氧合铵盐氧化醇

氧合铵盐氧化醇
1.介绍
这篇文章中的多数材料已经在最近一篇题为“氧合铵和氮氧催化氧化醇”的专题中进行了一个全面的讨论。1)两篇先前的关于氧合铵2)和这个项目中所用的实验方法3)也被使用过。这篇报道中所述的氧化反应(多数是醇的氧化)含有一个明显的氧合铵阳离子选择性。即便是有,极少数情况下可以观测到异构化现象,无论是双键(顺式-反式)还是手性中心,也不会发生碳碳键裂解。此外,这些试剂都是“绿色环保”的没有重金属铬或是锰的参与。
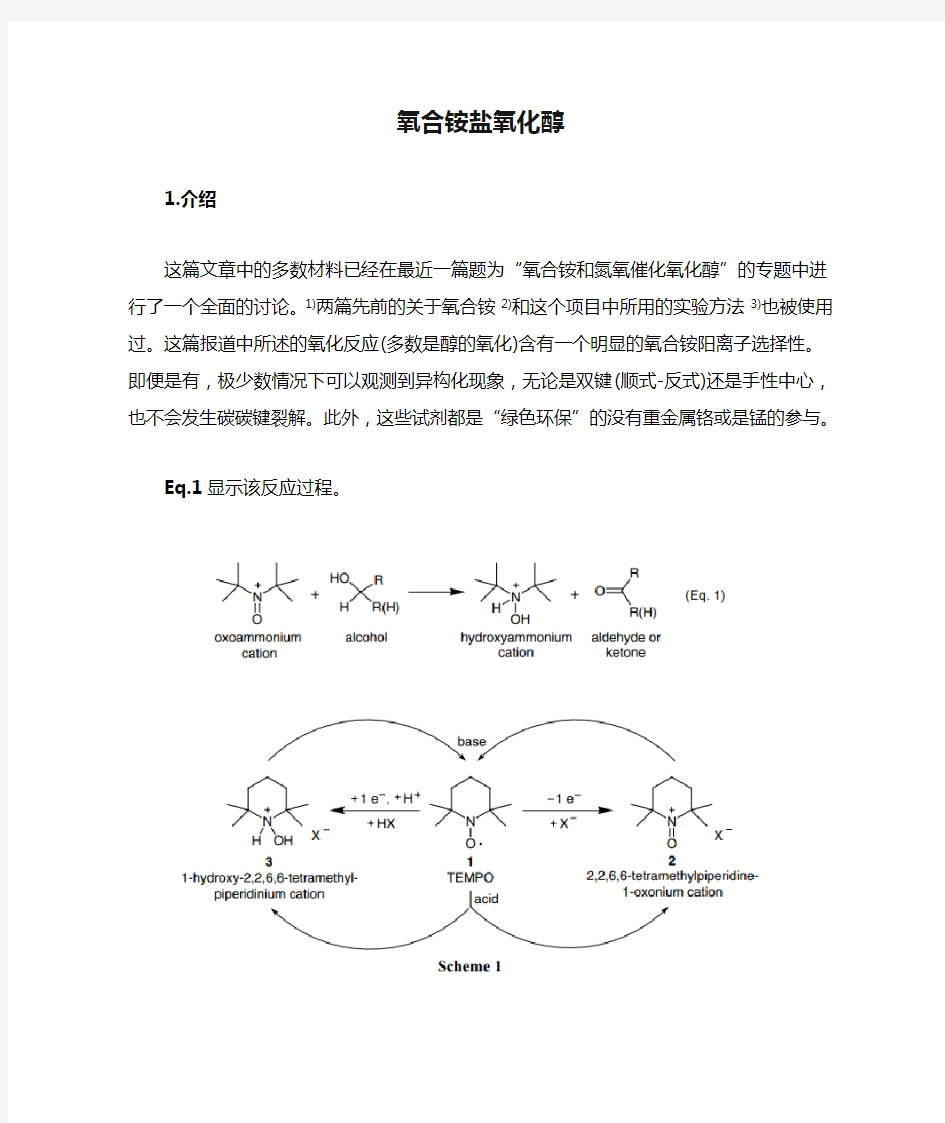
Eq.1显示该反应过程。
对于氧铵离子是稳定的,可以没有碳氢原子(α-氢原子)连接氮, 也灭有必要因为其他的原因在氮和相邻碳原子间形成一个双键。4)这里有很多这类盐的例子,2)但是常见的多数都是基于哌啶盐基础上的,如Scheme1所示。在Scheme1里面,氮氧1, 氧铵盐2, 羟铵或是其盐3的氧化还原性能和其制备、分解的常规方法被总体概括。
这个项目中的每一部分最知名的化合物就是TEMPO或者2,2,6,6-四甲基哌啶氧化物1。TEMPO是一个稳定的氧自由基,发现于1962。5)合适的阴离子的参与下,从TEMPO中移除一个电子生成氧铵盐,2 (2,2,6,6-四甲基哌啶-1-氧鎓或者2,2,6,6-四甲基-1-氧代哌啶鎓盐)。合适的阴离子和算条件下,TEMPO加入电子得到羟铵盐
3(1-羟基-2,2,6,6-四甲基哌啶鎓盐)。在强酸中发生显著的歧化反应,TEMPO被转化为2和3中的一个分子形式。这个反应由Golubev发现,并且作为制备氧铵盐的方法之一。6)该反应强大的驱动力几乎可以肯定来自于中性TEMPO离子产物的形成。如果2和3混合物被制成碱性,那么就会有相对应的反歧化反应发生,从而将化合物重新转换为1。
结合Eq.1和Scheme1, 很明显,醇氧化涉及了氧铵盐2中一个二电子反应,转换为羟铵盐3。氧铵离子氧化反应可以用四个途径(Scheme 2);在化学计量氧化反应中,酸(Eq.2)或是碱(Eq.3), 再或者利用酸-歧化反应(Eq. 4)如Scheme1所述,和利用辅助氧化剂比如次氯酸钠(漂白剂)氮氧催化氧化(Eq.5)。这些方法中,使用氮氧和酸的酸歧化反应(Eq.4)少有研究。7,8)氮氧催化反应(Eq.5)研究广泛,并可以用到很多的不同的二级氧化剂,1)但是这篇文章中也不做考虑。式2-5中,R基团可以是氢或者其他的基团。
我们主要对化学计量反应感兴趣,即预备氧铵盐反应中使用1mole/1mole当量的醇或者其他化合物被氧化。这些反应可以在碱的参与下于中性或者是微酸性条件下
进行,如Eqs. 2和3所示。Eq.4中所描述的酸歧化反应是一个化学计量法过程,在这个反应中氮氧化物和p–苯甲磺酸(两个当量)用于化学计量法中的量。
2.氧铵盐的制备及其性能研究
通过进一步氧化氮氧化物例如1来制备氧铵盐。反过来,氮氧化物的制备是通过氧化不含有α–氢或者是不能形成双键的胺来完成。很多已知的氮氧化物具有不同的结构,9-11)因此有很多种类的氧铵盐。2)然而,几乎所有已报道的氧化反应都是通过由2,2,6,6–四甲基哌啶衍生而得的氮氧化物(作为催化剂或者盐)来进行完成的。这主要归因于它们的商业可用性。最常用的盐如Scheme 3所示。相对应的氮氧化物被用于酸岐化或者是氮氧催化反应中。
