高等有机化学_第5章羰基的亲核加成及相关反应

第五章 羰基的亲核加成及相关反应
羰基化合物包括醛、酮、羧酸及衍生物和CO 2。 5.1 羰基的结构
C
O
δ+δ-
亲电中心羰基碳的活性较大,易被亲核试剂进攻而发生亲核加成反应和亲核取代反应。 5.2 亲核加成反应的历程及影响因素 5.2.1 HCN 的加成 反应为碱催化。
]CN ][CO [k v ->=
OH -
+HCN
CN -+ H 2O
快
-C
δ+δ-C
O -CN
C
OH
CN
+OH - 反应的平衡位置受电子效应和空间效应的影响。
二、亲核加成反应的一般特点 1.反应可以被酸或碱催化
酸催化可提高羰基的亲电活性。
C
O +H +
+
OH
碱催化提高亲核试剂的亲核性。
Nu
H +OH --+H 2O
Nu H ->
2.多数醛酮的亲核加成为可逆反应,用于分离与提纯。 5.2.2 影响羰基亲核加成反应活性的因素 一、羰基化合物的结构 1.电子效应
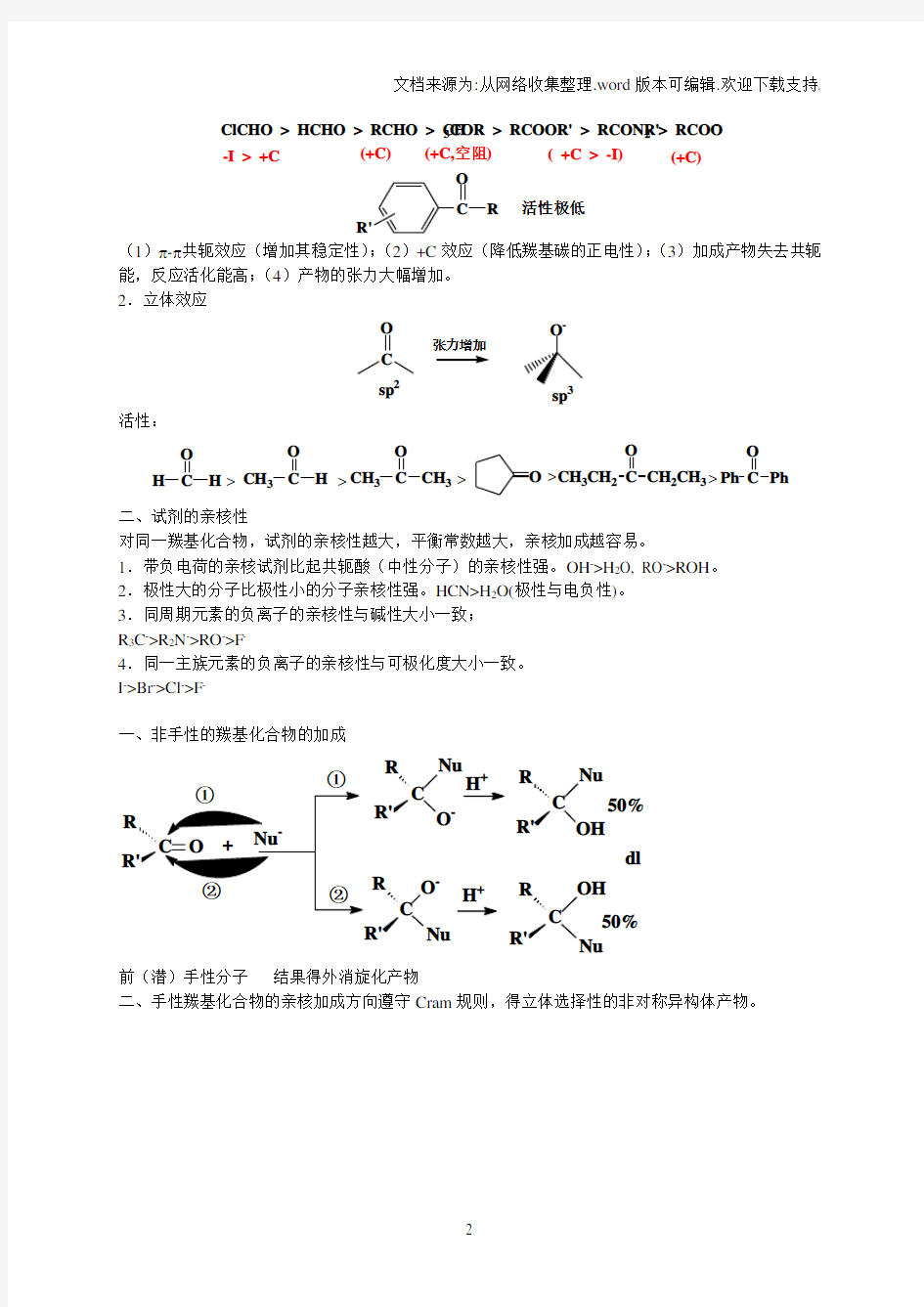
羰基碳的正电性越大,亲核加成速度越大,反应活性越大。羰基碳所连的吸电基(-I ,-C )使其亲核加成反应的活性增加,而供电基(+I ,+C )则使其活性降低。 活泼顺序:
ClCHO > HCHO > RCHO > CH 3COR > RCOOR' > RCONR'2 > RCOO --I > +C
(+C)
(+C,空阻)
( +C > -I)
(+C)
C
O R
R'
活性极低
(1)π-π共轭效应(增加其稳定性);(2)+C 效应(降低羰基碳的正电性);(3)加成产物失去共轭能,反应活化能高;(4)产物的张力大幅增加。 2.立体效应
C
O -
sp 2
活性:
O C
H
H O
C CH 3H O
C CH 3CH 3O O
C CH 3CH 2CH 2CH 3O
C Ph Ph
>>>>
>
二、试剂的亲核性
对同一羰基化合物,试剂的亲核性越大,平衡常数越大,亲核加成越容易。
1.带负电荷的亲核试剂比起共轭酸(中性分子)的亲核性强。OH ->H 2O, RO ->ROH 。 2.极性大的分子比极性小的分子亲核性强。HCN>H 2O(极性与电负性)。 3.同周期元素的负离子的亲核性与碱性大小一致; R 3C ->R 2N ->RO ->F -
4.同一主族元素的负离子的亲核性与可极化度大小一致。 I ->Br ->Cl ->F -
一、非手性的羰基化合物的加成
C
Nu O -
H
+
C Nu
OH
50%C
O -Nu
+
C
OH Nu 50%dl
前(潜)手性分子 结果得外消旋化产物
二、手性羰基化合物的亲核加成方向遵守Cram 规则,得立体选择性的非对称异构体产物。
高等有机化学习题教学内容
