内点法+外点法
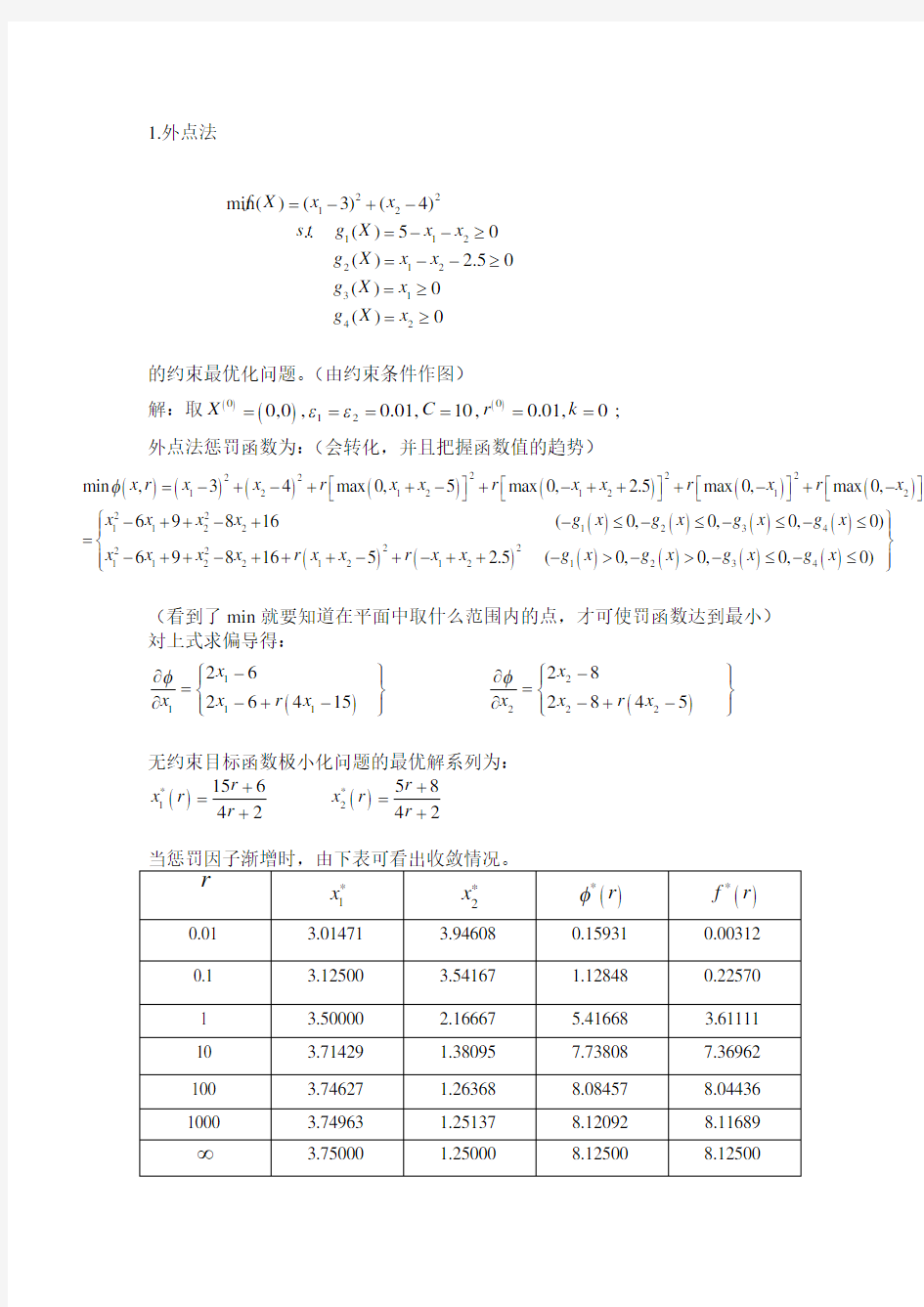

1.外点法
的约束最优化问题。(由约束条件作图)
解:取()()()00120,0,0.01,10,0.01,0;X C r k εε======
外点法惩罚函数为:(会转化,并且把握函数值的趋势)
(看到了min 就要知道在平面中取什么范围内的点,才可使罚函数达到最小) 対上式求偏导得:
()
()
1211221226
28
264152845x x x r x x r x x x φφ--????????
??
==????-+--+-????????
??
无约束目标函数极小化问题的最优解系列为:
()()**
12156584242
r r x r x r r r ++==
++
22
121122123142 min ()(3)(4) .. ()50 () 2.50
()0
()0
f X x x s t
g X x x g X x x g X x g X x =-+-=--≥=--≥=≥=≥()()()()()()()()()()()()()()()222222
1212121222
112212342222
11
22121212min ,34max 0,5max 0, 2.5max 0,max 0,69816(0,0,0,0)698165 2.5(0,0,x r x x r x x r x x r x r x x x x x g x g x g x g x x x x x r x x r x x g x g x g φ=-+-++-+-+++-+-????????????????-++-+-≤-≤-≤-≤=-++-+++-+-++->->-()()340,0)x g x ????
??≤-≤????
