Bioconductor基因芯片数据分析系列(一):数据的读取
Bioconductor基因芯片数据分析系列(一):R包中数据的读取
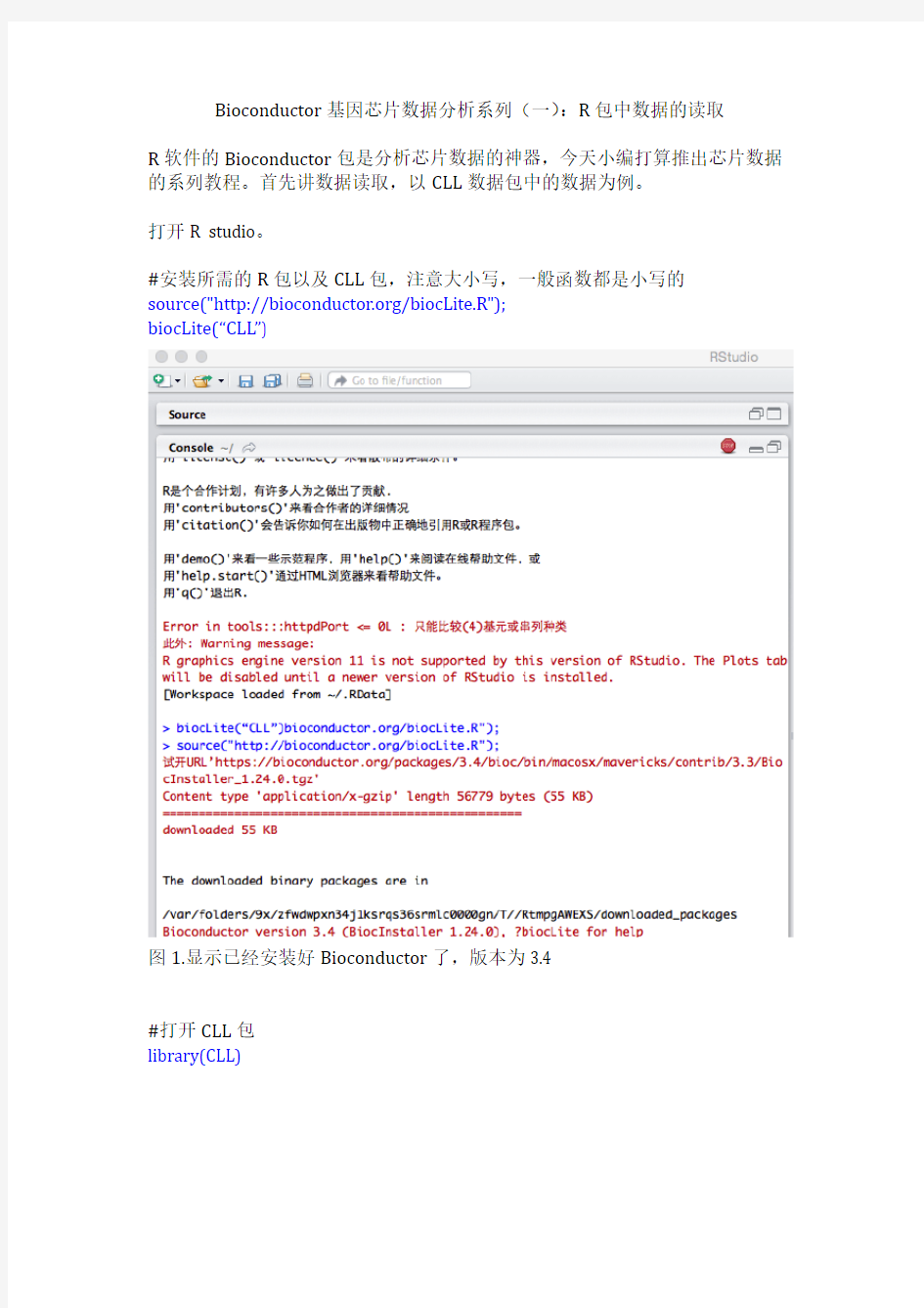
R软件的Bioconductor包是分析芯片数据的神器,今天小编打算推出芯片数据的系列教程。首先讲数据读取,以CLL数据包中的数据为例。
打开R studio。
#安装所需的R包以及CLL包,注意大小写,一般函数都是小写的
source("https://www.360docs.net/doc/3718211759.html,/biocLite.R");
biocLite(“CLL”)
图1.显示已经安装好Bioconductor了,版本为3.4
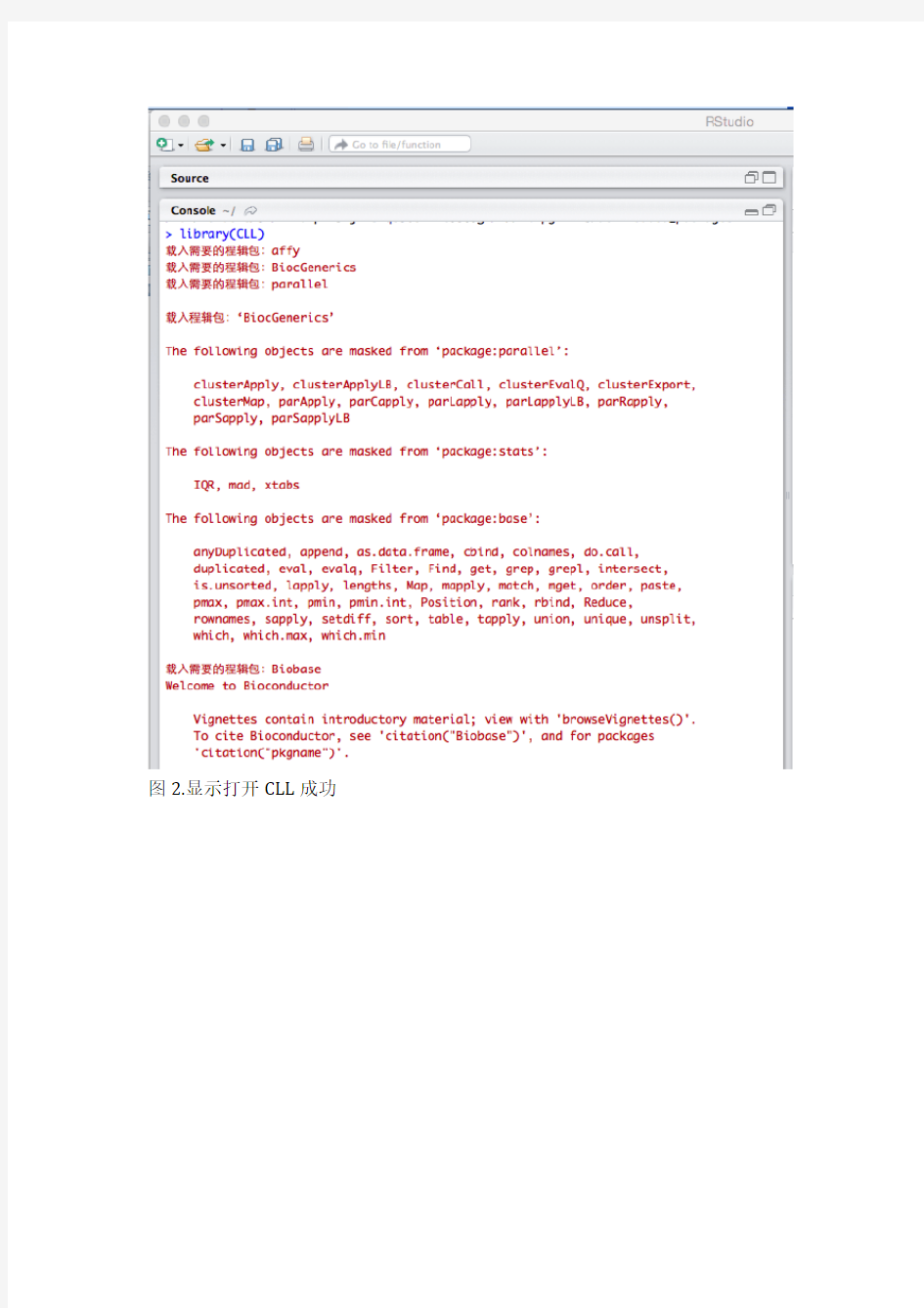
#打开CLL包
library(CLL)
图2.显示打开CLL成功
图3.右侧栏内可见看到目前载入的程序包
data(CLLbatch)
#调用RMA算法对数据预处理
CLLrma<-rma(CLLbatch)
#读取处理后所有样品的基因表达值
e<- exprs(CLLrma)
#查看数据
e
我们可以看到,CLL数据集中共有24个样品(CLL10.CEL, CLL11.CEL, CLL12.CEL, 等),此数据集的病人分为两组:稳定组和进展组,采用的设计为两组之间的对照试验(Control Test)。从上面的结果可知,Bioconductor具有强大的数据预处理能力和调用能力,仅仅用了6行代码就完成了数据的读取及预处理。
Bioconductor基因芯片数据分析系列(二):GEO下载数据CEL的读取首先得下载一个数据,读取GEO的CEL文件采用如下命令:
登陆pubmed,找到一个你感兴趣的数据库
在底下栏目下载CEL文件
打开R软件
#安装所需的R包以及CLL包,注意大小写,一般函数都是小写的