高等有机化学习题 第一章 化学键 一、用共振轮说明下列问题 1) 联本中的C 1-C 2键长为什么比乙烷中的键长短?联苯的硝化反应为什么主要发生在2-位 和4-位? 联苯的共振结构式可表是如下: (1) 由共振结构式可以看出C 1-C 2键有双键结构的贡献,故比乙烷的C 1-C 2键短。 (2) 由共振结构式可以看出邻对位负电荷相对集中,故有利于发生硝化反应。 2) 方酸为什么是强酸?(强于硫酸) 方酸的共振结构式可表是如下:对吗? 由方酸的共振结构式可以看出方酸的电子离域效果更好。 二、试推测6,6-二苯基富烯的亲电取代发生于哪个环,哪个位置?亲核取代发生于哪个环, 哪个位置? 6,6-二苯基富烯的共振式如下: 由6,6-二苯基富烯的共振式可以看出,亲电取代发生在五元环的2位上,而亲核取代 发生在苯环的2位上。 三、计算烯丙基正离子和环丙烯正离子π分子轨道的总能量,并比较两者的稳定性。 烯丙基正离子有两个电子在成键轨道上其总能量为 E 烯丙基正离子=2E 1=2(α+1.414β)=2α+2.828β 11' O HO O O O OH O O O OH O O O HO O O O S O O HO O S O O OH O S O O O S O O OH
环丙烯正离子有两个电子在成键轨道上其总能量为 E 环丙烯正离子=2E 1=2(α+2β)=2α+2β 能量差值为 E 烯丙基正离子- E 环丙烯正离子=(2α+2.828β)- (2α+2β)=0.828β 因此,环丙烯正离子比烯丙基正离子稳定。 四、用HMO 法分别说明烯丙基负离子和环丙烯负离子的电子排布和成键情况,并比较两者 稳定性。 五、简要说明 1)吡咯和吡啶分子的极性方向相反,为什么? 吡咯分子中氮原子给出一对为共用电子参与了共轭分子的大π键,也就是电子从氮原子流向五员环,而吡啶分子中氮原子只拿出一个电子参与共轭,并且氮原子的电负性大于碳原子使电子流向氮原子的方向。因此,两个分子的极性正好相反。 2)富烯分子为什么具有极性?其极性方向与环庚富烯的相反,为什么? 富烯分子中环外双键的流向五员环形成稳定的6π体系的去向,从而环外双键中的末端碳原子带有部分正电荷,五员环接受电子后变成负电荷的中心,因此分子具有极性。 N N H 能级 烯丙基负离子 环丙基负离子 α+1.414β α+2β E=2(α+1.414β)+2α-2(α+2β)-2(α-β) = 2α+2.828β+2α-2α-4β-2α+2β =1.172β
醛和酮亲核加成反应附加答案全解
醛和酮 亲核加成反应 一、基本要求 1.掌握醛酮的命名、结构、性质;醛酮的鉴别反应;不饱和醛酮的性质 2.熟悉亲核加成反应历程及其反应活性规律;醛酮的制备 二、知识要点 (一)醛酮的分类和命名 (二)醛酮的结构: 醛酮的官能团是羰基,所以要了解醛酮必须先了解羰基的结构。 C=O 双键中氧原子的电负性比碳原子大,所以π电子云的分布偏向氧原子,故羰基是极化的,氧原子上带部分负电荷,碳原子上带部分正电荷。 (三)醛酮的化学性质 醛酮中的羰基由于π键的极化,使得氧原子上带部分负电荷,碳原子上带部分正电荷。氧原子可以形成比较稳定的氧负离子,它较带正电荷的碳原子要稳定得多,因此反应中心是羰基中带正电荷的碳。所以羰基易与亲核试剂进行加成反应(亲核加成反应)。 此外,受羰基的影响,与羰基直接相连的α-碳原子上的氢原子(α-H )较活泼,能发生一系列反应。 亲核加成反应和α-H 的反应是醛、酮的两类主要化学性质。 