市面上唯一的氧铵盐(5作为四氟硼酸盐)的总合成如Scheme 4所示。Bobbitt12)文章中给出了5(BF4?)的具体说明和其有机合成的描述。13)
Scheme 4中的反应根本上是在Scheme 1 Golubev6)中工作基础上得来的。合成氧铵盐其他的主要方法涉及到用卤元素氧化氮氧化物从而得到卤代盐。14)化合物6(4-乙酰氨基-2,2,6,6-四甲基哌啶-1-氧化物)需要特殊的标注,尤其是与TEMPO进行比较时。它的熔点是143-145°C,相对于36-38°C的TEMPO,更加稳定。它很容易从4-氨基-2,2,6,6-四甲基哌啶(Scheme 4)中制备,并且代替TEMPO
广泛的应用在氮氧化物催化氧化反应中。它还具有十分有趣的溶解特征,极易溶于二氯甲烷,部分溶于水,微溶于乙醚。因此,它可以用二氯甲烷将其从水中提取出来,或是用水将其从乙醚中提取出来。可以从乙酸乙酯或水中重结晶(损失很少)。
3.醇在酸性或是近中性条件下的氧化
在由Bobbitt12)所报道前于所发现的高氯酸盐的一系列的醇类和5(ClO4?)的氧
化反应是不稳定的,很容易爆炸。15)然而在同样的一篇报道中市售四氟硼酸盐,似乎功效非常好。Scheme 5中显示了用硅胶作为催化剂催化氧化醇的整体反应方案。
醇浆,明黄色的氧铵盐和硅胶在二氯甲烷中搅拌直至颜色变白。将混合物过滤,通过硅胶薄垫,然后蒸发至干。通常,没有必要进一步的纯化。产率很高,并且二氯甲烷非常适合一些后续的反应(格氏反应, Wittig反应,Baylis-Hillman反应和其他更多中反应)而不需要产品隔离。该方法用于制备低分子量或者是不稳定的醛、酮十分理想。
这个反应具有很多优点。他在室温下二氯甲烷中进行,并且不要求严格的无水条件。就意义上而言,这个反应是比色反应,混合物从明黄色浆液变至白色浆液。这个盐充分可溶,并且在溶剂中反应,但是氧化还原剂7 , 极不易溶。硅胶和7混合物处理过后得到回收物6。12,13)
然而正常的从醇到羰基化合物的氧化反应,二醇有时候会得到内酯,尤其是在五元六元环形成的时候。16,17)在吡啶(碱反应)的参与下也可以生成内酯,有一些具有大环结构。18,19)
各种醇类氧化反应速率是,从最快到最慢依次是苄基或烯丙基醇,仲脂肪醇和炔醇,最后是伯脂肪醇。12)在硅胶的参与下,所有的反应速率都显著加快。
硅胶催化的原因尚不明确,但是可以通过想象来解释,极性硅胶吸收极性分子在其表面上,因此相对增加了反应物的局部浓度。有一个类似的催化剂被指出是氧化铝,而不是极性小的木炭。12)从实际应用的角度出发,50-50的盐和硅胶混合反应
就可以进行。将混合物或盐研磨,如果单独使用,反应速率似乎会加快。这可能是因为这个反应是一个表面反应或者仅仅是细碎的试剂更快的溶于溶液。硅胶吸附少量的副产物有助于产品的隔离。
这个方法的主要缺点是具有β氧功能的醇的氧化很慢使得反应不进行。7,12)尽管这个抑制作用的原因尚不明确,但是这样的分子内部氢键可以产生一个五元环,这就可能会抑制该反应反应机制中可视化氢键中间体的形成(参见酸性或中性介质反
应机制)。
除了少数情况例外(参阅非醇类-氧化反应), 胺与氧铵盐反应得到无法识别的