则得到最优解()*
*
123.75 1.25
min 8.125X X f x ===
2用内点法求解:
31211221
min ()(1)12
.. ()10 ()0
f X x x s t
g X x g X x =
++=-≥=≥ 的约束最优化问题。
解:取()()()0010,10,0.01,0.1,1,0;X C r k ε===== 外点法惩罚函数为:
()()()()()3
()1212121,ln 1ln()1ln[1()]12
k x r f x r x x x x r x x φ=--+-=++---????
対上式求偏导得:
2111122
114241x x r
r
x x x x φφ??=++-
=-?-?
令上式等于零:
2111122
10424110x x r
x x r
x x φφ?=++-=?-?=-=?
即:3
1
241
()x r x r r +=
无约束目标函数极小化问题的最优解系列为:
*
*
3
12()
41
()x r r x r r +=
当惩罚因子渐减时,由下表可看出收敛情况。
()**
12210
min 3
X X f x ===
(注:文档可能无法思考全面,请浏览后下载,供参考。可复制、编制,期待你的好评与关注!)
外标法测定乙醇含量
外标法测定乙醇含量 一、实验目的 1.熟悉气相色谱仪的操作步骤 2.学习外标法定量的基本原理和测定方法 二、基本原理 用欲测组分的纯物质加稀释剂(对液体试样用溶剂稀释,气体试样用载气或空气稀释)配制成不同质量分数的标准溶液,取固定量标准溶液进样分析,从所得色谱图上测得相应信号(峰面积或峰高),然后绘制响应信号(纵坐标)对质量分数(横坐标)标准曲线。分析时,取和绘制的标准曲线时同样量的试样(固定量进样),测得该试样的响应信号,由标准曲线即可查出其质量分数。 本实验以未知含量的乙醇样品为待测物,利用外标法,测定样品中乙醇的质量分数。 三、实验仪器与试剂 1.仪器: GC7700 气相色谱仪、TCD检测器 2)微量进样器 10μl 3)色谱柱试剂: 2.药品乙醇(分析纯),去离子水载气:高纯氮 四、实验步骤 1.标准溶液的配制: 配制标准系列(50–500ppb) 2.GC7700(TCD)的操作步骤: (1)打开载气总阀门,至0.4 MPa,保持半小时。 (2)打开设备电源,TCD设为“开”,设置各路温度参数,TCD电流为“0”。 3)开启T2000工作站,观测设备输出信号。 (3)15分钟后,气路稳定,确定TCD温度高于100o (4)C并初步稳定,根据分析条件设TCD电流。 (5)观测 TCD基线,调至可视范围,基本稳定后,调零(0-5mv). 6)进样分析,如出倒峰,则可通过修改TCD极性校正。 (6)进样结束后,进行降温设置,或调用关闭设置,关闭TCD电流,各温度设置降到80o C 以下,关电源,关载气。 3.标准曲线的绘制: 用微量进样器向色谱柱中进1号、2号、3号、4号、5号标准溶液各1μl,得到各标准溶液的色谱图,进行数据处理,绘制出标准曲线。 4.未知样的测定: 用微量进样器吸取与标准溶液相同体积的待测样,进样,得到未知样的色谱图,套用标准曲线,从标准曲线上查得其含量。 5.注意事项: (1) 开机时必须先通载气,再开色谱仪(升温);关机时,先关色谱仪(降温),后关载气。 (2) 进样技术 ①进样时要求注射器垂直于进样口,左手扶着针头以防弯曲,右手拿注射器,食指卡在注射器芯和管的交界处,这样就可以避免当进针到气路中由于载气压力较高把芯顶出,影响进样; ②注射器取样时,应先用被测试液洗涤5–6次,然后缓慢抽取一定量试液。若仍有空气带入注射器内,可将针头朝上,轻轻敲注射器管,待空气排尽后,再排除多余试液即可,用滤纸擦净针头; ③进样时要求操作稳当、连贯、迅速,进针位置、进针速度、针尖停留和推出速度都会影响进样重现性,一般要求进样相对误差为2–5%;
气相色谱仪操作步骤(做组分)
气相色谱仪操作步骤(做组分) 1、打开氮气钢瓶,减压阀输出压力调到0.4MPA,平时不需调整,只开关总阀。 2、打开氢气发生器,待发生器的压力达到0.4MPA以后,打开空气发生器。 3、待空气发生器的压力达到0.4MPA以后,观察色谱仪上空气Ⅱ的压力表指示应为0.12mp a(平时不需要调整),打开仪器总开关,仪器开始升温,待所有绿色指示灯闪烁后,按一下色谱仪上的桥流红色按钮,桥流指示灯亮。 4、打开色谱在线工作站,选择通道1,观察基线,基线平稳后开始进行分析。分析时,用上面的六通阀时,按参数键,TCD的极性设为1,检出CO2、C2H4、C2H6等,用下面的六通阀时,按参数键,TCD的极性设为0,检出O2、N2、CH4、CO等。 5、所有工作完成后,先关闭色谱仪总打开,再依次关掉氮、空、氢发生器。 二、求校正因子的操作步骤。 1、打开在线工作站,选择通道1点OK。 2、点击“实验信息”根据需要输入相关信息{也可以不输} 3、点击“方法”再点击屏幕左下方的“采样控制”,输入采样结束时间,选择文件保存方式,并点击“采用” 4、点击屏幕左下方的“积分”选择积分参量为面积,积分方法为“外标法”并点击“采用”。 5、点击“数据采集”“查看基线”待基线稳定后,开始做标准气样,
每做完一个标准气样,你要记住它所保存的文件名。 6、标准气体做完后,打开离线工作站。 7、点击“积分方法”选择积分参量为“面积”,积分方法为“外标法”并点击“采用”。 8、在离线工作站中,点击左上角的“打开”,选择一个你在在线工作站中所做的标样,并打开它。 9、点击“组分表”点击屏幕右边的“全选”在组分表中输入各组分的名字,点击“采用”。 10、点击“校正”,点击“标准含量”输入各组分的含量,选择重复次数,点击OK 11、点击加入标样,找到你在在线工作站所做的标样的文件名,并打开,再点击“加入标样”,直到把你所做的标样都加入一次,点击“校正完毕”点击“校正曲线”中个组分的名字,选择“强制过零”,就会出现校正曲线了。 12、点击屏幕上方的输出,把你所做的校正因子以一个方法文件的名称存在标样下 三、在线工作站中求样品含量的操作步骤 1、打开在线工作站,选择通道2,点击打开,找到你在离线工作站中所保存的相应的校正因子方法文件并打开它。 2、点击查看基线,待基线稳定后就可以进行样品分析。 3、样品出完后,点击预览,就可以看到结果了 四、在离线工作站中,查看所保存的样品的数据结果 1、打开离线工作站 2、点击屏幕左上方的打开,找到你想查看数据的样品的文件名并双