1.羰基上的亲核加成反应 醛,酮亲核加成反应的影响因素:羰基碳上正电性的多少有关,羰基碳上所连的烃基结构有关,亲核试剂的亲核性大小有关。 (1)与含碳的亲核试剂的加成 ○ 1氰氢酸: ○2 炔化物 C O C O H C H O 121.8116.5。。sp 2 杂化键 键近平面三角形结构πσC C R O H H ( )δδ 酸和亲电试剂进攻富电子的氧碱和亲核试剂进攻缺电子的碳 涉及醛的反应 氧化反应( ) αH 的反应羟醛缩合反应卤代反应C O C OH + HCN CN 羟基睛 α
○3 有机金属化合物: (2)与含氮的亲核试剂的加成 ○ 11o 胺 ○ 2 2o 胺 ○ 3氨的多种衍生物: (3)与含硫的亲核试剂的加成-------亚硫酸氢钠 产物α-羟基磺酸盐为白色结晶,不溶于饱和的亚硫酸氢钠溶液中,容易分离出来;与酸或碱共热,又可得原来的醛、酮。故此反应可用以提纯醛、酮。 反应范围: 醛、甲基酮、八元环以下的脂环酮。 反应的应用:鉴别化合物,分离和提纯醛、酮。 (4)与含氧的亲核试剂的加成 ○1水 ○ 2醇 醛较易形成缩醛,酮在一般条件下形成缩酮较困难,用12二醇或13-二醇则易生成缩酮。有机合成中用来保护羰基。 2.α-H 的反应(羟醛缩合、交叉缩合、卤仿反应) 醛、酮分子中由于羰基的影响,α-H 变得活泼,具有酸性,所以带有α-H 的醛、酮具有如下的性质: (1)羟醛缩合 有α-H 的醛在稀碱(10%NaOH )溶液中能和另一分子醛相互作用,生成β-羟基醛 ,故称为羟醛缩合反应。 (2)交叉缩合 C O δδ+ R MgX δδC OMgX R H 2O R C OH +HOMgX 无水乙醚C O NaO-S-OH C OH SO 3Na C ONa SO 3H +O 醇钠 强酸强酸盐 白( )R C H ( R' )R C OH H O ( R' ) O +R'' R''OH R C O H O ( R' ) R''OH R''R'' HCl HCl 无水干+H 2O 半缩醛 酮不稳定 一般不能分离出来缩醛 酮 ,双醚结构。 对碱、氧化剂、还原剂稳定,可分离出来。酸性条件下易水解 ( ) ( )NH 2-OH NH 2-NH 2NH 2-NH NH 2-NH-C-NH 2O NH 2-NH O 2N NO 2羟氨 肼苯肼二硝基苯肼氨基脲2,4
醛,酮结构对羰基亲核加成反应活性的影响
大学化学 第15卷 第2期2000年4月 醛、酮结构对羰基亲核加成反应活性的影响 许 申 鸿 (青岛大学化学系 山东266071) 醛、酮分子中都含有活泼的羰基,亲核加成是醛酮最重要、最典型的反应之一。其反应历程为: 式中R为H或烃基,Nu为亲核试剂。这两种历程,决定反应速率的关键步骤均为Nu对羰基的进攻[1~3]。因此,羰基化合物的结构以及Nu的性质对加成反应进行的难易程度均有影响。但在相同的条件下,同一亲核试剂对不同羰基化合物的加成反应,影响反应活性的因素就只有羰基化合物的结构了。国内有机化学教科书[3~7]一般都是从两方面论述羰基反应活性的:①电子因素:当羰基碳上连有给电性基团(如烷基、芳基等)时,由于中心碳原子的电正性减小,从而降低了它的亲电能力,使反应活性下降。