产品并且很好避免,尽管胺性质稳定。分子中具有一个三取代双键、苄氧基或者缩醛基团时与试剂反应十分缓慢。保护集团叔顶级二甲基硅烷氧基(TBDMS)缓慢裂解,但是叔丁基二苯基硅烷氧基(TBDPS)稳定。12)三取代基烯烃与乙腈的反应有了进一步的研究,20)同样的与苄基的反应也是。21,22)唯有与脂肪醇的反应是有问题的;苄基或烯丙基醇在二氯甲烷中反应迅速并且没有任何问题。通常在碱性介质中发生的氧化催化反应很显然不会存在这些缺点。
4.基础条件下氧铵盐的氧化
氧铵盐碱性条件下在水中反应得到过氧化物和氮氧化物。23)重要的是要注意,几乎所有的氮氧化物催化氧化反应都发生在水环境下的碱性介质中。1)在吡啶碱的参与下,反应在二氯甲烷中反应也会发生,然而这个反应少有研究。18,19,24)反之,具有β氧的醇类与氧铵盐在中性或酸性条件下不反应,在吡啶的参与下它们能够得到收益很好的二聚体酯。18)Eq. 6中显示了糖衍生物一个具体的例子。反应允许烯丙基双键、丙烯酸酯、苄氧基、环丙基醚和硫化物的存在。
该反应似乎取决于吡啶碱的性质,因为2,6-lutidine参与下形成了醛。25)这些反应正在研究中。
5.通过酸岐化方法的氧化
酸岐化反应中,如Eq.4所示,一当量的基质,两当量的氮氧化物6,和两个当量的p–甲苯磺酸加入二氯甲烷中搅拌。7)氮氧化物歧化得到一个当量的氧铵甲苯磺酸酯和一个当量的羟胺甲苯磺酸酯,如Scheme 1所示。氧铵盐甲苯磺酸酯氧化反应转换为一个两个当量的羟铵甲苯磺酸酯析出。混合物过滤,滤液用水和盐水洗涤,干燥,必要时进一步纯化。像氧铵盐氧化反应属于比色法,橙色的氮氧化物被转换为白色浆液,产率很高。7)此外,在氧铵盐反应中,滤去的氧化剂可以再生得到氮氧化物。
在澳大利亚,这个方法已经被Banwell团队广泛的开发研究。8,26)在Banwell的研究中,1,2-二醇很容易氧化成羟基酮或者1,2-二酮(Scheme 8)。很显然,这些反应中没有β氧抑制剂。
6. 机制
醇氧化为醛或酮是一个看似简单额转换反应。关于醇类的氧铵盐氧化反应和氮氧催化氧化反应机制已经有大量的研究公布出来,虽然两个质子和两个电子(Eq. 1)的方式的资料还没有完全的清楚。27-29)
在本节中,我们将只涉及包括氧铵阳离子和各种基质材料的机制么。催化机制竟在其他地方描述。1)
机制的某些不确定性源于氧铵阳离子可以配制出两个反应共振形式,2A和2B (Eq.7)。2A中有一个氧和氮完整的电子对,氮原子上面具有正电荷作为电负性原子。因此,这有助于实现其反应性能。然而,2B形式的存在也不能排除在外,代表了一个氧电子的少见的例子。30,31)2B可以用来解释氧铵盐与格氏试剂32)的反应和激活双键(将讨论在Miscellaneous副反应中不良反应),7,20,31,33),更重要的是很可能是醇氧化作用比迄今为止更容易实现。
酸或中性介质中的机制在最初的脂肪醇计量化学氧铵盐氧化反应中,基质的相对反应性能依次是仲醇>伯醇>甲醇,27,34)并且相应的反应速率与羟基轴碳上面已知的碳氢键的产度相吻合。抽象的氢化物被提了出来。
这种类型的机制,其中通过B3LYP/6-31+G*的能量计算,与异丙醇的氧化速率比甲醇的快是保持一致的。29)
环状中间体,很可能借助2B结构中醇和氮电子对中氢之间的氢键的形成来促进该反应(Eq. 8).