N2000色谱工作站的使用方法
校正归一法(单点多次校正) 在本例中,所测样品有四种组分,各组分浓度如下: 该样品所采用的方法为“面积校正归一”,且已对该标样进行了2次平行进样(重复进样)。现要求对方法进行单点二次的校正归一法校正。 参照本章第一节的叙述,其校正步骤如下: 1、选择方法和积分参数,在此例中我们使用“面积校正归一法”,如图所示: 2、编辑组分表,调开标样谱图,在本例中我们采用“全选”方式套用各组分保留时间及时间宽度,在峰名一栏中输入组分名“A”、“B”、“C”、“D”, 然后单击“采用”,工作站将跳出提交成功窗口: 点击OK,工作站将加亮“校正”按扭,单击“校正”,工作站将跳出校正窗口。 3、输入标准品含量:单击“组分含量”,在组分含量表中输入A、B、C、D的浓度,如图所示:
注:在输入组分含量时,请将输入法切换成英文形式,在中文输入法下,很容易将小数点“.”输成句号“。”。这是软件不能接受的格式,还则系统将提示您“请输入正确的数值”。 4、校正:在本例中我们采用“加入标样”来运行校正,单击“加入标样”,工作站将跳出“打开谱图文件”窗口,选择标样hplc1.dat,单击“打开”,以完成一次校正; 重复第一次校正步骤,即再点击一次“加入标样”,打开标样谱图hplc2.dat文件,并“确定”,单击“校正完毕”用以完成本次平行两点校正。 5、预览校正曲线:
单击“另存”,输入方法文件名“面积校正归一法(平行两次校正)测样.MTD”,用以保存校正曲线,以便分析样品使用。 面积外标法(单组分多点一次校正) 本例是测定气体CO2组分含量示例,其组分分浓度如下表: 该样品所采用的方法为“面积外标法”,且已对该标样进行了5次不同浓度进样。现要求对方法进行单点五次的面积外标法校正。 参照本章第一节的叙述,其校正步骤如下: 1、选择方法和积分参数,在此例中我们使用“面积外标法”,如图所示: 2、编辑组分表,调开标样谱图,在本例中我们采用“全选”方式套用各组分保留时间及时间宽度,在峰名一栏中输入组分名“CO2”,
归一化法,内标法,外标法
归一化法normalization method 一种常用的色谱定量方法。归一化法是把样品中各个组分的峰面积乘以各自的相对校正因子并求和,此和值相当于所有组分的总质量,即所谓“归一”,样品中某组分i的百分含量可用下式计算: pt%= Aifi/(A1f1+A2f2 + ....Anfn )*100 式中f1、f2、fn…为各组分的相对校正因子,A1、A2、…An为各组分的峰面积。如果操作条件稳定,也可以用峰高归一化法定量,此时组分i的百分含量可按下式计算:pt%= hifi/(h1f1+h2f2 + ....hnfn )*100 式中f1、f2、fn、…为各组分在该操作条件下特定的峰高相对校正因子,h1、h2、…hn为各组分的峰高。用归一化法定量时,必须保证样品中所有组分都能流出色谱柱,并在色谱图上显示色谱峰。 定量方法 色谱中常用的定量方法有: a.校正归一化法 当试样中各组分都能流出色谱柱且在检测器上均有响应,各组分的相对校正因子已知时,可用此法定量。组分i在混合物中的百分含量可由下式计算: 其中fi可为质量校正因子,也可为摩尔校正因子。 若各组分的定量校正因子相近或相同(如同系物中沸点接近的组分),则上式可 简化为: 该法简称为归一化法。 校正归一化法的优点是:简便、准确,当操作条件如进样量、流速变化时,对定量结果影响很小。缺点是:对该法的苛刻要求限制了该法的使用。该法适合于常量物质的定量。 b.内标法 所谓内标法是将一定量的纯物质作为内标物,加入到准确称量的试样中,根据被测物和内标物的质量及在色谱图上相应的峰面积比,求出某组分的百分含量。
当只需测定试样中某几各组分时,而且试样中所有组分不能全部出峰时,可用此法。此法适合于微量物质的分析。该法的计算公式如下: 其中,f 是被测组分相对于内标物的相对校正因子。 si 该法的优点是:受操作条件的影响较小,定量结果较为准确,使用上不象归一化法那样受到限制。该法的缺点是:每次分析必须准确称量被测物和内标物,不适合于快速分析。 内标物的选择十分重要。它应该是试样中不存在的物质;加入的量应接近于被测组分色谱;同时要求内标物的色谱峰位于被测组分色谱附近或几个被测组分色谱峰的中间,并与这些组分完全分离;内标物必须不与样品发生反应等。 c.外标法(定量进样-标准曲线法) 所谓外标法就是应用被测组分的纯物质来绘制浓度c对响应值A(h)的标准曲线,然后测试被测样品中被测组分的响应信号(峰面积或峰高),由标准曲线即可查出或通过线型回归计算出其百分含量。 该法必须定量进样,即测定标准曲线和测定未知物时,进入色谱中的样品量必须一致。 该法的优点是:操作简单,计算方便,缺点是:结果的准确度取决于进样量的重现性和操作条件的稳定性。 当被测试样中各组分的浓度变化范围不大时(如工厂的中间控制分析)可不必绘制标准曲线,而用单点校正法。即配制一个与被测组分含量十分接近的浓度为的标准溶液,定量进样,由下式计算被测物的含量。 C S 关于归一化法的一些问题?? 归一化法中的校正因子如何得到??? 如果不做,采用面积归一化法的误差大吗?? 一般那么多的校正因子不可能一一测出呀??
内标法及外标法方法、原理、优缺点
An internal standard should be used when performing MS quantitation. An appropriate internal standard will control for extraction, HPLC injection and ionization variability. In a complex matrix it is not uncommon for two different standard levels in SRM integrated plots, at the lower end of the standard curve, to give nearly an identical response. It is only when an internal standard is used that the two points can be differentiated. Some researchers attempt to prepare