另一方面,给电作用还强化了过渡态中氧上发展出来的负电荷,使过渡态能量增加而不利于反应的进行。相反的,当羰基碳上连有吸电基团(如F3C—等)时,则会使反应速度加快。②空间因素:由于从反应物到过渡态及产物,羰基碳由sp2杂化变为sp3杂化,反应中存在着明显的空间特性。在反应过程中,R基会被越来越近地挤在一起,非键张力使过渡态内能增加,不利于反应的进行。故当R基的体积增大时,反应速率迅速下降。当然,Nu体积增大,同样也会降低反应速率。综合上述两方面的影响,可以得出一般醛、酮亲核加成反应的活性次序: 以上论述,对于一般脂肪醛酮的亲核加成反应活性的比较是足够的。例如根据上述讨论,很容易给出下列各组醛酮的亲核加成反应活性:①CF3CH2CH O>CH3CH2CH O>CH3C OCH2CH3 >CH3CH2C OCH2CH3;②ArCH2C OR>ArC OR>Ar2C O。 可是对于环酮来说,前面的讨论就显得有些单薄、不完善。例如,如何比较下两组酮①环己酮与CH3C OCH2CH2CH3;②环己酮与环丁酮的反应活性呢?对此,学生在解答习题时常会感到困难,不知该如何去分析解答。因为仅从上述两方面的影响因素去考虑,显然是不够的,无法做出正确判断。我们先来讨论例①。由于烷基的供电子能力差别甚小,因此,这两个 54
高等有机化学课后习题
第一章习题 1.用共振式说明苯甲醚分子中甲氧基的邻/对位效应。 2.比较下列各组化合物的酸性大小并予以解释。 (1)HOCH2CH2COOH 和CH3CH(OH)COOH (2)乙酸、丙二酸、乙二酸和甲酸 (3)COOH NO 2 和 COOH HO (4) H 2C CH 2H 3C CH 3HC CH 、和 3.比较碱性。 H 2N (1) CH 3CH 2NH 2 和 (2)NH 2HO NH 2 O 2N 和(3) N H N 和 比较C-Br 键断裂的难易次序 和CH 3CH 2CHCH 2CH 3CH 3OCHCH 2CH 3CH 3CH 2CHCF 2CF 3在极性条件下,、4. 5.下列共振式哪一个贡献大,为什么? C C C C O A B 6.在亲核加成反应中ArCH 2COR 和ArCOR 哪一个活性高,为什么? 7.解释酸性大小。 COOH < COOH (1) COOH COOH (2) OH > 8.为什么ArCOR 被HI 断裂时,得到的产物是RI 和ArOH ,而不是ROH 和ArI 。 9.下列反应中几乎不会生成PhCH 2CBr(CH 3)2,为什么? PhCH 2CH(CH 3)2 + Br 2 PhCHBrCH(CH 3)2 + HBr hv 10.比较拗CH 3COCH 2COCH 3和CH 3COCH 2COOC 2H 5的酸性,并简要说明原因。 11.为什么胺的碱性大于酰胺? 12.羧酸及其衍生物的亲核取代反应活性为什么是RCOCl>(RCO)2O>RC OOR’~ RCOOH>RCONH 2。 13.为什么顺丁烯二酸的p K a1比反丁烯二酸小,p K a2却正相反?