氧铵盐的反向阴离子对氧化反应速率有影响。14,35)氯离子的出现导致反应速率与高氯酸和四氟硼酸盐离子相比反应速率更快,其中相应的,比溴离子速率也相对加快。Eq. 8中间体忽略了任何阴离子的影响并且无法解释所观测到的反应速率的差异。在硅胶的存在下,使用高氯酸或者四氟硼酸盐使得反应速率明显提高。12)酸性介质中,含有一个β氧的醇对于氧铵盐的反应性的缺失,可能涉及到上面所提到的氢键。7,12,36)如果被氧化的羟基集团之间的氢键和氧化剂通过β氧化合物中的β氧和分子内氢键建合作用而被抑制,反应将会被制止或者显著放慢速度。另一方面,这样一个机制是否也将贯穿在强酸或者水性介质中是有疑问的。这个问题主要在碱性介质中进行的话似乎并没有发生催化反应。
获得的结果与氧铵盐进行酸岐化反应增加了反应的混乱度。氧化反应中使用氮氧化物与p–甲苯磺酸作为歧化试剂(Eq. 4),β氧抑制简单的分子,7)虽然他不一定适合更为复杂的案例。8)这一切表明,β氧效果还是一个谜。
碱性介质中的机制几乎所有的氮氧化物催化氧化反应都在碱性介质中进行。氧铵盐与稀碱水溶液反应缓慢生成氮氧化物和过氧化氢。37,38)
在碱中的氧化反应被认为是含有作为一个亲和物种的醇化物。27-29)醇的形成和反应进一步细节如Scheme 6所示。这种机制已经通过计算结果得到支撑。29)这些化合物形成的平衡常数减少了醇盐空间体积的增加,并且合成体的稳定性差异可以用来解释所观测到的伯醇比仲醇氧化速度快的现象。这是试验中观测到27),在氮氧催化反应中尤其重要。1)吡啶碱的存在下氧化反应化学计量研究已经被报道,但是机制尚不明确。18,24)
7.氧化反应的具体实例
很多氧铵盐氧化反应(至少1,500种),包括化学计量和催化都已经有报道。1,2,39,40)在我们的有机合成反应章节中至08年7月,已经编目了270个化学计量氧化的例子(Eqs.2-4)。1)由于高氯酸盐易爆,15)我们建议使用四氟硼酸盐。
氧铵盐氧化反应(Eqs. 2和3) Scheme 7中给出了一些更为复杂的基质的结构。氧化位置如箭头所示,氧化得到羰基,可以是醛也可以是酮。反应显示了在氧铵盐氧化反应中可观测到的某些选择性。
氮氧化物酸歧化氧化反应(Eq.4) Scheme 8中给出了几个不均衡型氧化反应的例子。所用的酸是p-甲苯磺酸,反应在二氯甲烷中进行。
8.非醇类氧化化学计量
这里有几个不是醇类氧化的氧铵盐反应。在某种情况下,它们和醇类氧化可能会产生问题,但是多数发生在其他溶剂中,比如乙腈,在二氯甲烷中反应缓慢。
胺类与氧铵盐的反应最好避免,且没有实际用处。34,50,51)还有更多的工作需要去完成。
单酮可以被氧化为α-二酮(Eq.9),33,52-55)1,3-二酮可以被氧化成1,2,3-三酮(Eq.10)。53)后者反应生成1,2,3-三羰基化合物,不需要碳碳键的裂解,反应独特,并没有被进一步开发研究。
烯醇醚和烯胺与氧铵盐反应得到加成产物。56,57)
三烷基和四烷基二锡氧烷与氧铵盐在二氯甲烷中反应缓慢,但是在乙腈中反应迅速并且得到加成产物(Eq. 11)。20)在一个独特的反应中,一个活化的双键生成不饱和酮(Eq. 12)。58)这个反应将得到进一步的探讨。
在另一个独特的反应中,氧铵盐氧化一个碳附着于吲哚3位置处(像四氢咔唑和其类似物)得到酮(Eq. 13)。59)这个反应包含一个水分子的加成,但是不确定。
酚与苯基醚与氧铵盐反应得到醌或者碳碳耦合二聚体(Eq.14)。51,60-63)
苄基醚在乙腈中反应迅速,得到苯甲醛,进一步氧化得到醇(Eq.15)。21,22)该反应在二氯甲烷中反应非常缓慢。在二氯甲烷中苄基醇的氧化速度非常的快使得苄醚(如果存在)不会受到影响。
硫化物与氧铵盐的反应是有争议的。硫醇与氧铵盐反应得到二硫化物。56)据报道硫化物是具有抗氧化性的。60)二甲亚砜与氧铵盐反应快速(比如在NMR tube中), 但是反应产物没有明确。64)