standard curves and run samples without an internal standard and find moderate success. Often without an internal standard % RSDs of replicates can be as high as 20%. Using an internal standard the % RSDs can be brought down to approximately 2%. We run triplicates at each level of our standard curve. How do I choose an internal standard? The best internal standard is an isotopically labeled version of the molecule you want to quantify. An isotopically labeled internal standard will have a similar extraction recovery, ionization response in ESI mass spectrometry, and a similar chromatographic retention time. If you are performing non-clinical PK quantitation it may be difficult to justify such a standard since a special synthesis of an isotopically labeled standard can be expensive and time consuming. Often if you are working with medicinal chemists they will have a library of compound analogs that can be used as internal standards. These analogs were made in the evolution of the compound to be tested and will be similar to the compound to be quantified and more importantly will be slightly different by parent mass. Try to avoid using de-methylated (-14) or hydroxylated (+16) analogs as internal standards since these are the most common mass shifts observed in naturally occuring metabolites of the parent compound. A common internal standard is a chlorinated version of the parent molecule. A chlorinated version of the parent molecule will commonly have a similar chromatographic retention time which is an important characteristic of an internal standard. We have found that one of the most important characteristics of an internal standard is that it co-elutes with the compound to be quantified.
内标法与外标法(定义及应用).
内标法与外标法 一、内标法 什么叫内标法?怎样选择内标物? 内标法是一种间接或相对的校准方法。在分析测定样品中某组分含量时,加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。 内标法在气相色谱定量分析中是一种重要的技术。使用内标法时,在样品中加入一定量的标准物质,它可被色谱拄所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。采用内标法定量时,内标物的选择是一项十分重要的工作。理想地说,内标物应当是一个能得到纯样的己知化合物,这样它能以准确、已知的量加到样品中去,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质(如化学结构、极性、挥发度及在溶剂中的溶解度等)、色谱行为和响应特征,最好是被分析物质的一个同系物。当然,在色谱分析条什下,内标物必须能与样品中各组分充分分离。需要指出的是,在少数情况下,分析人员可能比较关心化台物在一个复杂过程中所得到的回收率,此时,他可以使用一种在这种过程中很容易被完全回收的化台物作内标,来测定感兴趣化合物的百分回收率,而不必遵循以上所说的选择原则。 在使用内标法定量时,有哪些因素会影响内标和被测组分的峰高或峰面积的比值? 影响内标和被测组分峰高或峰面积比值的因素主要有化学方面的、色谱方面的和仪器方面的三类。 由化学方面的原因产生的面积比的变化常常在分析重复样品时出现。 化学方面的因素包括: 1、内标物在样品里混合不好; 2、内标物和样品组分之间发生反应, 3、内标物纯度可变等。 对于一个比较成熟的方法来说,色谱方面的问题发生的可能性更大一些,色谱上常见的一些问题(如渗漏)对绝对面积的影响比较大,对面积比的影响则要小一些,但如果绝对面积的变化已大到足以使面积比发生显著变化的程度,那么一定有某个重要的色谱问题存在,比如进样量改变太大,样品组分浓度和内标浓度之间有很大的差别,检测器非线性等。进样量应足够小并保持不变,这样才不致于造成检测器和积分装置饱和。如果认为方法比较可靠,而色谱固看来也是正常的话,应着重检查积分装置和设置、斜率和峰宽定位。对积分装置发生怀疑的最有力的证据是:面积比可变,而峰高比保持相对恒定, 在制作内标标准曲线时应注意什么?