高等有机化学_第5章羰基的亲核加成及相关反应
第五章 羰基的亲核加成及相关反应 羰基化合物包括醛、酮、羧酸及衍生物和CO 2。 5.1 羰基的结构 C O δ+δ- 亲电中心羰基碳的活性较大,易被亲核试剂进攻而发生亲核加成反应和亲核取代反应。 5.2 亲核加成反应的历程及影响因素 5.2.1 HCN 的加成 反应为碱催化。 ]CN ][CO [k v ->= OH - +HCN CN -+ H 2O 快 -C δ+δ-C O -CN C OH CN +OH - 反应的平衡位置受电子效应和空间效应的影响。 二、亲核加成反应的一般特点 1.反应可以被酸或碱催化 酸催化可提高羰基的亲电活性。 C O +H + + OH 碱催化提高亲核试剂的亲核性。 Nu H +OH --+H 2O Nu H -> 2.多数醛酮的亲核加成为可逆反应,用于分离与提纯。 5.2.2 影响羰基亲核加成反应活性的因素 一、羰基化合物的结构 1.电子效应 羰基碳的正电性越大,亲核加成速度越大,反应活性越大。羰基碳所连的吸电基(-I ,-C )使其亲核加成反应的活性增加,而供电基(+I ,+C )则使其活性降低。 活泼顺序:
ClCHO > HCHO > RCHO > CH 3COR > RCOOR' > RCONR'2 > RCOO --I > +C (+C) (+C,空阻) ( +C > -I) (+C) C O R R' 活性极低 (1)π-π共轭效应(增加其稳定性);(2)+C 效应(降低羰基碳的正电性);(3)加成产物失去共轭能,反应活化能高;(4)产物的张力大幅增加。 2.立体效应 C O - sp 2 活性: O C H H O C CH 3H O C CH 3CH 3O O C CH 3CH 2CH 2CH 3O C Ph Ph >>>> > 二、试剂的亲核性 对同一羰基化合物,试剂的亲核性越大,平衡常数越大,亲核加成越容易。 1.带负电荷的亲核试剂比起共轭酸(中性分子)的亲核性强。OH ->H 2O, RO ->ROH 。 2.极性大的分子比极性小的分子亲核性强。HCN>H 2O(极性与电负性)。 3.同周期元素的负离子的亲核性与碱性大小一致; R 3C ->R 2N ->RO ->F - 4.同一主族元素的负离子的亲核性与可极化度大小一致。 I ->Br ->Cl ->F - 一、非手性的羰基化合物的加成 C Nu O - H + C Nu OH 50%C O -Nu + C OH Nu 50%dl 前(潜)手性分子 结果得外消旋化产物 二、手性羰基化合物的亲核加成方向遵守Cram 规则,得立体选择性的非对称异构体产物。
(完整版)羰基的亲核加成及相关反应
羰基的亲核加成及相关反应 羰基化合物包括醛、酮、羧酸及衍生物和CO 2。 5.1 羰基的结构 C O δ+δ- 亲电中心羰基碳的活性较大,易被亲核试剂进攻而发生亲核加成反应和亲核取代反应。 5.2 亲核加成反应的历程及影响因素 5.2.1 HCN 的加成 反应为碱催化。 ]CN ][CO [k v ->= OH - +HCN CN -+ H 2O 快 -C δ+δ-C O -CN C OH CN +OH - 反应的平衡位置受电子效应和空间效应的影响。 酮正向反应的趋势较小(空阻大)。 二、亲核加成反应的一般特点 1.反应可以被酸或碱催化 酸催化可提高羰基的亲电活性。 C O +H + + OH 碱催化提高亲核试剂的亲核性。 Nu H +OH --+H 2O Nu H -> 2.多数醛酮的亲核加成为可逆反应,用于分离与提纯。 5.2.2 影响羰基亲核加成反应活性的因素 一、羰基化合物的结构 1.电子效应 羰基碳的正电性越大,亲核加成速度越大,反应活性越大。羰基碳所连的吸电基(-I ,-C )使其亲核加成反应的活性增加,而供电基(+I ,+C )则使其活性降低。 活泼顺序:
ClCHO > HCHO > RCHO > CH 3COR > RCOOR' > RCONR'2 > RCOO --I > +C (+C) (+C,空阻) ( +C > -I) (+C) C O R R' 活性极低 (1)π-π共轭效应(增加其稳定性);(2)+C 效应(降低羰基碳的正电性);(3)加成产物失去共轭能,反应活化能高;(4)产物的张力大幅增加。 2.立体效应 C O - sp 2 活性: O C H H O C CH 3H O C CH 3CH 3O O C CH 3CH 2CH 2CH 3O C Ph Ph >>>> > 二、试剂的亲核性 对同一羰基化合物,试剂的亲核性越大,平衡常数越大,亲核加成越容易。 1.