9. 结语
氧铵盐氧化和酸岐化氧化表现出高产率醇的简单氧化和产品的隔离。此外,这个反应不要严格的反应条件,绿色且没有重金属。当产物有挥发性和不稳定时,这个反应尤其的合适。
References
1 J. M. Bobbitt, C. Brückner, N. Merbouh, Org. React. 2009, 74, 103-424.
2 J. M. Bobbitt, M. C. L. Flores,Heterocycles 1988, 27 , 509-533.
3 N. Merbouh, J. M. Bobbitt, C. Brückner, Org. Prep. Proc. Int. 2004, 36 , 3-31.
4 M. Shibuya, M. Tomizawa, I. Suzuki, Y. Iwabuchi, J. Am. Chem. Soc. 2006, 128, 8412-8413.
5 M. B. Neiman, é. G. Rozantzev, Y. G. Mamedova, Nature 1962, 196, 472.
6 V. A. Golubev, R. I. Zhdanov, V. M. Gida, E. G. Rozantsev, Russ. Chem. Bull. 1971, 20, 768-770. (English translation)
7 Z. Ma, J. M. Bobbitt, J. Org. Chem. 1991, 56 , 6110-6114.
8 M. G. Banwell, V. S. Bridges, J. R. Dupuche, S. L. Richards, J. M. Walter, J. Org. Chem. 1994 , 59 , 6338-6343.
9 é. G. Rozantsev, V. D. Sholle, Synthesis1971, 401-414.
10 é. G. Rozantsev, V. D. Sholle, Synthesis1971, 190-202.
11 J. F. W. Keana, Chem. Rev. 1978, 78 , 37-64.
12 J. M. Bobbitt, J. Org. Chem. 1998, 63 , 9367-9374.
13 J. M. Bobbitt, N. Merbouh, Org. Synth. 2005, 82 , 80-86.
14 T. Miyazawa, T. Endo, S. Shiihashi, M. Okawara, J. Or
g. Chem. 1985, 50 , 1332-1334.
15 J. M. Bobbitt, Chem. Eng. News 1999, 77 , 6.
16 T. Miyazawa,T. Endo, J. Am. Chem. Soc. 1985, 50 , 3930-3931.
17 P. L. Anelli, S. Banfi, F. Montanari, S. Quici, J. Org. Chem. 1989, 54 , 2970-2972.
18 N. Merbouh, J. M. Bobbitt, C. Brückner, J. Org. Chem. 2004, 69 , 5116-5119.
19 A. Hassannia, G. Piercy, N. Merbouh, Lett. Org. Chem. 2009, 6, 478-480.
20 P. P. Pradhan, J. M. Bobbitt, W. F. Bailey, Org. Lett. 2006, 8, 5485-5487.
21 T. Miyazawa, T. Endo, Tetrahedron Lett. 1986, 27 , 3395-3398.
22 P. P. Pradhan, J. M. Bobbitt, W. F. Bailey,
J. Org. Chem. 2009, 74 , 9524-9527.
23 T. Endo, T. Miyazawa, S. Shiihashi, M. Okawara, J. Am. Chem. Soc. 1984, 106, 3877-3878.
24 N. Merbouh, J. M. Bobbitt, C. Brückner, Tetrahedron Lett. 2001, 42 , 8793-8796.
25 A. L. Bartelson, J. M. Bobbitt, W. F. Bailey, 2009, Unpublished material.
26 A. D. Findlay, A. Gebert, I. A. Cade, M. G. Banwell, Aust. J. Chem. 2009, 62 , 1173-1180.
27 V. A. Golubev, V. N. Borislavskii, A. L. Aleksandrov, Russ. Chem. Bull. 1977, 9, 1874-1881. (English translation)
28 M . F . S e m m e l h a c k , C . R . S c h m i
d , D . A . C o r t és , Tetrahedron Lett. 1986, 27 , 1119-1122.
29 W. F. Bailey, J. M. Bobbitt, K. B. Wiberg,
J. Org. Chem. 2007, 72 , 4504-4509.
30 L. O. Atovmyan, V. A. Golubev, N. I. Golovina, G. A. Klitskaya,J. Struct. Chem. 1975, 16 , 79-83. (English translation)