source("https://www.360docs.net/doc/3718211759.html,/biocLite.R");
biocLite(“CLL”)
>library(affy)
>affybatch<- ReadAffy(celfile.path = "GSE36376_RAW")
请注意目录的路径,在window下,反斜杠‘\’要用转义字符“\\”表示。
然后可以使用RMA或者MAS5等方法对数据进行background.correction, normaliztion, pm.correct等等一系列处理。如果你一切用默认参数,则可以使用如下命令:
>eset<- rma(affybatch),or eset<- mas5(affybatch)
>exp<- exprs(eset)
exp就是数字化的表达谱矩阵了
请注意,rma只使用匹配探针(PM)信号,exp数据已经进行log2处理。mas5综合考虑PM和错配探针(MM)信号,exp数据没有取对数。
下一期就得等到2017年春节期间啦,敬请期待~
另外一种是直接利用GEO上面的GEO2R按钮里面的R script下载文件:
# Version info: R 3.2.3, Biobase 2.30.0, GEOquery 2.40.0, limma 3.26.8
# R scripts generated Mon Dec 26 06:54:42 EST 2016 Server: https://www.360docs.net/doc/3718211759.html,
Query:
acc=GSE36376&platform=GPL10558&type=txt&groups=&color s=&selection=XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXX&padj=fdr&logtransform=auto&col umns=ID&columns=adj.P.Val&columns=P.Value&columns=F&c
