的起始构型是进行分子模拟的基础,一般分子的其实构型主要是来自实验数据或量子化学计算。在确定起始构型之后要赋予构成分子的各个原子速度,这一速度是根据玻尔兹曼分布随机生成,由于速度的分布符合玻尔兹曼统计,因此在这个阶段,体系的温度是恒定的。另外,在随机生成各个原子的运动速度之后须进行调整,使得体系总体在各个方向上的动量之和为零,即保证体系没有平动位移。由上一步确定的分子组建平衡相,在构建平衡相的时候会对构型、温度等参数加以监控。
分子动力学作业概要
分子动力学(MD) 1 分子动力学(MD)基础 1.1 MD分类 1.2 MD简介 1.3 MD适用范围 2 分子动力学运动方程数值求解 2.1 基础知识 2.1.1 运动方程 2.1.2 空间描述 2.1.3 最小作用量原理 2.1.4 拉格朗日(Lagrange)方程 2.1.5 哈密顿(Hamilton)方程 2.2 粒子运动方程的数值解法 2.2.1 Verlet算法 2.2.2 欧拉(Euler)预测—矫正公式 2.2.3 Gear预测—矫正方法 3 分子动力学原胞与边界条件 3.1 分子动力学原胞 3.2 边界条件 3.2.1 自由表面边界 3.2.2 固定边界 3.2.3 柔性边界 3.2.4 周期性边界 4 势函数与分子力场 4.1 势函数 4.1.1 两体势 4.1.2 多体势 4.2 分子力场 4.2.1 分子力场函数的构成
4.2.2 常用力场函数和分类 5 分子动力学模拟的基本步骤 5.1 设定模拟所采用的模型 5.2 给定初始条件 5.3 趋于平衡计算 5.4 宏观物理量的计算 6 平衡态分子动力学模拟 6.1 系综 6.2 微正则系综的分子动力学模拟6.3 正则系综的分子动力学模拟
1 分子动力学(MD)基础 1.1MD分类 微正则系综(VNE) 正则系综(VNP) 平衡态MD 等温等压系综(NPT) 经典MD 等焓等压系综(NPH) 巨正则系综(VTμ) 非平衡态MD 量子MD 1.2分子动力学(MD)简介 分子动力学是在原子、分子水平上求解多体问题的重要的计算机模拟方法。分子动力学方法为确定性模拟方法,广泛地用于研究经典的多粒子体系的研究中,是按该体系内部的内禀动力学规律来计算并确定位形的转变。 分子动力学方法是通过建立一组分子的运动方程,并通过直接对系统中的一个个分子运动方程进行数值求解,得到每个时刻各个分子的坐标与动量,即在相空间的运动轨迹,再利用统计计算方法得到多体系统的静态和动态特性, 从而得到系统的宏观性质。 在分子动力学中,粒子的运动行为是通过经典的Newton运动方程所描述。系统的所有粒子服从经典力学的运动规律,它的动力学方程就是从经典力学的运动方程——拉格朗日(lagrange)方程和哈密顿(Hamilton)方程导出。 1.3适用范围 原则上,分子动力学方法所适用的微观物理体系并无什么限制。这个方法适用的体系既可以是少体系统,也可以是多体系统;既可以是点粒子体系,也可以是具有内部结构的体系;处理的微观客体既可以是分子,也可以是其它的微观粒子。 实际上,分子动力学模拟方法和随机模拟方法一样都面临着两个基本限制:
分子动力学模拟在材料科学与生命科学的应用听后感悟
分子动力学模拟在材料科学与生命科学的应用听后感悟 当实验研究方法不能满足研究工作的需求时,用计算机模拟却可以提供实验上尚无法获得或很难获得的重要信息;虽然计算机模拟不能完全取代实验,但可以用来指导实验,并验证某些理论假设,从而促进理论和实验的发展。特别是材料形成过程中许多与原子有关的微观细节,在实验中无法获得,而在计算机模拟中即可以方便地得到。这种优点使分子动力学模拟在材料科学研究中显得非常有吸引力。 分子动力学(Molecular Dynamics,MD)模拟是指对于原子核和电子所构成的多体系统,求解运动方程(如牛顿方程、哈密顿方程或拉格朗日方程),其中每一个原子核被视为在全部其它原子核和电子作用下运动,通过分析系统中各粒子的受力情况,用经典或量子的方法求解系统中各粒子在某时刻的位置和速度,以确定粒子的运动状态,进而计算系统的结构和性质。20世纪80年代后期,计算机技术飞速发展,加上多体势函数的提出与发展,使分子动力学模拟技术有了进一步的发展。 根据对原子间作用势不同的简化处理方法,分子动力学可划分为经典分子动力学和现代分子动力学。经典分子动力学计算量较小,可以解决较大规模的问题,但针对不同的问题,可能需要确定不同的经验参数。