31 T. Takata, Y. Tsujino, S. Nakanishi, K. Nakamura, E. Yoshida, T. Endo, Chem. Lett. 1999, 937-938.
32 V. A. Golubev, E. V. Kobylyanskii, J. Org. Chem. USSR 1972, 8, 2657-2662. (English translation)
33 V. A. Golubev, T. S. Rudyk, V. D. Sen’, A. L. Aleksandrov, Russ. Chem. Bull. 1976, 25, 744-750. (English translation)
34 V. A. Golubev, é.G. Rozantsev, M. B. Neiman, Russ. Chem. Bull. 1965, 14, 1898-1904. (English translation)
35 Y. Liu, H. Guo, Z. Liu, Huaxue Xuebao 1991, 49 , 187-192. (in Chinese)
36 M. Yamaguchi, T. Takata, T. Endo, Tetrahedron Lett. 1988, 29 , 5671-5672.
37 T. Miyazawa, T. Endo, M. Okawara, J. Org. Chem. 1985, 50, 5389-5391.
38 J. M. Bobbitt, Unpublished work.
39 T. Miyazawa, T. Endo, Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.) 1986 , 44 , 1134-1144. (in Japanese)
40 J. M. Bobbitt, Z. Ma, D. Bolz, T. Osa, Y. Kashiwagi, Y. Yanagisawa,
F. Kurashima, J. Anzai, J. E. Tacorante-Morales, in
Oxoammonium salt oxidations of alcohols in the presence of, and on the surfaces of, an electrode, an ion exchange resin, and silica gel, London, UK, 1998.
41 D. M. Philipp, R. Muller, W. A. Goddard, K. A. Abboud, M. J. Mullins, R. V. Snelgrove, P. S. Athey, Tetrahedron Lett. 2004, 45, 5441-5444.
42 A. Abad, C. Agulló, A. C. Cunat, R. H. Perni, Tetrahedron: Asymmetry 2000, 11, 1607-1615.
43 T. V. Ovaska, J. A. Sullivan, S. I. Ovaska, J. B. Winegrad, J. D. Fair, Org. Lett. 2009, 11 , 2715-2718.
44 T. R. Hoye, M. Hu, J. Am. Chem. Soc. 2003, 125, 9576-9577.
45 J. Hudon, T. A. Cernak, J. A. Ashenhurst, J. L. Gleason, Angew. Chem. Int. Ed. 2008, 47 , 8885-8888.
46 D. M. Troast, J. Yuan, J. A. Porco Jr, Adv.Synth. Catal. 2008, 350, 1701-1711.
47 K. A. B. Austin, M. G. Banwell, G. J. Harfoot, A. C. Willis, Tetrahedron Lett. 2006, 47 , 7381-7384.
48 P. T. Gulyas, S. J. Langford, N. R. Lokan, M. G. Ranasinghe, M. N. Paddon-Row, J. Org. Chem. 1997, 62 , 3038-3039.
49 L. W. Habel, S. De Keersmaecker, J. Wahlen, P. A. Jacobs, D. E. De Vos, Tetrahedron Lett. 2004, 45 , 4057-4059.
50 D. H. Hunter, J. S. Racok, A. W. Rey, Y. Z. Ponce, J. Am. Chem. Soc. 1988, 53 , 1278-1281.
51 J. M. Bobbitt, Z. Ma, Heterocycles 1992, 33 , 641-648.
52 V. A. Golubev, R. I. Zhdanov, I. T. Protsishin, é.G. Rozantsev, Russ. Chem. Bull. 1970, 19, 2043. (English translation)
53 V. A. Golubev, R. V. Miklyush, J. Org. Chem. USSR1972, 8, 1376-1377. (English translation).
54 Y. Liu, T. Ren, Q. Guo, Chin. J. Chem. 1996, 14 , 252-258.
55 S. Weik, G. Nicholson, G. Jung, J. Rademann, Angew. Chem. Int. Ed. 2001, 40 , 1436-1439.
56 Z. Ma, Ph.D. Dissertation, University of Connecticut, Storrs, CT, 1991.
57 M. Sch?mann,H. J. Sch?fer, Electrochim. Acta2005, 50 , 4956-4972.
58 T. Breton, D. Liaigre, E. M. Belgsir, Tetrahedron Lett. 2005, 46 , 2487-2490.
59 J. M. Bobbitt, M. C. F. Guttermuth, Z. Ma, H. Tang, Heterocycles 1990, 30 , 1131-1140.
60 D. H. Hunter, D. H. R. Barton, W. J. Motherwell, Tetrahedron Lett. 1984, 25 , 603-606.
61 H.-X. Guo, Y.-C. Liu, Z.-L. Liu, C.-L. Li, Res. Chem. Intermed. 1992, 17 , 137.
62 Y. B. Ding, L. Yang, Z. H. Liu, Y. C. Liu, J. Chem. Res., Synop. 1994, 328-329.
63 Y. Liu, W. Wang, Q. Guo, Chin. Chem. Lett. 1996, 7, 790-793.
64 P. Pradhan, J. M. Bobbitt, Unpublished work.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有
问题可以查找原文。
醇的催化氧化精选.