olumns=Gene+symbol&columns=Gene+title&num=250&annot=n cbi
# Unable to generate script analyzing differential expression.
# Invalid input: at least two groups of samples should be selected.
##################################################### ###########
# Boxplot for selected GEO samples
library(Biobase)
library(GEOquery)
# load series and platform data from GEO
gset<- getGEO("GSE36376", GSEMatrix =TRUE,
getGPL=FALSE)
if (length(gset) > 1) idx<- grep("GPL10558", attr(gset, "names")) else idx<- 1
gset<- gset[[idx]]
# set parameters and draw the plot
dev.new(width=4+dim(gset)[[2]]/5, height=6)
par(mar=c(2+round(max(nchar(sampleNames(gset)))/2),4, 2,1))
title<- paste ("GSE36376", '/', annotation(gset), " selected samples", sep ='')
boxplot(exprs(gset), boxwex=0.7, notch=T, main=title, outline=FALSE, las=2)
基因芯片实验中的细胞和组织制备规程
一个完整地基因芯片实验包括以下几个步骤:研究课题地提出、芯片设计、样品制备、杂交反应、数据分析和处理.本文主要介绍其中地样品制备(细胞标本采集和组织标本采集)地建议做法 一个完整地基因芯片实验包括以下几个步骤:研究课题地提出、芯片设计、样品制备、杂交反应、数据分析和处理.本文主要介绍其中地样品制备(细胞标本采集和组织标本采集)地建议做法. 细胞标本采集操作建议规程 . 所有样品均应有样品标签(注明样品编号),同时有一张样品登记表,写明样品名称、种类、编号、取样日期、样品处理情况等. . 一张芯片实验一般要求细胞数在,建议设计实验和收获细胞时可考虑多收集一些. . 贴壁与悬浮细胞培养诱导结束后,去除培养液,保留地细胞用缓冲液洗一下,除去缓冲液,加溶液*充分溶解细胞,放入液氮运输.样品量以实际得到地为准. . 血液:将白细胞分离出来,加溶液充分溶解细胞,放入液氮运输.样品量以实际得到地为准. . 如果是细胞未经溶液处理,直接冻入液氮罐(不推荐).工作人员会对细胞作相关处理,以便为细胞记数. . 以上提到地均是新鲜细胞,对一些已老化或质量不明地细胞,工作人员有权提出疑义,并要求退回或重新取样. 不同组织抽提μ所需组织量
考虑到个体差异以及样品在研磨、匀浆等过程中地损失,客户提供地样品量应在上述基础上增加倍. 组织标本采集操作建议规程 注: · 以下步骤应在冰上进行且不超过分钟,超过时间会导致样品地降解. · 对肿瘤组织地取材,要求尽可能准确地判定肿瘤和正常组织,例如对于手术切除地整个或部分前列腺,可能要根据冰冻切片报告地结果来判定要进行研究地取材部位. . 离体新鲜组织,切成多个小块,剔除结缔组织和脂肪组织.胃、肠组织应剪除外膜;肝、肾、脾应剪除门部血管神经,肿瘤组织应将周围地正常组织切除干净(正常组织也应将周围地肿瘤组织切除干净). . 在生理盐水中漂洗样品,以去除血渍和污物. . 用铝箔包裹组织,或用冻存管装载组织(但最好统一采用铝箔).用记号笔在铝箔或冻存管外表写明样品编号,并贴上标签,迅速投入液氮冷却. . 填写样品登记表,写明样品名称、种类、编号、取样日期、样品处理情况等 . . 将液氮冷却地组织放入样品袋(每个样品袋只保存同样地组织),袋口留一根编号绳,绳上粘一张标签纸(标签上注明:样品名称、编号、日期),迅速转入便携式液氮罐. . 保留张取材部位地病理切片. 基因芯片样品地制备要点: .目地地选择对于大规模地研究基因表达问题,需要分析每一个基因地表达.因此,选择地目地必须能够代表要研究地各个基因.对于整个基因组序列全部已知地 生物,最直接地方法是用扩增基因组中每个已知地或预测地开放阅读框架(),亦可以选择自己感兴趣地部分序列.对于没有测序,或只有部分测序地基因组,或
基因芯片的数据分析
基因表达谱芯片的数据分析 基因芯片数据分析就是对从基因芯片高密度杂交点阵图中提取的杂交点荧光强度信号进行的定量分析,通过有效数据的筛选和相关基因表达谱的聚类,最终整合杂交点的生物学信息,发现基因的表达谱与功能可能存在的联系。然而每次实验都产生海量数据,如何解读芯片上成千上万个基因点的杂交信息,将无机的信息数据与有机的生命活动联系起来,阐释生命特征和规律以及基因的功能,是生物信息学研究的重要课题[1]。基因芯片的数据分析方法从机器学习的角度可分为监督分析和非监督分析,假如分类还没有形成,非监督分析和聚类方法是恰当的分析方法;假如分类已经存在,则监督分析和判别方法就比非监督分析和聚类方法更有效率。根据研究目的的不同[2,3],我们对基因芯片数据分析方法分类如下。(1)差异基因表达分析:基因芯片可用于监测基因在不同组织样品中的表达差异,例如在正常细胞和肿瘤细胞中;(2)聚类分析:分析基因或样本之间的相互关系,使用的统计方法主要是聚类分析;(3)判别分析:以某些在不同样品中表达差异显著的基因作为模版,通过判别分析就可建立有效的疾病诊断方法。 1 差异基因表达分析(difference expression, DE) 对于使用参照实验设计进行的重复实验,可以对2样本的基因表达数据进行差异基因表达分析,具体方法包括倍数分析、t检验、方差分析等。 1.1倍数变化(fold change, FC) 倍数分析是最早应用于基因芯片数据分析的方法[4],该方法是通过对基因芯片的ratio值从大到小排序,ratio 是cy3/cy5的比值,又称R/G值。一般0.5-2.0范围内的基因不存在显著表达差异,该范围之外则认为基因的表达出现显著改变。由于实验条件的不同,此阈值范围会根据可信区间应有所调整[5,6]。处理后得到的信息再根据不同要求以各种形式输出,如柱形图、饼形图、点图等。该方法的优点是需要的芯片少,节约研究成本;缺点是结论过于简单,很难发现更高层次功能的线索;除了有非常显著的倍数变化的基因外,其它变化小的基因的可靠性就值得怀疑了;这种方法对于预实验或实验初筛是可行的[7]。此外倍数取值是任意的,而且可能是不恰当的,例如,假如以2倍为标准筛选差异表达基因,有可能没有1条入选,结果敏感性为0,同样也可能出现很多差异表达基因,结果使人认为倍数筛选法是在盲目的推测[8,9]。 1.2 t检验(t-test) 差异基因表达分析的另一种方法是t检验[10],当t超过根据可信度选择的标准时,比较