四元低压液相色谱仪操作步骤
四元低压液相色谱仪操作步骤 目录 一、开机步骤 二、基线不稳的原因以及解决方法 三、如何维护保护柱 四、进样操作结束后的信息填写 五、关机步骤 六、故障及排除方法
一、开机步骤: 1、根据药典的要求配置好流动相,过滤和超声30分钟流动相。更换好所需流动相;开电脑主机,然后开四元液相机器,待检测器稳定的时候,打开伍丰液相色谱工作站 2、置换流动相排气泡其步骤如下:设置压力下限为0MPa,打开排空阀. 分别把更换好的流动相通道比例设置100和流速5ml/min按“泵冲洗”,冲洗管路约5分钟左右,排出气泡;然后根据药典上的要求将流动相的比例设置好以流速5ml/min按“泵启动”,一起冲洗管路约5分钟左右,排出气泡;泵停止,关闭排空阀。(例如:药典要求流动相:A泵为甲醇,B泵为水,比例为75:25;打开排空阀,设置压力下限为0MPa.,A泵的流量设置为100和5ml/min流速大概冲洗管路5分钟左右;B泵的流量设置为100和5ml/min流速大概冲洗管路5分钟左右;然后按照要求,将A泵流动相设置为75,B泵流动相设置为25和5ml/min 流速一起大概冲洗管路5分钟左右,排出气泡;泵停止,关排空阀。) 3、然后按照药典在色谱工作站上设置波长、所需各泵的流量,流速一般为1ml/min(例如:A泵:75,B泵:25,C和D自动为0;流量:1.000)、柱温(一般30℃-35℃)、压力下限和压力上限(一般为0.3MPa和25-42MPa),按泵启动。 4、如果药典方法需要梯度洗脱,则在上面界面的左下角时间表设置梯度流量 例如:桑叶的高效流动相:以甲醇为流动相A,以0.5%磷酸溶液为流动相B (因为实验室有一个共识:A为甲醇,B为水,C为乙腈,D为含有酸、碱或缓
高效液相色谱使用详细操作步骤
For personal use only in study and research; not for commercial use 高效液相色谱使用操作步骤 一、泵操作面板 PUM P:运行键 START:梯度运行键 PURGE:快冲键RESET:复位键 HOLD:程序暂停键 STOP:泵停止键TIME:程序开启键 FLOW:流速: RSVR: A B C PROG:程序 DEL:删除程序 PMAX:最大压力PMIN:最小压力 ENTER:确定 TIME 0 FLOW ENTER (设置流速) TIME 0 RSVR ABC ENTER (设置溶剂) TIME 0 PROG ENTER (设置程序) TIME 0 % A 30 ENTER (设置比例) TIME 0 % B 30 ENTER (设置比例) TIME 0 % C 40 ENTER (设置比例) DEL PROG 1 ENTER (删除程序) 1.对于梯度泵设定举例如下:(人参皂苷的梯度) TIME 0 PROG 1 ENTER TIME 0 % A 81 ENTER TIME 15 % A 81 ENTER TIME 60 % A 65 ENTER
TIME 65 % A 0 ENTER TIME 80 % A 0 ENTER TIME 81 % A 81 ENTER TIME 100 % A 81 ENTER 因为A+B之和始终是100%,所以B相就不用设了。7.对于对照品在这种条件下只要对照品的峰全部出完后按下RESET键等20分钟即可进第二针进完针后按START 键运行梯度程序。但是对样品来说一定要等峰出完后再按下RESET键等基线基走直才可进下一针。 二、检测器操作面板 RESET:复位键 A/Z:调零键 WL:调波长 ENTER:确定 三、实验前的准备工作 1、实验前流动相提前配好过滤脱气。 2、置换流动相时把泵的滤头从原来的流动相中换到新的流 动相中滤头要轻拿轻放。 3、液路排气顺序:首先打开排气阀—按PURGE键进行排气 结束后—按STOP键停止—再拧紧排气阀—按PUM P键让泵运行。 4、打开检测器首先观察右上角氘灯指示灯确认氘灯是否点 亮,按WL键调好实验波长后按A/Z键调零。 5、打开电脑打开在线工站选择对应的通道,输入实验信息、方法、包括采样控制,积分和仪器条件,积分面积外标法测
色谱定量计算三种方法,归一化法,内标法和外标法