带负电荷的亲核试剂比起共轭酸(中性分子)的亲核性强。OH ->H 2O, RO ->ROH 。 2.极性大的分子比极性小的分子亲核性强。HCN>H 2O(极性与电负性)。 3.同周期元素的负离子的亲核性与碱性大小一致; R 3C ->R 2N ->RO ->F - 4.同一主族元素的负离子的亲核性与可极化度大小一致。 I ->Br ->Cl ->F - 5.2.3亲核加成反应的立体化学 一、非手性的羰基化合物的加成 C Nu O - H + C Nu OH 50%C O -Nu + C OH Nu 50%dl 前(潜)手性分子 结果得外消旋化产物 二、手性羰基化合物的亲核加成方向遵守Cram 规则,得立体选择性的非对称异构体产物。
第八章 烯烃 亲核加成 自由基加成 共轭加成复习过程
1. 烯烃的分类:累积二烯烃(H 2C=C=CH 2)、孤立二烯烃、共轭二烯烃 2. 烯烃的结构特征:未参与杂化的p 轨道与烯烃平面垂直。 如果吸收一定的能量,克服了p 轨道的结合力,顺式或反式可以互转。 C=C 键的平均键能为610.9kJ ·mol -1,C-C σ键的平均键能为347.3 kJ ·mol -1, 因此 键的键能大约为263.6 kJ ·mol -1。 二元取代烯烃比一元取代烯烃稳定8.3~12.5 kJ ·mol -1。所以烯烃取代越多越稳定。 1,3-丁二烯是一个平面型分子。键长均匀化是共轭烯烃的共性。 3. 烯烃的物理性质 含2~4个碳原子的烯烃是气体,含5~15个碳原子的烯烃为液体,高级烯烃 为固体。所有烯烃都不溶于水,所有烃(C 、H )都不溶于水。燃烧时,火焰明亮。 在sp n 杂化轨道中,n 数值越小,s 性质越强。由于s 电子靠近原子核,它比p 电子与原子核结合得更紧,轨道的电负性越大,所以电负性大小次序为s>sp>sp 2>sp 3>p 。即碳原子的电负性随杂化时s 成分的增大而增大。烯烃由 于sp 2碳原子的电负性比sp 3碳原子的大,比烷烃容易极化,成为有偶极矩的分子。以丙烯为例,甲基与双键碳原子相连的键易于极化,键电子偏向于sp 2碳原子,形成偶极,负极指向双键,正极位于甲基一边。因此当烷烃和不饱和碳原子相连时,由于诱导效应与超共轭效应成为给电子基团。 第八章 烯烃 亲核加成 自由基加成 共轭加成
①在abC=Cab类型的烯烃中,顺型异构体总是偶极分子,而且沸点较高。这对于识别顺反异构体是很有用的。②也可以通过X射线衍射的方法测定 相同基团之间的距离,以确定顺反异构体。③核磁共振也是测定顺反异构体的有效方法。 共轭烯烃物理性质的特点:①紫外(电子)吸收光谱——向长波方向移动②易极化——折射率增高③趋于稳定——氢化热(烯烃催化加氢生成烷烃放出的热)降低。 4.烯烃的反应 (1)烯烃的加成:离子型(亲电加成、亲核加成)、自由基型、协同
亲核加成反应
1. 重要的亲核加成反应 (1) 加氰化氰 醛、脂肪族甲基酮和含8个碳以下的脂环酮都可以加氰化氢,生成氰醇(α-羟基腈)。 C O +C O H C H N CN α-羟基腈 实验证明碱对这个反应的影响颇大。例如,丙酮和氰化氢作用,不加任何催化剂,3至4小时内只有50%的丙酮起反应;当加入一滴氢氧化钠溶液,反应在两分钟内完成。若加入酸,反应速度减慢;加入较多的酸,放置几个星期也不反应;因为氢氰酸是弱酸,酸或碱的存在将直接影响它的电离平衡。 + H +--C O H H H N CN + 加入碱,平衡向右移动,CN -的浓度增加;加入酸,平衡向左移动,CN -的浓度降低。这些事实说明在丙酮与氰化氢的反应中起决定作用的是CN -本身的性质和浓度。 醛、酮加氰化氢的反应是可逆的,亲核试剂是CN -,其历程可以表示如下: 反应分两步进行,第一步是CN -进攻羰基碳,生成氧负离子中间体。这是个慢步骤,也是决定速度的步骤。第二步是氧负离子中间体和质子结合,形成氰醇,这是个快步骤。 醛、酮和氰化氢直接加成反应的产率较好,但是氰化氧有剧毒,且挥发性大(沸点26.5℃)。使用起来不安全。为了避免反应中直接使用氰化氢,一般采用醛或酮与氰化钾(钠)的水溶液混合,然后加入无机酸,使氰化氢一旦生成立即和醛或酮作用。.但在加酸时应控制溶液的pH 值,使之始终偏于碱性(pH ≌8),以利于反应的进行。 醛、酮加氰化氢在有机合成中很有实用价值。它是增长碳链的一种方法;此外加成物含 有双官能团,是一类较活泼的化合物,可进一步转化为多种其它化合物。例如: CH 3CH 3CH 2 CH 3 CH 3 CH 3CH 3 CH 3(CH 3)2C CH 2NH 2 CH 3 H H 2SO 4CH 3OH C O H O H O +C H C O O O H N CN C O C C H O C H 3+O 浓,Δ α-甲基丙烯酸甲酯(90%) α-甲基丙烯酸甲酯是合成有机玻璃——聚α-甲基丙烯酸甲酯的单体。 C R R R`δδ+ O H ()O + --慢快C H C H N CN + R`)H (-C R O CN R`)H (