基因芯片数据功能分析
生物信息学在基因芯片数据功能分析中的应用 2009-4-29 随着人类基因组计划(Human Genome Project)即全部核苷酸测序的即将完成,人类基因组研究的重心逐渐进入后基因组时代(Postgenome Era),向基因的功能及基因的多样性倾斜。通过对个体在不同生长发育阶段或不同生理状态下大量基因表达的平行分析,研究相应基因在生物体内的功能,阐明不同层次多基因协同作用的机理,进而在人类重大疾病如癌症、心血管疾病的发病机理、诊断治疗、药物开发等方面的研究发挥巨大的作用。它将大大推动人类结构基因组及功能基因组的各项基因组研究计划。生物信息学在基因组学中发挥着重大的作用, 而另一项崭新的技术——基因芯片已经成为大规模探索和提取生物分子信息的强有力手段,将在后基因组研究中发挥突出的作用。基因芯片与生物信息学是相辅相成的,基因芯片技术本身是为了解决如何快速获得庞大遗传信息而发展起来的,可以为生物信息学研究提供必需的数据库,同时基因芯片的数据分析也极大地依赖于生物信息学,因此两者的结合给分子生物学研究提供了一条快捷通道。 本文介绍了几种常用的基因功能分析方法和工具: 一、GO基因本体论分类法 最先出现的芯片数据基因功能分析法是GO分类法。Gene Ontology(GO,即基因本体论)数据库是一个较大的公开的生物分类学网络资源的一部分,它包含38675 个Entrez Gene注释基因中的17348个,并把它们的功能分为三类:分子功能,生物学过程和细胞组分。在每一个分类中,都提供一个描述功能信息的分级结构。这样,GO中每一个分类术语都以一种被称为定向非循环图表(DAGs)的结构组织起来。研究者可以通过GO分类号和各种GO数据库相关分析工具将分类与具体基因联系起来,从而对这个基因的功能进行描述。在芯片的数据分析中,研究者可以找出哪些变化基因属于一个共同的GO功能分支,并用统计学方法检定结果是否具有统计学意义,从而得出变化基因主要参与了哪些生物功能。 EASE(Expressing Analysis Systematic Explorer)是比较早的用于芯片功能分析的网络平台。由美国国立卫生研究院(NIH)的研究人员开发。研究者可以用多种不同的格式将芯片中得到的基因导入EASE 进行分析,EASE会找出这一系列的基因都存在于哪些GO分类中。其最主要特点是提供了一些统计学选项以判断得到的GO分类是否符合统计学标准。EASE 能进行的统计学检验主要包括Fisher 精确概率检验,或是对Fisher精确概率检验进行了修饰的EASE 得分(EASE score)。 由于进行统计学检验的GO分类的数量很多,所以EASE采取了一系列方法对“多重检验”的结果进行校正。这些方法包括弗朗尼校正法(Bonferroni),本杰明假阳性率法(Benjamini falsediscovery rate)和靴带法(bootstraping)。同年出现的基于GO分类的芯片基因功能分析平台还有底特律韦恩大学开发的Onto-Express。2002年,挪威大学和乌普萨拉大学联合推出的Rosetta 系统将GO分类与基因表达数据相联系,引入了“最小决定法则”(minimal decision rules)的概念。它的基本思想是在对多张芯片结果进行聚类分析之后,与表达模式
基因芯片数据处理流程与分析介绍
基因芯片数据处理流程与分析介绍 关键词:基因芯片数据处理 当人类基因体定序计划的重要里程碑完成之后,生命科学正式迈入了一个后基因体时代,基因芯片(microarray) 的出现让研究人员得以宏观的视野来探讨分子机转。不过分析是相当复杂的学问,正因为基因芯片成千上万的信息使得分析数据量庞大,更需要应用到生物统计与生物信息相关软件的协助。要取得一完整的数据结果,除了前端的实验设计与操作的无暇外,如何以精确的分析取得可信数据,运筹帷幄于方寸之间,更是画龙点睛的关键。 基因芯片的应用 基因芯片可以同时针对生物体内数以千计的基因进行表现量分析,对于科学研究者而言,不论是细胞的生命周期、生化调控路径、蛋白质交互作用关系等等研究,或是药物研发中对于药物作用目标基因的筛选,到临床的疾病诊断预测,都为基因芯片可以发挥功用的范畴。 基因表现图谱抓取了时间点当下所有的动态基因表现情形,将所有的探针所代表的基因与荧光强度转换成基本数据(raw data) 后,仿如尚未解密前的达文西密码,隐藏的奥秘由丝丝的线索串联绵延,有待专家抽丝剥茧,如剥洋葱般从外而内层层解析出数千数万数据下的隐晦含义。 要获得有意义的分析结果,恐怕不能如泼墨画般洒脱随兴所致。从raw data 取得后,需要一连贯的分析流程(图一),经过许多统计方法,才能条清理明的将raw data 整理出一初步的分析数据,当处理到取得实验组除以对照组的对数值后(log2 ratio),大约完成初步的统计工作,可进展到下一步的进阶分析阶段。
图一、整体分析流程。基本上raw data 取得后,将经过从最上到下的一连串分析流程。(1) Rosetta 软件会透过统计的model,给予不同的权重来评估数据的可信度,譬如一些实验操作的误差或是样品制备与处理上的瑕疵等,可已经过Rosetta error model 的修正而提高数据的可信值;(2) 移除重复出现的探针数据;(3) 移除flagged 数据,并以中位数对荧光强度的数据进行标准化(Normalized) 的校正;(4) Pearson correlation coefficient (得到R 值) 目的在比较技术性重复下的相似性,R 值越高表示两芯片结果越近似。当R 值超过0.975,我们才将此次的实验结果视为可信,才继续后面的分析流程;(5) 将技术性重复芯片间的数据进行平均,取得一平均之后的数据;(6) 将实验组除以对照组的荧光表现强度差异数据,取对数值(log2 ratio) 进行计算。 找寻差异表现基因 实验组与对照组比较后的数据,最重要的就是要找出显著的差异表现基因,因为这些正是条件改变后而受到调控的目标基因,透过差异表现基因的加以分析,背后所隐藏的生物意义才能如拨云见日般的被发掘出来。 一般根据以下两种条件来筛选出差异表现基因:(i) 荧光表现强度差异达2 倍变化(fold change 增加2 倍或减少2倍) 的基因。而我们通常会取对数(log2) 来做fold change 数值的转换,所以看的是log2 ≧1 或≦-1 的差异表现基因;(ii) 显著值低于0.05 (p 值< 0.05) 的基因。当这两种条件都符合的情况下所交集出来的基因群,才是显著性高且稳定的差异表现基因。