色谱法是根据色谱峰的面积或高度进行定量分析的。色谱定量计算方法很多,目前比较广泛应用的有归一化法、内标法和外标法。 1. 归一化法 如果试样中所有组分均能流出色谱柱并显示色谱峰,则可用此法计算组分含量。设试样中共有n个组分,各组分的量分别为m1,m2,……,m n,则i种组分的百分含量为: 归一化法的优点是简便、准确,进样量的多少不影响定量的准确性,操作条件的变动对结果的影响也较小,对组分的同时测定尤其显得方便。缺点是试样中所用的组分必须全部出峰,某些不需定量的组分也需测出其校正因子和峰面积,因此应用受到一些限制。 2. 内标法 当试样中所有组分不能全部出峰,或只要求测定试样中某个或几个组分时,可用此法。 准确称取m(g)试样,加入某种纯物质ms(g)作为内标物,根据试样和内标物的质量比m s/m及相应的色谱峰面积之比,基于下式可求组分i的百分含量W i%: 因为 所以 内标物的选择条件是:内标物与试样互溶且是试样中不存在的纯物质;内标物的色谱峰既处于待测组分峰附近,彼此又能很好地分开且不受其它峰干扰;加入量宜与待测组分量相近。 内标法的优点是定量准确,操作条件不必严格控制,且不象归一化法那样在使用上有所限制。缺点是必须对试样和内标物准确称重,比较费时。
3. 外标法(亦称标准曲线法) 该法是在一定色谱操作条件下,用纯物质配制一系列不同的浓度的标准样,定量进样,按测得的峰面积对标准系列的浓度作图绘制标准曲线。进行试样分析时,在与标准系列严格相同的条件下定量进样,由所得峰面积从标准曲线上即可查得待测组分的含量。 外标法的优点是操作和计算简便,不需要知道所有组分的相对校正因子,其准确度主要取决于进样量的准确和重现性,以及操作条件的稳定性。
高效液相色谱使用详细操作步骤
高效液相色谱使用操作步骤 一、泵操作面板 PUM P:运行键 START:梯度运行键 PURGE:快冲键RESET:复位键 HOLD:程序暂停键 STOP:泵停止键TIME:程序开启键 FLOW:流速: RSVR: A B C PROG:程序 DEL:删除程序 PMAX:最大压力PMIN:最小压力 ENTER:确定 TIME 0 FLOW ENTER (设置流速) TIME 0 RSVR ABC ENTER (设置溶剂) TIME 0 PROG ENTER (设置程序) TIME 0 % A 30 ENTER (设置比例) TIME 0 % B 30 ENTER (设置比例) TIME 0 % C 40 ENTER (设置比例) DEL PROG 1 ENTER (删除程序) 1.对于梯度泵设定举例如下:(人参皂苷的梯度) TIME 0 PROG 1 ENTER TIME 0 % A 81 ENTER TIME 15 % A 81 ENTER TIME 60 % A 65 ENTER TIME 65 % A 0 ENTER TIME 80 % A 0 ENTER TIME 81 % A 81 ENTER
TIME 100 % A 81 ENTER 因为A+B之和始终是100%,所以B相就不用设了。7.对于对照品在这种条件下只要对照品的峰全部出完后按下RESET键等20分钟即可进第二针进完针后按START 键运行梯度程序。但是对样品来说一定要等峰出完后再按下RESET键等基线基走直才可进下一针。 二、检测器操作面板 RESET:复位键 A/Z:调零键 WL:调波长 ENTER:确定 三、实验前的准备工作 1、实验前流动相提前配好过滤脱气。 2、置换流动相时把泵的滤头从原来的流动相中换到新的流 动相中滤头要轻拿轻放。 3、液路排气顺序:首先打开排气阀—按PURGE键进行排气 结束后—按STOP键停止—再拧紧排气阀—按PUM P键让泵运行。 4、打开检测器首先观察右上角氘灯指示灯确认氘灯是否点 亮,按WL键调好实验波长后按A/Z键调零。 5、打开电脑打开在线工站选择对应的通道,输入实验信息、方法、包括采样控制,积分和仪器条件,积分面积外标法测绝对含量归一法相对含量内标法测绝对含量;后点数据采集,将电压调到-20~20,时间调到0~30,点零点校正后再点查看基线,大约1个小时左右基线基本稳定(就是基本成直
岛津液相色谱工作站外标法使用解析
岛津液相色谱工作站外标法使用解析
————————————————————————————————作者: ————————————————————————————————日期: ?