高等有机化学_谢斌_第一章有机化学反应概论
第一章 有机化学反应概论 反应物转变为产物的具体途径叫反应历程或反应机理,研究和确定一个新的有机反应历程时一般经过如下步骤:首先,要提出一个与已有的实验结果及理论相符合的可能的反应历程;然后通过实验来验证所提出的历程。如果新的实验结果与提出的历程相符合,即可对最初提出的历程加以肯定;如果新的实验结果与假设的历程不相符合,则需重新提出历程;如果部分符合,则需要罪提出的历程进行修正。 1.1 有机化学反应的分类 1.1.1按反应历程分类 按化学键断裂和形成方式可将有机化学反应分为三类: 一、离子反应(异裂历程) 共价键发生异裂形成了正负离子,有离子参与的反应叫离子反应。 R 3C R 3C ++Br -慢 异裂 R 3C + 2 R 3C OH 2 -H +R 3C OH + 这是 S N 1反应 二、自由基反应(均裂反应) 共价键发生均裂形成两个自由基,如烯的反马氏加成即过氧化反应。 均裂2RO 快 ROH + 慢 2CH CH BrCH 23 +HBr + Br BrCH 23 BrCH 2CH 2CH 3 三 分子反应(协同反应,周环反应) 共价键的断裂与形成是同时(协同) 进行的,反应一步完成反应叫协同反应。如S N 2,E2,Diels-Alder 均叫协同反应。 如果经过一个环状过渡态,一步形成产物,过程无任何中间体的反应叫周环反应。 S N 2,E2,Diels-Alder 均叫协同反应。但只有 Diel-Alder 反应叫周环反应。 环转过渡态
周环反应的特点:1一般不受溶剂极性、酸性、催化剂、自由基引发剂或抑制剂的影响,而受加热或光照的影响,而且光照和加热的结果相反。2具有高度的立体专一性。 3周环反应通过环状过渡态而实现的协同反应。 周环反应分类:电环化、环加成和σ-迁移。 1.1.2按反应物与产物之间的关系分类 不饱和度计算:UN=n 4+1+1/2(n 3-n 1) 一、取代反应 反应产物的不饱和度不发生变化,根据进攻试剂的类型分为亲核取代,亲电取代和自由基取代。 RCH 2Br +OH - RCH 2 OH +Br -亲核取代 +NO 2+ NO 2 + H + 亲电取代 RCH(CH 3)2++HCl Cl 2 RCCl(CH 3)2自由基取代 二、加成反应 反应物不饱和度减少,分为亲核加成,亲电加成和自由基加成。 亲核加成 R - C H CN O - H + R C H CN HO 亲电加成RCH 2 +H + R CH CH 3 Cl -R CH CH 3 Cl 自由基加成 RCH CH 2 + Br. R CH 2Br 三、消除反应 反应物不饱和度减小,分为离子消去及协同消去或α-消除,β—消除 RCHCH 2X H OH - RCH=CH 2+HX 离子消去和β—消除 (CH 3)3COK CCl 2+(CH 3)3COH +KCl +CHCl 3 α-消除 四、重排反应 碳骨架发生变化,分子的不饱和度不变,有离子重排、自由基重排和协同重排
(完整版)羰基的亲核加成及相关反应
羰基的亲核加成及相关反应 羰基化合物包括醛、酮、羧酸及衍生物和 5.1羰基的结构 CO 2。 C O 亲电中心羰基碳的活性较大,易被亲核试剂进攻而发生亲核加成反应和亲核取代反应。 5.2亲核加成反应的历程及影响因素 5.2.1 HCN 的加成 反应为碱催化。 v k[ CO ][CN ] 快 OH - + HCN CN - + H 2O 、f 慢 \ H 2O \ /OH CNJ / = O — /C 、b +0H - / CN / CN 酮正向反应的趋势较小(空阻大) 二、亲核加成反应的一般特点 1 .反应可以被酸或碱催化 酸催化可提高羰基的亲电活性。 碱催化提高亲核试剂的亲核性。 活性.Nu - > Nu — H 2?多数醛酮的亲核加成为可逆反应,用于分离与提纯。 5.2.2影响羰基亲核加成反应活性的因素 一、羰基化合物的结构 1 .电子效应 羰基碳的正电性越大,亲核加成速度越大,反应活性越大。羰基碳所连的吸电基( 加成反应的活性增加,而供电基( +I , +C )则使其活性降低。 活泼顺序: Nu —H + OH Nu - + H 2O C=O + C = O H — C +—OH -I , -C )使其亲核