Bioconductor基因芯片数据分析系列(一):数据的读取
Bioconductor基因芯片数据分析系列(一):R包中数据的读取 R软件的Bioconductor包是分析芯片数据的神器,今天小编打算推出芯片数据的系列教程。首先讲数据读取,以CLL数据包中的数据为例。 打开R studio。 #安装所需的R包以及CLL包,注意大小写,一般函数都是小写的 source("https://www.360docs.net/doc/3718211759.html,/biocLite.R"); biocLite(“CLL”) 图1.显示已经安装好Bioconductor了,版本为3.4 #打开CLL包 library(CLL)
图2.显示打开CLL成功
图3.右侧栏内可见看到目前载入的程序包 data(CLLbatch) #调用RMA算法对数据预处理 CLLrma<-rma(CLLbatch) #读取处理后所有样品的基因表达值 e<- exprs(CLLrma) #查看数据 e 我们可以看到,CLL数据集中共有24个样品(CLL10.CEL, CLL11.CEL, CLL12.CEL, 等),此数据集的病人分为两组:稳定组和进展组,采用的设计为两组之间的对照试验(Control Test)。从上面的结果可知,Bioconductor具有强大的数据预处理能力和调用能力,仅仅用了6行代码就完成了数据的读取及预处理。
Bioconductor基因芯片数据分析系列(二):GEO下载数据CEL的读取首先得下载一个数据,读取GEO的CEL文件采用如下命令: 登陆pubmed,找到一个你感兴趣的数据库
在底下栏目下载CEL文件 打开R软件 #安装所需的R包以及CLL包,注意大小写,一般函数都是小写的 source("https://www.360docs.net/doc/3718211759.html,/biocLite.R"); biocLite(“CLL”) >library(affy) >affybatch<- ReadAffy(celfile.path = "GSE36376_RAW") 请注意目录的路径,在window下,反斜杠‘\’要用转义字符“\\”表示。 然后可以使用RMA或者MAS5等方法对数据进行background.correction, normaliztion, pm.correct等等一系列处理。如果你一切用默认参数,则可以使用如下命令: >eset<- rma(affybatch),or eset<- mas5(affybatch) >exp<- exprs(eset) exp就是数字化的表达谱矩阵了 请注意,rma只使用匹配探针(PM)信号,exp数据已经进行log2处理。mas5综合考虑PM和错配探针(MM)信号,exp数据没有取对数。 下一期就得等到2017年春节期间啦,敬请期待~ 另外一种是直接利用GEO上面的GEO2R按钮里面的R script下载文件: # Version info: R 3.2.3, Biobase 2.30.0, GEOquery 2.40.0, limma 3.26.8 # R scripts generated Mon Dec 26 06:54:42 EST 2016 Server: https://www.360docs.net/doc/3718211759.html, Query: acc=GSE36376&platform=GPL10558&type=txt&groups=&color s=&selection=XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXX&padj=fdr&logtransform=auto&col umns=ID&columns=adj.P.Val&columns=P.Value&columns=F&c
基因芯片制备
中南大学 基因芯片探针 实验报告 学院: 班级: 姓名: 学号:
一、实验背景 随着人类基因组(测序)计划(Human genome project )的逐步实施以及分子生物学相关学科的迅猛发展,越来越多的动植物、微生物基因组序列得以测定,基因序列数据正在以前所未有的速度迅速增长。然而, 怎样去研究如此众多基因在生命过程中所担负的功能就成了全世界生命科学工作者共同的课题。为此,建立新型杂交和测序方法以对大量的遗传信息进行高效、快速的检测、分析就显得格外重要了。基因芯片(又称DNA 芯片、生物芯片)技术就是顺应这一科学发展要求的产物,它的出现为解决此类问题提供了光辉的前景。该技术系指将大量(通常每平方厘米点阵密度高于400 )探针分子固定于支持物上后与标记的样品分子进行杂交,通过检测每个探针分子的杂交信号强度进而获取样品分子的数量和序列信息。 二、基因芯片 基因芯片又称为DNA微阵列(DNA microarray),基因芯片的制备主要有两种基本方法,一是在片合成法,另一种方法是点样法。 1)片合成法是基于组合化学的合成原理,它通过一组定位模板来决定基片表面上不同化学单体的偶联位点和次序。在片合成法制备DNA芯片的关键是高空间分辨率的模板定位技术和固相合成化学技术的精巧结合。 目前,已有多种模板技术用于基因芯片的在片合成,如光去保护并行合成法、光刻胶保护合成法、微流体模板固相合成技术、分子印章多次压印原位合成的方法、喷印合成法。在片合成法可以发挥微细加工技术的优势,很适合制作大规模DNA探针阵列芯片,实现高密度芯片的标准化和规模化生产。美国Affymetrix公司制备的基因芯片产品在
基因芯片与高通量测序