而现代分子动力学直接从量子力学轨道理论出发获取原子间作用势,不需要经验参数,准确性高,但计算量比较大,一般用来解决较小规模的问题。分子动力学模拟在深入研究液体结构,揭示金属熔体的结构演变、非晶倾向及热力学性质计算等方面具有很大的发展和应用前景。金属熔体结构是一个重要的研究领域,采用分子动力学方法从原子层次上描述熔体系统的原子组态及其凝固过程中的演变过程,进行了微观和宏观的良好结合。进一步扩大分子动力学在该领域的应用显然是凝聚态物理的一个热门发展方向。 生物大分子的具体功能正不断的被科学家们解析。借助于一些新表征方法,诸如X 射线晶体衍射技术、电子晶体学技术、多维核磁共振波谱、冷冻电子显微镜等,人类甚至已经观测到了生物分子中最小的氢原子,因此,更多的注意力被放在研究生物大分子之间的相互关系上。与此同时,具有跨时代意义的“人类基因组计划”以及其后续的多种科研计划导致了海量生物学数据的产生,这些数据迫切的需要进一步的处理与分析。高性能计算机的发展为生物学中海量数据的处理提供了必要条件,信息时代的来临也为现代生物学的发展提供了最广阔的信息交互平台。高性能计算机与网络也成为现代生物学必不可少的一部分,不断推进着更多,更细分的交叉学科的发展。
分子动力学模拟I
Gromacs中文教程 淮海一粟 分子动力学(MD)模拟分为三步:首先,要准备好模拟系统;然后,对准备好的系统进行模拟;最后,对模拟结果进行分析。虽然第二步是最耗费计算资源的,有时候需要计算几个月,但是最耗费体力的步骤在于模拟系统准备和结果分析。本教程涉及模拟系统准备、模拟和结果分析。 一、数据格式处理 准备好模拟系统是MD最重要的步骤之一。MD模拟原子尺度的动力学过程,可用于理解实验现象、验证理论假说,或者为一个待验证的新假说提供基础。然而,对于上述各种情形,都需要根据实际情况对模拟过程进行设计;这意味着模拟的时候必须十分小心。 丢失的残基、原子和非标准基团 本教程模拟的是蛋白质。首先需要找到蛋白质序列并选择其起始结构,见前述;然后就要检查这个结构是否包含所有的残基和原子,这些残基和原子有时候也是模拟所必需的。本教程假定不存在缺失,故略去。 另一个需要注意的问题是结构文件中可能包含非标准残基,被修饰过的残基或者配体,这些基团还没有力场参数。如果有这些基团,要么被除去,要么就需要补充力场参数,这牵涉到MD的高级技巧。本教程假定所有的蛋白质不含这类残基。 结构质量 对结构文件进行检查以了解结构文件的质量是一个很好的练习。例如,晶体结构解析过程中,对于谷氨酰胺和天冬酰胺有可能产生不正确的构象;对于组氨酸的质子化状态和侧链构象的解析也可能有问题。为了得到正确的结构,可以利用一些程序和服务器(如 WHATIF)。本教程假定所用的结构没有问题,我们只进行数据格式处理。 二、结构转换和拓扑化 一个分子可以由各个原子的坐标、键接情况与非键相互作用来确定。由于.pdb 结构文件只含有原子坐标,我们首先必须建立拓扑文件,该文件描述了原子类型、电荷、成键情况等信息。拓扑文件对应着一种力场,选择何种力场对于拓扑文件的建立是一个值得仔细考虑的问题。这里我们用的是GROMOS96 53a6连接原子力场,该力场对于氨基酸侧链的自由能预测较好,并且与NMR试验结果较吻合。
分子动力学模拟方法的基本原理与应用
分子动力学模拟方法的基本原理与应用 摘要: 介绍了分子动力学模拟的基本原理及常用的原子间相互作用势, 如Lennard-Jones势; 论述了几种常用的有限差分算法, 如Verlet算法; 说明了分子动力学模拟的几种系综及感兴趣的宏观统计量的提取。 关键词: 分子动力学模拟; 原子间相互作用势; 有限差分算法; 分子学是一门结合物理,和化学的综合技术。分子学是一套方法,该方法主要是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的量和其他宏观性质。 从统计物理学中衍生出来的分子动力学模拟方法(Molecular Dynamics Simulation, MDS) , 实践证明是一种描述纳米科技研究对象的有效方法, 得到越来越广泛的重视。所谓分子动力学模拟, 是指对于原子核和电子所构成的多体系统, 用计算机模拟原子核的运动过程, 从而计算系统的结构和性质, 其中每一个原子核被视为在全部其他原子核和电子所提供的经验势场作用下按牛顿定律运动。