岛津液相色谱工作站外标法使用解析 外标法又称工作曲线法,首先配制含待测组分浓度不同的系列标准工作溶液,如果待测组分不止一个,我们也把该标准溶液称为混合标准溶液,然后进样得浓度不同的系列标准工作溶液的色谱图。通常利用浓度不同的系列标准工作溶液的色谱数据建立工作曲线和线性方程,然后根据试样的色谱数据既可以利用工作曲线查得试样的浓度数据,也可利用线性方程计算试样中各组分的浓度。在相同色谱条件下,可以用已建立的工作曲线或线性方程进行批量样品分析。 一、进样 首先进浓度不同的系列标准工作溶液,然后进试样,得标样色谱图和试样色谱图并保存。如果是单次分析(手动进样模式),在设置单次分析条件时,应将样品瓶架设置为“-1”,如图所示,然后按电脑提示进样,待采样结束后停在分析。 启动或停止单次分析,既可以用助手栏“主项目”下的功能按钮,也可用数据采集窗口中的功能按钮,如图所示。 通常使用数据采集窗口的功能按钮启动和停止单次分析更为高效。无论是启动或停止单次分析,系统均有“请稍后……”的等待提示,务必等待足够时间,方可进行下一步操作。 二、色谱图解析 得到色谱图后,套取组分的保留时间,确定组分的名称,输入标样中各组分的浓度是最基本的工作。有时还要根据色谱图的具体情况对色谱图进行手动处理,色谱图处理通常放在进样完成后,停机或在流动相置换期间进行。点击“LabSolutions Essentia主项目”窗口下的“处理工具”图标,再双击右边窗口中的“再解析”图标,弹出“再解析”(即数据处理)窗口。在数据管理器选择文件所在的文件夹,切换到数据标签,打开所需的数据文件,这里我们选择打开“化妆品抗菌剂_Std11”数据文件,过程如图所示。 1.编辑色谱图
CLASS-VP多点(外标法)校正标准曲线制作
岛津class-vp外标法标准曲线制作流程 1.定义峰。打开目标数据文件,点击define peaks或define Single peak 查看定义时间范围,选择合适时间窗(一般默认5%)等,点击OK。
2.编辑峰组表。 2.1点击method菜单下的peaks/groups。 2.2 弹出对话框(下图)中给出峰名, fit type项下选择linear,多点不过零点。Level1、level2等输入标准溶液浓度级别。 2.3保存方法文件。点击file\method\save as…命名该方法文件。
3通过后处理方式建立标准曲线(多点校正时需建立序列表) 3.1建立序列表 File/sequence/new 点击OK 3.2编辑sequence表:批表中调入方法文件(2.3中保存的方法文件)、数据文件(按照浓度顺序)、level(从上到下依次1、2、3等级别),Run type项第一行选择clear all Calibration 和begin calibration,最后一行end calibration
3.3 保存sequence表:file/sequence/save as,命名并保存序列表。 3.4运行sequence :sequence菜单下process。执行Start。
4.查看标准曲线。Method/Review Calibration 弹出标准曲线及相关信息
5.定量未知样品 5.1 后处理未知样品定量 5.1.1打开未知样品数据文件(File/data/open) 点击打开 5.1.2查看外标报告。Reports/View/External Standard (Det A)
利巴韦林检验标准操作规程(外标法)
利巴韦林检验标准操作规程(外标法)
文件名称利巴韦林检验标准操作规程 文件编码版本号00 制定人部门审核质量保证 部审核 批准人 制定日期审核日期审核日期批准日期 颁发部门质量保证部生效日期年月日 分发部门质量保证部、质量控制部 1.目的:指导检验人员掌握正确的操作方法,以确保检测结果的准确性和可靠性。 2.适用范围:仅适用于本公司。 3.责任:质量控制部经理、化验室主任、化验员。 4.标准依据:利巴韦林内控质量标准 5.内容: 5.1.【性状】 5.1.1.标准规定:本品为白色或类白色结晶性粉末;无臭,无味。 5.1.1.1.仪器:偏光显微镜,载玻片 5.1.1.2.试液:液状石蜡 5.1.1.3.操作方法:取本品少许,置载玻片上,加液状石蜡1滴使悬浮,在偏光显微镜下 检视。观察转动载物台时,是否呈现消光位和双折射现象,进行结晶性判断。 5.1.1.4.结果与判定:根据所观察的供试品外观与转动载物台时,是否呈现消光位和双折 射现象进行判断,与上述描述相符者,判为结晶性粉末。 5.1.2.标准规定:本品在水中易溶,在乙醇中微溶,在乙醚或二氯甲烷中不溶。 5.1.2.1.用具:小试管 5.1.2.2.试剂:乙醚、乙醇、二氯甲烷 5.1.2.3.操作方法:称取研成细粉的供试品,置于 25℃±2℃一定容量的溶剂中,每隔5 分钟强力振摇30秒钟;观察30分钟内的溶解情况,如无目视可见的溶质颗粒时,即视为完全溶解。 5.1.2.4.结果与判定:本品在水中易溶,在乙醇中微溶,在乙醚或二氯甲烷中不溶,判为 符合规定;否则,判为不符合规定。