已知: 1、 如何鉴别1-丙醇与2-丙醇。 2、 分子式为C 5H 12O 的醇,其中能被氧化为醛的结构是哪几种?能被氧化为酮的是哪几种?不能被氧化的是哪几种? 3、分子式为C 4H 8的烃可以发生如图转化: 其中E 、F 均呈酸性。 写出下列物质的结构简式: C 4H 8: C : D : E : F : C 4H 8 R —CH 2OH R —CHO ; 氧化 R’ R CH —OH 氧化 R —C —R ’ O (酮); 则很难被氧化(R 、R ’、R ’’表示烃基)。 R —C —OH R ’ R ’’
4、(2012房山期末1,16分)已知:Ⅰ. 质谱分析测得有机化合物A的相对分子质量为92.5 ;其含碳、氢的质量分数分别为51.89% 、9.73% ,其余为氯。 Ⅱ. A有如下转化关系: Ⅲ. 与羟基相连的碳上没有氢原子的醇(结构: )不能发生催化氧化反应。 Ⅳ. F的核磁共振氢谱有两种峰,峰高比值为1:9 , 不能发生催化氧化反应。 Ⅴ. E和G都能和新制的Cu(OH)2悬浊液反应, H是一种有果香味的液体。 写出下列物质的结构简式: F:C: B:A: D:E: G:H: 5、由C= CH3 CH2 选择合适的途径制备 CH2 C= COOH(C COOH CH2 )。(写流程图) 6. (11东城期末)23.(14分)上海世博会英国馆――种子圣殿,由六万多根透明的亚克力[其
分子式是(C 5H 8O 2)n ]杆构建而成。某同学从提供的原料库中选择一种原料X ,设计合成高分子亚克力的路线如下图所示: 原料库: a 、CH 2=CHCH 3 b 、CH 2=CHCH 2CH 3 c 、CH 2=CCH 3 , d 、CH 2CHCH 3 已知:① (不易被氧化成羧酸) ② 不易被氧化成醛或酮 ③ (R 、R ’、R ’’表示烃基) 写出下列物质的结构简式: X : A : B : C : D : E : F : 亚克力: R —C —R ’(H) O HCN R —C —R ’(H) OH H +/H 2O R —C —R ’(H) OH R ’’ R —C —R ’ OH R —C —R ’ O R —CH —R ’ OH [O] CH 3 CH 3
醇的催化氧化
为你成材 尽我所能 - 37 - 师生同心 金石为开 已知: 1、 如何鉴别1-丙醇与2-丙醇。 2、 分子式为C 5H 12O 的醇,其中能被氧化为醛的结构是哪几种?能被氧化为酮的是哪几种?不能被氧化的是哪几种? 3、分子式为C 4H 8的烃可以发生如图转化: 其中E 、F 均呈酸性。 写出下列物质的结构简式: C 4H 8: C : D : E : F : C 4H 8 R —CH 2OH R —CHO ; 氧化 R’ R CH —OH 氧化 R ——R ’ O (酮); 则很难被氧化(R 、R ’、R ’’表示烃基)。 R —C —OH R ’ R ’’
为你成材 尽我所能 - 38 - 师生同心 金石为开 4、(2012房山期末1,16分)已知:Ⅰ. 质谱分析测得有机化合物A 的相对分子质量为92.5 ;其含碳、氢的质量分数分别为51.89% 、9.73% ,其余为氯。 Ⅱ. A 有如下转化关系: Ⅲ. 与羟基相连的碳上没有氢原子的醇(结构: )不能发生催化氧化反应。 Ⅳ. F 的核磁共振氢谱有两种峰,峰高比值为1:9 , 不能发生催化氧化反应。 Ⅴ. E 和G 都能和新制的Cu(OH)2悬浊液反应, H 是一种有果香味的液体。 写出下列物质的结构简式: F : C : B : A : D : E : G : H : 5、由选择合适的途径制备 ( C COOH CH 2 )。(写流程图) C=CH 3 CH 2 CH 2C=COOH
为你成材 尽我所能 - 39 - 师生同心 金石为开 6. (11东城期末)23.(14分)上海世博会英国馆――种子圣殿,由六万多根透明的亚克力[其分子式是(C 5H 8O 2)n ]杆构建而成。某同学从提供的原料库中选择一种原料X ,设计合成高分子亚克力的路线如下图所示: 原料库: a 、CH 2=CHCH 3 b 、CH 2=CHCH 2CH 3 c 、CH 2=CCH 3 , d 、CH 2CHCH 3 已知:① (不易被氧化成羧酸) ② 不易被氧化成醛或酮 ③ (R 、R ’、R ’’表示烃基) 写出下列物质的结构简式: X : A : B : C : D : E : F : 亚克力: R ——R ’(H) O HCN R ——R ’(H) OH CN H + /H 2O R ——R ’(H) OH COOH ’’ R —C —R ’ OH R ——R ’ O R ——R ’ OH [O] CH 3 CH 3