基因芯片: 将大量(通常每平方厘米点阵密度高于400 )探针分子固定于支持物上后与标记的样品分子进行杂交,通过检测每个探针分子的杂交信号强度进而获取样品分子的数量和序列信息。通俗地说,就是通过微加工技术,将数以万计、乃至百万计的特定序列的DNA 片段(基因探针),有规律地排列固定于2cm2 的硅片、玻片等支持物上,构成的一个二维DNA探针阵列,与计算机的电子芯片十分相似,所以被称为基因芯片。当溶液中带有荧光标记的核酸序列TATGCAATCTAG,与基因芯片上对应位置的核酸探针产生互补匹配时,通过确定荧光强度最强的探针位置,获得一组序列完全互补的探针序列。据此可重组出靶核酸的序列。基因探针是人工合成的碱基序列。,所谓基因探针只是一段人工合成的碱基序列,在探针上连接一些可检测的物质,根据碱基互补的原理,利用基因探针到基因混合物中识别特定基因。它将大量探针分子固定于支持物上,然后与标记的样品进行杂交,通过检测杂交信号的强度及分布来进行分析。基因芯片通过应用平面微细加工技术和超分子自组装技术,把大量分子检测单元集成在一个微小的固体基片表面,可同时对大量的核酸和蛋白质等生物分子实现高效、快速、低成本的检测和分析 基因芯片制作 、芯片制备 目前制备芯片主要以玻璃片或硅片为载体,采用原位合成和微矩阵的方法将寡核苷酸片段或cDNA作为探针按顺序排列在载体上。芯片的制备除了用到微加工工艺外,还需要使用机器人技术。以便能快速、准确地将探针放置到芯片上的指定位置。 2、样品制备 生物样品往往是复杂的生物分子混合体,除少数特殊样品外,一般不能直接与芯片反应,有时样品的量很小。所以,必须将样品进行提取、扩增,获取其中的蛋白质或DNA、RNA,然后用荧光标记,以提高检测的灵敏度和使用者的安全性。 3、杂交反应 杂交反应是荧光标记的样品与芯片上的探针进行的反应产生一系列信息的过程。选择合适的反应条件能使生物分子间反应处于最佳状况中,减少生物分子之间的错配率。 4、信号检测和结果分析 杂交反应后的芯片上各个反应点的荧光位置、荧光强弱经过芯片扫描仪和相关软件可以分析图像,将荧光转换成数据,即可以获得有关生物信息。基因芯片技术发展的最终目标是将从样品制备、杂交反应到信号检测的整个分析过程集成化以获得微型全分析系统(micro total analytical system)或称缩微芯片实验室(laboratory on a chip)。使用缩微芯片实验室,就可以在一个封闭的系统内以很短的时间完成从原始样品到获取所需分析结果的全套操作。
基因芯片数据分析中的标准化算法和聚类算法
基因芯片数据分析中的标准化算法和聚类算法 北京大学生命科学院 生物信息专业 王向峰 学号:10211058 摘要: 基因芯片技术已经广泛的应用于各种模式生物的功能基因组的研究中,应用芯片技术可以高效,高通量的检测基因表达行为。芯片数据分析中的标准化主要分为芯片内标准化和芯片间标准化,芯片内标准化根据目的不同可分为消除染色偏差的Lowess Normalization ,消除点样针头引起的空间差异的Print-tip Normalization 。常用的芯片间标准化有Quantile Normalization ,Global Normalization 。芯片数据分析中常见的聚类算法有分层聚类(Hierarchical clustering)、K 均值聚类(K-means clustering)、自组织图谱SOM (self organizing map)、PCA (principle component analysis)等等。所有的聚类方法归结为有监督的学习和无监督的学习两种方法。 第一部分 基因芯片的数据标准化(Normalization) 对基因芯片数据的标准化处理,主要目的是消除由于实验技术所导致的表达量(Intensity)的变化,并且使各个样本(sample)和平行实验的数据处于相同的水平,从而使我们可以得到具有生物学意义的基因表达量的变化。标准化的方法根据芯片的种类、数据处理的阶段和目的不同而有所差异。这里主要讨论一下双荧光染色(Red and Green Chip)的cDNA 微列阵(cDNA microarray)的标准化方法。 一、实验数据的预处理(data transformation ) 的细胞进行培养(Cultured Cell),以保证绝大部分的基因可以表达。样本基因是根据试验设计的目的从不同组织,不同发育阶段,不同条件下培养的细胞中提取的cDNA 样本。通过样本基因对参照基因的比值,而判断不同条件下的基因表达量的变化。 扫描仪对基因芯片的图像进行扫描,根据每个点的光密度值尝试相对应的绝对表达量(intensity)。然后图像分析软件通过芯片的背景噪音以及杂交点的光密度分析,对每个点的intensity 校准,然后取样本基因和参照基因的比值(R/G ratio ),作为每个样本基因的相对表达量(relative intensity)。选择相对表达量,可以在一定程度上减少芯片之间,荧光染色,扫描所产生的系统偏差。然后对比值取对数,2log 10 =,选择以2为底的对数方便于对 基因表达量变化的研究,比如R/G=1 ,则2log 10=,即认为表达量没有发生变化,当R/G=2 或者,R/G=0.5,则log 值为1 或 –1,这是可以认为表达量都发生两倍的变化,只是一个是
基因芯片与高通量测序.
基因芯片: 将大量(通常每平方厘米点阵密度高于 400 )探针分子固定于支持物上后与标记的样品分子进行杂交,通过检测每个探针分子的杂交信号强度进而获取样品分子的数量和序列信息。通俗地说,就是通过微加工技术,将数以万计、乃至百万计的特定序列的DNA片段(基因探针),有规律地排列固定于2cm2 的硅片、玻片等支持物上,构成的一个二维DNA探针阵列,与计算机的电子芯片十分相似,所以被称为基因芯片。当溶液中带有荧光标记的核酸序列TATGCAATCTAG,与基因芯片上对应位置的核酸探针产生互补匹配时,通过确定荧光强度最强的探针位置,获得一组序列完全互补的探针序列。据此可重组出靶核酸的序列。基因探针是人工合成的碱基序列。,所谓基因探针只是一段人工合成的碱基序列,在探针上连接一些可检测的物质,根据碱基互补的原理,利用基因探针到基因混合物中识别特定基因。它将大量探针分子固定于支持物上,然后与标记的样品进行杂交,通过检测杂交信号的强度及分布来进行分析。基因芯片通过应用平面微细加工技术和超分子自组装技术,把大量分子检测单元集成在一个微小的固体基片表面,可同时对大量的核酸和蛋白质等生物分子实现高效、快速、低成本的检测和分析 基因芯片制作 、芯片制备 目前制备芯片主要以玻璃片或硅片为载体,采用原位合成和微矩阵的方法将寡核苷酸片段或cDNA作为探针按顺序排列在载体上。芯片的制备除了用到微加工工艺外,还需要使用机器人技术。以便能快速、准确地将探针放置到芯片上的指定位置。 2、样品制备 生物样品往往是复杂的生物分子混合体,除少数特殊样品外,一般不能直接与芯片反应,有时样品的量很小。所以,必须将样品进行提取、扩增,获取其中的蛋白质或DNA、RNA,然后用荧光标记,以提高检测的灵敏度和使用者的安全性。 3、杂交反应 杂交反应是荧光标记的样品与芯片上的探针进行的反应产生一系列信息的过程。选择合适的反应条件能使生物分子间反应处于最佳状况中,减少生物分子之间的错配率。 4、信号检测和结果分析 杂交反应后的芯片上各个反应点的荧光位置、荧光强弱经过芯片扫描仪和相关软件可以分析图像,将荧光转换成数据,即可以获得有关生物信息。基因芯片技术发展的最终目标是将从样品制备、杂交反应到信号
基因芯片数据功能分析