文件编码版本号00 5.1.3.比旋度 5.1.3.1.标准规定:40mg/ml的水溶液比旋度为-35.0o至-37.0o。 5.1.3.2.仪器与用具:分析天平、旋光仪、温度计 5.1.3.3.操作方法 5.1.3.3.1.取本品2.0g,精密称定,置50ml容量瓶中,加水稀释至刻度,制成每1ml中约 含40mg的溶液,摇匀,使供试品溶液的温度控制在20℃±0.5℃。用空白溶剂校正 仪器零点,供试品溶液与空白溶液同一测定管,每次测定应保持测定管方向,位置 不变,按比旋度测定法SOP测定,记录所测数据,旋光度读数应重复3次,取其平 均值。 5.1.3.4.计算公式: [α]t D = 100×α L×C = α×V样 2×W样×(1-干燥失重%) 式中:[α]为比旋度 α为测定的旋光度 L为测定管的长度,dm W为供试品重量,g 5.1.3.5.结果与判定:计算所得数值按有效数字修约规则修约为标准规定的有效数字,若 此数在-35.0o至-37.0o。范围内,判为符合规定;否则,判为不符合规定。 5.1.3. 6.注意事项:供试品溶液配制后应及时测定;通电开机之前应取出仪器样品室内的 物品。开机预热约20分钟后再进去测定。测定时注意环境温度。 5.2.【鉴别】 5.2.1.(1) 5.2.1.1.试剂与试液:氢氧化钠试液、 5.2.1.2.操作方法:取本品约0.1g,加水10ml使溶解,加氢氧化钠试液5ml,加热至沸, 即发生氨臭,能使湿润的红色石蕊试纸变蓝色。 5.2.1.3.结果与判定:若变蓝色,判为符合规定;否则,判为不符合规定。 5.2.2.(2)
外标法测定乙醇含量
乙醇中少量水分的测定 一、实验目的 1、熟悉气相色谱仪的组成及各部分的作用。 2、了解外标法定量的原理和方法。 3、学会用外标法测定样品含量的方法。 4、熟悉热导池检测器的特点及使用方法。 二、基本原理 外标法又称标准曲线法或直接比较法,是一种简便、快速的绝对定量方法(归一化法是相对定量方法)。用标准样品配置不同浓度的系列标准溶液,在与欲测组分相同的色谱条件下,等体积准确进样,测量每次进样标准物的峰面积或峰高,用峰面积或峰高对标准物有效浓度绘制标准曲线,标准曲线的斜率为绝对校正因子。在相同色谱条件下等体积进样品溶液,由其峰面积或峰高在标准曲线上找出对应的样品溶液组分浓度。 在一些工厂的常规分析中,样品中各组份中的浓度一般变化不大,可不必做校正曲线,而用单点校正法来分析。即配置一个和被测组份含量十分接近的标准样,定量进样,由被测组份与外标组份峰面积或峰高比求被测组份的含量。 该方法的优点是操作简单和计算方便,准确性较高。缺点是仪器和操作条件对分析结果影响很大,不象归一化法和内标法定量操作中可相互抵消。 三、仪器、试剂和色谱条件 1、仪器 配有热导池检测器和色谱工作站的SP6800A气相色谱仪,10μL微量注射器,GDX102型填充色谱柱。 2、试剂 5%水乙醇标准溶液:精密移取5.0mL蒸馏水用无水乙醇稀释并定容至100mL。95%乙醇样品。 3、色谱条件 柱温:120℃;进样室温度150℃;检测器温度150℃;载气流速:50mL/min;热导桥流100mA;衰减:1;进样量为1μL。
四、操作步骤 1、打开载气(H2)钢瓶总阀,通载气10min后,打开电源开关。 2、设置柱温为120℃、气化温度为150℃和检测温度为150℃,启动加热。 3、待恒温后,设定桥流及衰减。 4、打开工作站,选择通道。用仪器面板上的TCD调零电位器将基线调至约0.5mv处,让色谱仪走基线,待基线稳定。 5、用水乙醇标准溶液润洗微量注射器,准确吸取1μL进样,启动色谱工作站采集数据,记录水的峰面积。 6、按相同方法进乙醇试样,记录水的峰面积。 7、实训完毕后先设置桥电流数值为0.0。 8、设置气化室温度、柱温、检测室温度在室温以上约10℃。 9、待温度达到设定值时关闭气相色谱仪电源开关。 10、关闭载气钢瓶和减压阀。 五、数据记录与处理 乙醇中少量水分的测定 标准样tR(min) A水 样品 乙醇中水分含量X i按下式计算,即: X i=E i·A i/A E 式中: X i为乙醇试样溶液中组份水的含量%;E i为水乙醇标准溶液中水的含量%;A i 为乙醇试样溶液中水的峰面积;A E为水乙醇标准溶液中水的峰面积。 六、思考题 1、为什么TCD检测器在使用时一定要先通气,再通电?关机时,一定要先断电,待柱温等降下来后再断气? 2、简述外标法测定乙醇中少量水分的原理和方法?
Clarity 中文色谱工作站操作指导_定量篇_外标法
Clarity 中文色谱工作站操作指导_定量篇_外标法 本资料以含乙醇的样品为例,介绍了如何使用Clarity 工作站进行外标法定量。 校准样品: 样品1:含0.4mg/g 的乙醇。 样品2:含1.4mg/g 的乙醇。 样品3:含2.4mg/g 的乙醇。 三个校准样品分析完成后得到的校准色谱图文件存在clatiy 安装目录下,work1目录内的calib 子目录中,文件名分别是cal04.prm 、cal14.prm 和cal24.prm 。 未知样品:含未知量乙醇。 分析完成后得到的色谱图文件存在cal 子目录中,为cal09.prm 。 外标法定量步骤 1、进入校准窗口 1.1双击桌面上的clarity 工作站图标,启动工作站 1.2以任意用户名登陆工作站,默认用户为“Administrator ” 双击图标
1.3点击“校准窗口”图标进入校准视图 2、制作第一个浓度级别的校准曲线 2.1选择“文件”命令下拉菜单中的“新建”命令或者直接点击工具栏上的“新校准”图标新建一个校准文件。 单击“确定”
或者 2.2选择“校准”命令下拉菜单中的“OPTION ”命令或者工具栏上的“校准选项”图标打开校准选项设置界面。 或者 单 击“新校 准”图标 单击“新建” 单击“OPTIONS ”
2.3设置校准选项 2.4选择“文件”命令下拉菜单中的“OPEN STANDARD ”命令或者直接点击工具栏上的“打开标准”图标打开第一个级别的校准色谱图文件cal04.prm 。 ①显示模式设为“ESTD ” ②工作模式设为“校准” ③校准方式设为“自动” ④输入样品的实际浓度单位 ⑤设置完毕后, 单击“OK ”确认 “校准选项”