它被认为是本世纪以来除理论分析和实验观察之外的第三种科学研究手段, 称之为“计算机实验”手段, 在物理学、化学、生物学和材料科学等许多领域中得到广泛地应用。 科学工作者在长期的科学研究实践中发现,当实验研究方法不能满足研究工作的需求时,用计算机模拟却可以提供实验上尚无法获得或很难获得的重要信息;尽管计算机模拟不能完全取代实验,但可以用来指导实验,并验证某些理论假设,从而促进理论和实验的发展。特别是在材料形成过程中许多与原子有关的微观细节,在实验中基本上是无法获得的,而在计算机模拟中即可以方便地得到。这种优点使分子动力学模拟在材料研究中显得非常有吸引力。 分子动力学模拟就是用计算机方法来表示统计力学,作为实验的一个辅助手段。分子模拟就是对于原子核和电子所构成的多体系统,求解运动方程(如牛顿方程、哈
分子动力学模拟Word版
分子动力学模拟 分子动力学是一门结合物理,数学和化学的综合技术。分子动力学是一套分子模拟方法,该方法主要是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。 这门技术的发展进程是: 1980年:恒压条件下的动力学方法(Andersenの方法、Parrinello-Rahman法) 1983年:非平衡态动力学方法(Gillan and Dixon) 1984年:恒温条件下的动力学方法(能势‐フーバーの方法) 1985年:第一原理分子动力学法(→カー?パリネロ法) 1991年:巨正则系综的分子动力学方法(Cagin and Pettit). 最新的巨正则系综,即为组成系综的系统与一温度为T、化学势为μ的很大的热源、粒子源相接触,此时系统不仅同热源有能量交换,而且可以同粒子源有粒子的交换,最后达到平衡,这种系综称巨正则系综。 进行分子动力学模拟的第一步是确定起始构型,一个能量较低的起始构型是进行分子模拟的基础,一般分子的其实构型主要是来自实验数据或量子化学计算。在确定起始构型之后要赋予构成分子的各个原子速度,这一速度是根据玻尔兹曼分布随机生成,由于速度的分布符合玻尔兹曼统计,因此在这个阶段,体系的温度是恒定的。另外,在随机生成各个原子的运动速度之后须进行调整,使得体系总体在各个方向上的动量之和为零,即保证体系没有平动位移。 由上一步确定的分子组建平衡相,在构建平衡相的时候会对构型、温度等参数加以监控。进入生产相之后体系中的分子和分子中的原子开始根据初始速度运动,可以想象其间会发生吸引、排斥乃至碰撞,这时就根据牛顿力学和预先给定的粒子间相互作用势来对各个例子的运动轨迹进行计算,在这个过程中,体系总能量不变,但分子内部势能和动能不断相互转化,从而体系的温度也不断变化,在整个过程中,体系会遍历势能面上的各个点,计算的样本正是在这个过程中抽取的。 用抽样所得体系的各个状态计算当时体系的势能,进而计算构型积分。 作用势的选择与动力学计算的关系极为密切,选择不同的作用势,体系的势能面会有不同的形状,动力学计算所得的分子运动和分子内部运动的轨迹也会不同,进而影响到抽样的结果和抽样结果的势能计算,在计算宏观体积和微观成分关系的时候主要采用刚球模型的二体势,计算系统能量,熵等关系时早期多采用Lennard-Jones、morse势等双体势模型,对于金属计算,主要采用morse势,但是由于通过实验拟合的对势容易导致柯西关系,与实验不符,因此在后来的模拟中有人提出采用EAM等多体势模型,或者采用第一性原理计算结果通过一定的物理方法来拟合二体势函数。但是对于二体势模型,多体势往往缺乏明确的表达式,参量很多,模拟收敛速度很慢,给应用带来很大困难,因此在一般应用中,通过第一性原理计算结果拟合势函数的L-J,morse等势模型的应用仍非常广泛。 分子动力学计算的基本思想是赋予分子体系初始运动状态之后,利用分子的自然运动在相空间中抽取样本进行统计计算,时间步长就是抽样的间隔,因而时间步长的选取对动力学模拟非常重要。太长的时间步长会造成分子间的激烈碰撞,体系数据溢出;太短的时间步长会降低模拟过程搜索相空间的能力,因此一般选取的时间步长为体系各个自由度中最短运动周期的十分之一。但是通常情况下,体系各自由度中运动周期最短的是各个化学键的振动,而这种运动对计算某些宏观性质并不产生影响,因此就产生了屏蔽分子内部振动或其他无关运动的约束动力学,约束动力学可以有效地增长分子动力学模拟时间步长,提高搜索相空间的能力。