生物信息学在基因芯片数据功能分析中的应用2009-4-29 随着人类基因组计划(Human Genome Project)即全部核苷酸测序的即将完成,人类基因组研究的重心逐渐进入后基因组时代(PostgenomeEra),向基因的功能及基因的多样性倾斜。 通过对个体在不同生长发育阶段或不同生理状态下大量基因表达的平行分析,研究相应基因在生物体内的功能,阐明不同层次多基因协同作用的机理,进而在人类重大疾病如癌症、心血管疾病的发病机理、诊断治疗、药物开发等方面的研究发挥巨大的作用。它将大大推动人类结构基因组及功能基因组的各项基因组研究计划。生物信息学在基因组学中发挥着重大的作用,而另一项崭新的技术——基因芯片已经成为大规模探索和提取生物分子信息的强有力手段,将在后基因组研究中发挥突出的作用。基因芯片与生物信息学是相辅相成的,基因芯片技术本身是为了解决如何快速获得庞大遗传信息而发展起来的,可以为生物信息学研究提供必需的数据库,同时基因芯片的数据分析也极大地依赖于生物信息学,因此两者的结合给分子生物学研究提供了一条快捷通道。 本文介绍了几种常用的基因功能分析方法和工具: 一、GO基因本体论分类法 最先出现的芯片数据基因功能分析法是GO分类法。Gene Ontology(GO,即基因本体论)数据库是一个较大的公开的生物分类学网络资源的一部分,它包含38675个Entrez Gene注释基因中的17348个,并把它们的功能分为三类: 分子功能,生物学过程和细胞组分。在每一个分类中,都提供一个描述功能信息的分级结构。这样,GO中每一个分类术语都以一种被称为定向非循环图表(DAGs)的结构组织起来。研究者可以通过GO分类号和各种GO数据库相关分析工具将分类与具体基因联系起来,从而对这个基因的功能进行描述。在芯片的数据分析中,研究者可以找出哪些变化基因属于一个共同的GO功能分支,并用统计学方法检定结果是否具有统计学意义,从而得出变化基因主要参与了哪些生物功能。
基因芯片制造技术的比较研究
基因芯片制造技术的比较 摘要:基因芯片是电子学和生命科学相结合的产物,和计算机芯片有着奇妙的相似性,也储藏着极其丰富的信息量。基因芯片也称作DNA微阵列,目前制备芯片主要以玻璃片或者硅片为载体,采用原位合成和微距阵的方法将寡聚甘酸片段或cDNA作为探针按顺序排列在载体上【2】。芯片的制备除了用到微加工工艺外,还需要使用机器人技术。以便能快速、准确地讲探针防止到芯片上的指定位置。基因芯片主要包括芯片的制备、样品制备、生物分子反应、芯片信号的检测。本文主要从芯片的样品的制造入手,分析其常见的制备技术及优劣特性【3】。 关键词:基因探针,基因芯片,制造技术 一引言 基因芯片从实验室走向工业化直接得益于探针固相原味合成技术和照相平板印刷技术的有机结合以及激光共聚焦显微技术的引入。它使得合成、固定高密度的数以万计的探针分子切实可行,而且借助激光共聚焦显微扫描技术使得可以对杂交信号进行实时、灵敏、准确的检测和分析。目前已有多种方法可以将寡核苷酸或短肽固定到固相支持物上。这些方法总体上有两种,即原位合成(in situ synthesis)与合成点样两种【3】。原位合成法主要以光引导聚合技术(light-directed synthesis)和压电打印技术(piezoelectric printing)为主;合成点样主要是借助传统的DNA或多肽固相合成仪完成,只是合成后用特殊的自动化微量点样装置将其以比较高的密度涂布于硝酸纤维膜、尼龙膜或玻片上。 二芯片技术要点 2.1芯片技术的基本要点 芯片技术主要包括四个基本要点:芯片方阵的构建、样品的制备、生物分子反应和信号的检测。 2.1.1芯片制备 目前制备芯片主要以玻璃片或硅片为载体,采用原位合成和微矩阵的方法将寡核苷酸片段或cDNA作为探针按顺序排列在载体上。芯片的制备除了用到微加工工艺外,还需要使用机器人技术。以便能快速、准确地将探针放置到芯片上的指定位置。 2.1.2样品制备
神奇的基因芯片 教学设计
一、教学设计理念 根据17年版的新课程理念,高中生物教学着眼于学生适应未来社会发展和个人生活的需要,从生命 观念、科学思维、科学探究和社会责任等方面发展学生的学科核心素养,本课主要采用探究研讨法进行教 学,创设教学情境,在活动中潜移默化地渗透科学研究过程,训练学生科学的思维方法,培养学生的科学 素质,增强学生的社会责任感,让学生始终能参与到学习中来,成为学习的主人。 二、教材分析及学情分析 本节课节选自高中生物人教版选修三专题一中视野拓展部分,本节课主要包含了基因芯片的概述及基 因芯片的应用领域,学生在学习本节课知识之前,已经通过前面的学习已经对基因有了一定的了解,知道 什么是基因、DNA分子是如何进行杂交及杂交过程中遵循的原则等生物学知识,这无疑为本节课的学习奠 定了一定的基础;同时本节课节选的内容在生物学上都是比较前沿的知识,所列举的亲子鉴定、输血性艾 滋病、高科技犯罪等例子更能加深学生对本部分的了解,同时激发学生的学习兴趣。 三、教学目标及学业要求 1、能够设计简单的物理模型,介绍什么是基因芯片。(科学探究) 2、结合生活或生产实例,了解基因芯片在基因检测方面的操作过程,及基因芯片的应用领域。(生命观念) 3、面对日常生活或社会热点话题中与基因芯片技术有关的话题,能够表明自己的观点并展开讨论。(科学 思维、社会责任) 四、教学方法 探究研讨法 五、课时安排 1课时 六、教学过程
七、板书设计
八、教学反思 本节课涉及的教学内容均为生物学上比较前沿的知识,如果运用传统的教学方法,由老师滔滔不绝地讲,学生默默地听,课堂气氛比较沉闷,教学效率低下,更谈不上从生命观念、科学思维、科学探究和社会责任等方面发展学生的学科核心素养。本课主要采用探究研讨法进行教学,创设问题情境,在小组活动中潜移默化地渗透科学研究过程,训练学生科学的思维方法,培养学生的科学素质,增强学生的社会责任感,让学生始终能参与到学习中来,成为学习的主人。
R语言在基因芯片数据处理中的应用
1.R语言安装:官方网站安装软件。 2. 所需要的软件包: 2.1 affy数据处理相关的程序包 在R中复制source("/biocLite.R") biocLite("affy") 2.2 热度图相关程序包 Gplots():install.packages("gplots") 3.获取基因表达数据 3.1 读取基因芯片数据(cel.files) the.filter <- matrix(c("CEL file (*.cel)", "*.cel", "All (*.*)", "*.*"), ncol = 2, byrow = T) cel.files <- choose.files(caption = "Select CEL files", multi = TRUE, filters = the.filter, index = 1) raw.data <- ReadAffy( = cel.files) 3.2 sampleNames(raw.data)ang #先看看原样品名称的规律