化学乙醇催化氧化实验
化学乙醇催化氧化实验https://www.360docs.net/doc/041448172.html,work Information Technology Company.2020YEAR
某实验小组用下列装置进行乙醇催化氧化的实验。 (1)实验过程中铜网出现黑色和红色交替的现象,请写出相应的化学方程式、。在不断鼓入空气的情况下,熄灭酒精灯,反应仍能继续进行,说明该乙醇催化反应 是反应。(填吸热或放热) (2)甲和乙两个水浴作用不相同.甲的作用是;乙的作用是。 (3)反应进行一段时间后,试管a中收集到的主要有机生成物是。(写名称)若要检验试管a中能收集的该物质,进行的操作为。 (4)若试管a中收集到的液体用紫色石蕊试纸检验,试纸显红色,说明液体中还含有。(写结构简式) 要除去该物质,可向混合液中加入(填写序号)。再通 过(填试验操作名称)即可除去。 A.水B.苯C.碳酸氢钠溶 液 D.四氯化碳
(1)Cu+O 22CuO、CH3CH2OH+CuO CH3CHO+Cu+H2O;放热; (2)加热乙醇,便于乙醇的挥发;冷却,便于乙醛的收集;(3)乙醛;加入新制的氢氧化铜悬浊液,加热煮沸若有砖红色沉淀,证明产物是乙醛;(4)CH3COOH;C;蒸馏。 试题分析:(1)在加热Cu丝时发生反应Cu+O22CuO。当把热的Cu丝遇到乙醇蒸气时发生反应: CH 3CH2OH+CuO CH3CHO+Cu+H2O。在不断鼓入空气的情况下,熄灭酒精灯,反应仍能继续进行,说明该乙醇催化反应是放热反应,反应放出的热量就足够后面发生反应需要消耗的能量。(2)甲的水浴加热作用是产生乙醇蒸气,便于乙醇的挥发;而乙用的是冷水浴。目的是冷却降温,便于乙醛的收集。(3)由(1)的反应方程式可知:反应进行一段时间后,试管a中收集到的主要有机生成物是乙醛。若要检验试管a中能收集的该物质,可以利用醛基的性质进行的操作是加入新制的氢氧化铜悬浊液,加热煮沸若有砖红色沉淀产生,证明产物是乙醛;(4)若试管a中收集到的液体用紫色石蕊试纸检验,试纸显红色,说明液体中还含有酸性物质。在该该反应中产生的酸性物质只有乙酸CH3COOH。为除去乙酸。可以利用乙酸不同与乙醛的性质:有酸性,而且酸性比碳酸强,乙酸能跟碳酸盐发生反应消耗,同时生成的碳酸不温度,会分解,以
乙醇催化氧化
乙醇的催化氧化 一、选题来源 高一第二学期(上海教育出版社)p43.酒精的催化氧化。教材上是这样做的这个实验: “把铜丝烧成螺旋状,在火焰上加热后,铜丝表面发黑生成黑色的氧化铜,把它迅速插入酒精中,待黑色退去后,取出铜丝再加热,再插入酒精中,反复数次后嗅闻气味。” 反应的方程式为2Cu + O2→2CuO CuO + CH3CH2OH→CH3CHO + Cu + H2O 总方程式为:CH3CH2OH+ O2→CH3CHO +H2O 反应中起催化作用的是Cu,表面的氧化铜是中间产物。乙醇直接和氧化铜粉末反应生成的是什么?反应条件是什么? 二.实验步骤:实验.1加热乙醇使乙醇蒸汽通入氧化铜粉末中,检验收集到的液体并不是乙醛。 2.先加热氧化铜一段时间,再加热乙醇使乙醇蒸汽通入氧化铜粉末中,很快黑色的氧化铜变为红色。最后用希夫试剂检验生成的液体,显紫色。说明生成了乙醛。 三、实验中的问题: 在做上述实验的前两次试验中检验生成的物质是否为乙醛时,分别用了新制的氢氧化铜和银氨溶液来检验。但均未出现砖红色沉淀和银镜现象。最后用希夫试剂来检验,立即显现出浅紫色。证明生成了乙醛。
用新制的氢氧化铜和银氨溶液检验没现象的原因是乙醛中混有大量的乙醇。以下分别是希夫试剂与醛、所制生成物、乙醇反应的颜色对比图 出现这种不理想现象的可能原因是:乙醇过量,反应后没有立即撤去加热乙醇的酒精灯,致使乙醇蒸汽进入生成物中。 注意:1、该反应较快,氧化铜粉末很快都变成了红色的铜,在操作中必须先加热氧化铜,然后再加热乙醇,而且乙醇加热过程必须一直持续。反应结束后,必须先停止加热乙醇然后再停止加热氧化铜。 2、为减少生成物混入乙醇,应使氧化铜过量。 总结:乙醇和热的氧化铜粉末发生反应。反应的条件是氧化铜粉末必须先加热。该反应氧化铜被还原成铜,做的是氧化剂。 而铜作催化剂时,氧气做的是氧化剂,氧化铜是中间产物。 四、乙醇的催化氧化机理