50% ClCHO > HCHO > RCHO > CH 3COR > RCOOR' > RCONR' 2 > RCOO (1)-共轭效应(增加其稳定性);(2) +C 效应(降低羰基碳的正电性);(3)加成产物失去共轭 能,反应活化能高;(4 )产物的张力大幅增加。 2.立体效应 、试剂的亲核性 对同一羰基化合物,试剂的亲核性越大,平衡常数越大,亲核加成越容易。 1 .带负电荷的亲核试剂比起共轭酸(中性分子)的亲核性强。 OH ->H 2O, RO ->ROH 。 2?极性大的分子比极性小的分子亲核性强。 HCN>H 2O (极性与电负性)。 3?同周期元素的负离子的亲核性与碱性大小一致; R 3C ->R 2N ->RO ->F - 4?同一主族元素的负离子的亲核性与可极化度大小一致。 l ->Br ->CI ->F - 5.2.3亲核加成反应的立体化学 一、非手性的羰基化合物的加成 前(潜)手性分子 结果得外消旋化产物 、手性羰基化合物的亲核加成方向遵守 Cram 规则,得立体选择性的非对称异构体产物。 -I > +C 什C ) 什C,空阻) (+C > -I) 什 C) 活性极低 O II H > CH 3—C —H O II >CH 3—C —CH 3 > O O II II O >CH 3CH 2-C-CH 2CH 3> Ph-C —Ph R Nu R#、o - R Nu R'/ OH 50% dl R R ,Z Q C x Nu R R'Z OH C ? Nu R O ) 张力增加 O
最新高等有机化学教学大纲教学提纲
《高等有机化学》教学大纲 课程名称:高等有机化学 学时/学分:54/4 先修课程:无机化学、有机化学 适用专业:化学 开课教研室:有机化学 一、课程的性质和任务 1.课程性质:本课程是化学专业师范方向本科生的专业选修课程。 2.课程任务:本课程基本任务是在学习四大基础化学的基础上,对《有机化学》课程的进一步深化,为有关后继课程《精细化学品化学》和《有机合成化学》的学习以及毕业论文打下良好的理论基础。通过本门课程的学习,要求学生掌握有机反应历程的分类和测试方法。熟悉各类基本有机反应的历程、立体化学关系、影响因素和在有机合成上的应用。理解一些基本的有机反应理论,并能够用所学的知识解决一些有机化学问题和指导专业有机实验。 二、课程教学基本要求 本课程的教学环节包括课堂讲授,学生自学,习题讨论课,习题,答疑,质疑,期中测验和期末考试。通过上述基本教学步骤,要求同学们能用现代化学的理论知识,认识有机化学中化学键的本质,深刻认识有机化学分子结构与物理、化学性质的内在联系和变化规律。掌握高等有机化学的基本原理、动态学原理及其有机化学的五大反应原理。掌握研究反应机理和设计合成方法。从微观电子结构层次上认识有机化学动态反应过程。通过有机化合物的结构可推测其物理性质和化学反应性质。学会并领悟分析问题、解决问题的方法和技能,为继续学习相关课程奠定理论基础,为从事相应专业的工作提供必要的理论知识。本课程课堂讲授(包括自学、讨论)54学时,以便于每学期根据实际情况调整教学,考试方式为闭卷考试,总评成绩:平时成绩占40%,期末考试占60%。 三、课程教学内容
第一章化学键. (一)主要内容 1.偶极矩、氢键、氢键在有机化学中的应用 2.共振论与分子轨道,共振论在有机化学中的应用,分子轨道理论简介 (二)基本要求 掌握偶极矩、氢键、氢键在有机化学中的应用。共振论与分子轨道,共振论在有机化学中的应用,分子轨道理论简介。解决难点:共振论与分子轨道,共振论在有机化学中的应用,分子轨道理论简介。 第二章有机化学中的电子效应和空间效应 (一)主要内容 1.诱导效应、共轭效应与超共轭效应、场效应、烷基的电子效应 2.有机化合物的空间效应、空间效应对反应活性的影响、空间效应对酸碱性的影响 3.利用堵位基团的空间效应进行选择性反应 (二)基本要求 掌握诱导效应、共轭效应与超共轭效应、场效应、烷基的电子效应、有机化合物的空间效应、空间效应对反应活性的影响、空间效应对酸碱性的影响。解决难点:空间效应对酸碱性的影响。 第三章反应机理及研究方法 (一)主要内容 1.有机反应的类型如:取代反应、加成反应、消除反应、重排反应、氧化还原反应、协同反应 2.研究反应机理的方法 (二)基本要求 掌握研究反应机理的方法。解决难点:研究反应机理的方法。 第四章氧化与还原反应 (一)主要内容 1.催化氧化、催化脱氢、二甲基亚砜氧化 2.氨氧化、过氧化物氧化、锰化合物氧化、四氧化锇氧化、铬酸及其衍生物氧化、