7. 选取目的基因 在上确定探针,选取数据;汇总到excel表格中,保存为csv格式。 8.热度图 cipk=read.csv("c:/users/suntao/desktop/TaCIPK affx arry log.csv") https://www.360docs.net/doc/3718211759.html,s(cipk)=cipk$genename cipk <- cipk[,-1] cipk_matrix=data.matrix(cipk) library(gplots) heatmap.2(cipk_matrix,Rowv=FALSE,Colv=FALSE,col=greenred(75),key=TRUE,keysize=0.8,trace="n one",https://www.360docs.net/doc/3718211759.html,="none",symkey=FALSE,revC=FALSE,margins=c(10,10),denscol=tracecol,distfun=dist, hclustfun=hclust,dendrogram="none",symm=FALSE) heatmap.2颜色选择函数col=colorRampPalette(c("black","red")) 中10个是当地固有个体(old),另外10个是新迁入的个体(new),old和new个体两两随机配对,分别用不同颜色染料(波长分别为555和647nm)标记后,在同一张基因芯片上杂交;此外,每个基因在每张芯片上都重复点样3次,因此此数据是有3个replicates及10张芯片的双通道芯片。数据是样点的信号强度值,没有经过标准化处理的。
基因芯片的必备知识和操作流程
基因芯片技术的诞生为生物技术工作人员打开了一道科研的便利之门,曾被评为1998年年度十大科技进展之一。本文对基因芯片的实验原理、技术基础、分类、用途、操作主要环节等内容做详细的介绍。 基因芯片技术的诞生为生物技术工作人员打开了一道科研的便利之门,曾被评为1998年年度十大科技进展之一。本文对基因芯片的实验原理、技术基础、分类、用途、操作主要环节等内容做详细的介绍。 1.基本原理和技术基础 基因芯片以DNA杂交为基本原理,基于A和T、G和C的互补关系。它是在探针的基础上研制出的。所谓探针是一段人工合成或筛选出的已知顺序的碱基序列,样品分子上连接有一些cy3、cy5等可检测的物质。经激光共聚焦荧光显微镜检出杂交或反应信号,通过计算机处理、分析,即可获得所需信息。例如,用红、绿荧光分别标记实验样本和对照样本的cDNA,混合后与微阵列杂交,可显示实验样本和对照样本基因的表达强度(显示红色、绿色或黄色),由此可在同一微阵列上同时检测两样本的基因表达差异。在基因芯片工作过程中,固定位点使用不同分子生物学技术和碱基互补配对原则与待测基因片段杂交,并通过自动阅读设备分析杂交结果,达到定性、定量分析的目的。基因芯片通过应用平面微细加工技术和超分子自组装技术,把大量分子检测单元集成在一个微小的固体基片表面,可同时对大量的核酸等生物分子实现高效、快速、低成本的检测和分析。 基因芯片的检测主要建立在放射标记技术、荧光标记技术、质谱分析、化学发光等技术上。使用荧光标记的基因芯片需要专用的荧光扫描仪。对于高密度的基因芯片,目前最常用的是激光共聚焦显微镜和高性能的冷却CCD。目前专用于荧光扫描的扫描仪大致分为两类:一类是基于CCD(charge-coupled device,电荷耦合装置)的检测光子;另一类则是基于PMT(photomultiplier tube,光电倍增管)的检测系统。 生物芯片的发展得益于微细加工技术和现代分子生物技术的结合。随着人类基因组计划的实施,现代分子生物技术也取得了巨大突破:体外基因扩增技术实现了核酸样品的快速制备和放大,基因扩增后可进行基因测序,而基因探针技术使得基因测序的检测自动化成为可能,极大地促使了生物芯片的发展。微阵列的加工借助的是微电子工业中应用十分成熟的光学光刻技术和微机电系统加工中所采用的各种方法,根据要求在玻璃、塑料、硅片等基底材料上加工出用于生物样品分离、反应的各种微细结构,然后在微结构上施加表面化学处理。光学光刻的第一步是通过光学制板照相制备掩模。制备好的掩模通常是镀有铬层的石英玻璃板,该铬层事先已按微细结构的图形被刻蚀成透光和不透光两个部分。第二步是光刻,让掩模与表面涂有光刻胶的硅片接触,然后让光源通过掩模照射到硅片上。通过显影光刻胶中的图形,使其上未被掩模遮掩的部分被溶解除去。上述工作完成之后,通过对无胶保护的硅片用腐蚀剂蚀刻,再去除剩余的光刻胶便可获得所需生物芯片的微细结构。微米级甚至纳米级的微细加工技术使得数以
DNA芯片的制备详细步骤
第一部分:基片的制备 第二部分:点样 第三部分:杂交 第四部分:显色系统与信号检测 第一部分:基片的制备 基片是生物芯片的基础,是生化反应的载体。生物芯片以其基片的不同可以分为无机基片和有机合成物基片。前者主要包括玻璃片、塑料、硅胶晶片、微型磁珠,其上的探针主要以原位聚合的方法合成;后者主要有特定孔径硝酸纤维膜、尼龙膜和凝胶块等,其上的探针主要是预先合成后通过特殊的微量点样装置或仪器滴加到基片上去(20-22)。在选择固相载体时,应考虑其荧光背景的大小、化学稳定性、结构复杂性、介质对化学修饰作用的反应、介质表面积及其承载能力以及非特异吸附的程度等因素。因此制作生物芯片的载体材料必须以下特征:良好的生物兼容性;足够的稳定性,能够抵抗一定热、酸、碱等条件;表面活性,表面应具有各种活性基团,以便与各种生物分子连接;良好的光学性质。 基片在使用前一般需活化处理。活化试剂EDC、NHS等通过化学反应在载体表面键合上活性基团如氨基、环氧化物、巯基、醛基、肼基,以便于与配基相结合,形成具有生物特异性的亲和载体,用于固定生物活性分子,如蛋白质、核酸、多肽等。不同的载体就有不同的活化方式,玻璃基片常采用氨基、醛基、巯基这三种修饰方式。 主要材料:玻璃片:普通无色玻璃,规格75×25×1mm 质量要求: 主要设备仪器:通风厨一个、烘箱一台 氨基修饰基片的处理流程: 1)玻片的清洗: 取普通载玻片放入95%的浓硫酸与5%重铬酸钾的溶液中浸泡过夜。用蒸馏水洗净玻片,浸入25%的氨水中,过夜。取出玻片,超纯水洗净,晾干。 2)玻片的硅烷化: 将清洗后的玻片放入10mM 氨基丙基三乙氧基硅烷(3-aminopropyltriethoxysilane APES)的无水乙醇溶液浸泡30分钟。用无水乙醇反复清洗几次,晾干后放入烘箱(100-110℃)烘干。