1-VASP计算教程第一课-认识VASP的输入和输出

V ASP计算教程
第一课认识V ASP的输入和输出
课程目标:
通过计算孤立氧原子的能量,初步认识V ASP的输入和输出。
课程正文:
一、V ASP的输入文件(lecture1-01)
V ASP的基本输入文件共有四个:POSCAR,INCAR,KPOINTS,POTCAR。其中POSCAR是结构文件(计算的体系是什么);INCAR是参数文件(怎么计算);KPOINTS是K点文件,决定了在体系的哪些点进行计算;POTCAR是赝势文件,包含了相应体系的元素的基本信息。
1、POSCAR
以孤立氧原子为例,创建相应的结构文件。V ASP要求计算的结构必须是周期体系,因此我们的结构可以描述为“一个足够大的晶胞(盒子)中存在一个氧原子”,之所以强调“足够大”,是因为晶胞具有周期性,晶胞足够大,氧原子之间的相互作用才可以忽略不计。POSCAR文件内容如下:
----------------------------------------------------------------------------------------------------------------------
1 O atom
2 1.0
3 8.00 0.00 0.00
4 0.00 8.00 0.00
5 0.00 0.00 8.00
6 O
7 1
8 Cartesian
9 0.00 0.00 0.00
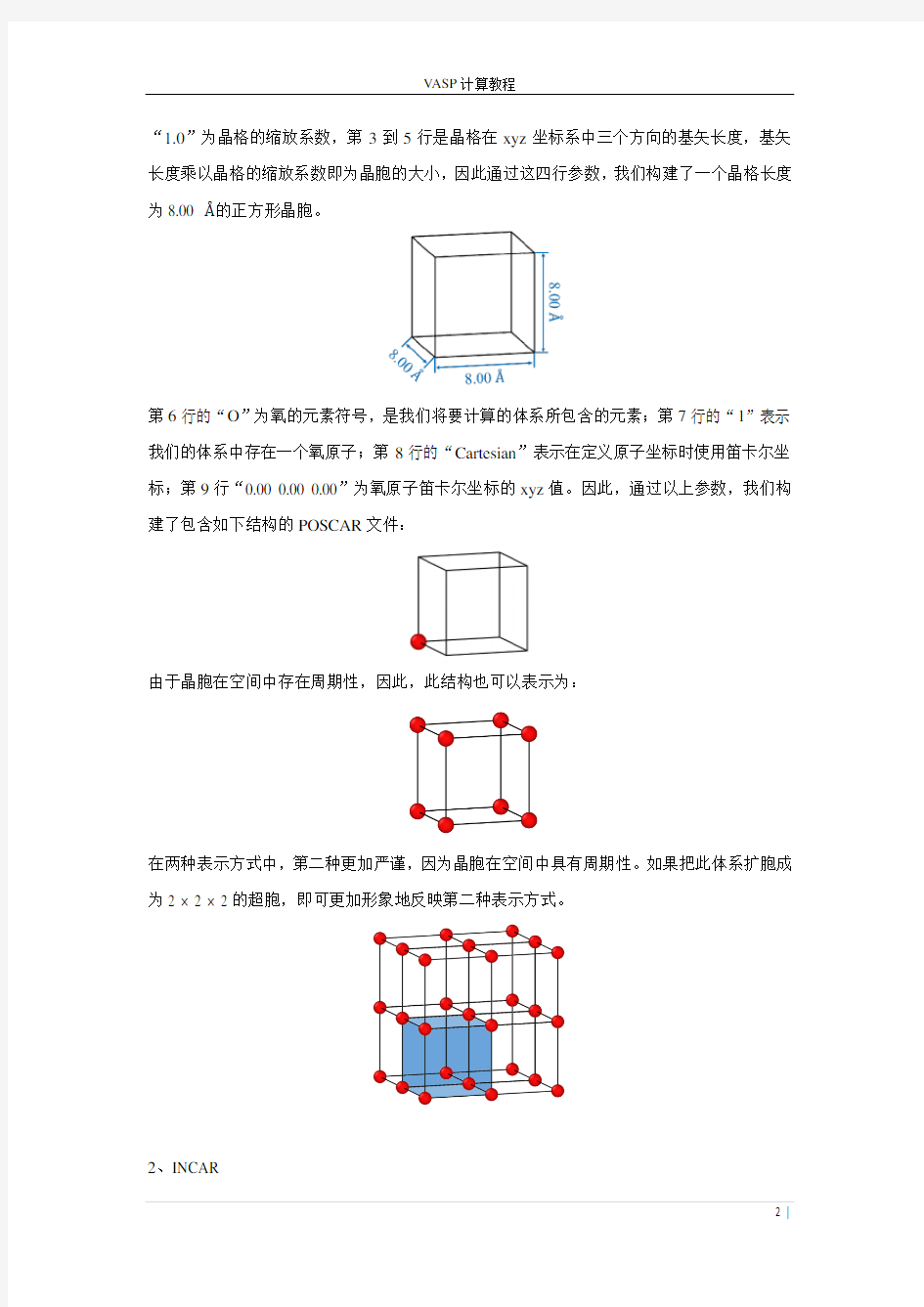
---------------------------------------------------------------------------------------------------------------------- 在这里,灰色背景及其中的数字为行号,POSCAR文件中并不包含。其中,第1行的“O atom”是体系的名称,可以根据个人的喜好进行命名(如可以替换为isolated O,single O atom,one Oxygen atom等),方便对计算任务的记忆与理解,不同的命名不会影响计算;第2行的
“1.0”为晶格的缩放系数,第3到5行是晶格在xyz坐标系中三个方向的基矢长度,基矢长度乘以晶格的缩放系数即为晶胞的大小,因此通过这四行参数,我们构建了一个晶格长度为8.00 ?的正方形晶胞。
第6行的“O”为氧的元素符号,是我们将要计算的体系所包含的元素;第7行的“1”表示我们的体系中存在一个氧原子;第8行的“Cartesian”表示在定义原子坐标时使用笛卡尔坐标;第9行“0.00 0.00 0.00”为氧原子笛卡尔坐标的xyz值。因此,通过以上参数,我们构建了包含如下结构的POSCAR文件:
由于晶胞在空间中存在周期性,因此,此结构也可以表示为:
在两种表示方式中,第二种更加严谨,因为晶胞在空间中具有周期性。如果把此体系扩胞成为2 × 2 × 2的超胞,即可更加形象地反映第二种表示方式。
2、INCAR
在本例中,我们只需要计算氧原子的能量。内容如下:
----------------------------------------------------------------------------------------------------------------------
1 SYSTEM = O atom in a box
2 ISMEAR = 0
---------------------------------------------------------------------------------------------------------------------- 如上所示,INCAR的书写语法为“参数名称= 参数值”。同POSCAR一样,第1行可以依据个人喜好命名,但不同的是,命名必须要在“SYSTEM = ”之后书写,因此本行也可以写为“SYSTEM = O atom”或“SYSTEM = O energy”等;第2行“ISMEAR = 0”表示使用高斯展开来描述每个原子轨道的占据。
拓展阅读:
ISMEAR
ISMEAR可用取值有-5,-4,-3,-2,-1,0,以及大于零的整数N。默认值为1。它定义了每个轨道中电子的局域占据方式(determines how the partial occupancies f nk are set for each orbital.)ISMEAR取大于零的整数N时,使用的是N阶Methfessel-Paxton方法(Phys. Rev. B 1989, 40, 6, 3616.)。需要注意的是,使用MP方法可能导致电子局域占据为负值或者大于1,在计算绝缘体时可能得到错误的结果。
ISMEAR = 0:高斯展开(Gaussian smearing)
ISMEAR = -1:费米展开(Fermi smearing)
ISMEAR = -2:电子的局域占据由WA VECAR或INCAR中读取,并且在计算的过程中保持不变。在INCAR中分配电子占据的方式为FERWE = f(1) f(2) f(3) ... f(NBANDS×N k),如果体系带有磁性,则需要添加FERDO = f(1) f(2) f(3) ... f(NBANDS×N k)。通过INCAR分配电子占据情况时必须对所有能带和K点进行分配,且其取值范围为0至1。电子占据数值会输出于OUTCAR 结果文件中,但在OUTCAR中,其数值是分配值的二倍(0至2)。
ISMEAR = -3:对展开参数进行循环。此时需要在INCAR中通过SMEARINGS = ismear1 sigma1 ismear2sigma2 ... 分配不同的展开数值。另外IBRION必须设置为1,NSW必须通过ismear i/sigma i确定。ISMEAR = -3时,循环的第一步使用布洛赫修正的四面体方法。
ISMEAR = -4:四面体方法(使用Γ为中心的K点网格)
ISMEAR = -5:布洛赫修正的四面体方法(使用Γ为中心的K点网格)
计算体相材料的总能时,推荐使用布洛赫修正的四面体方法(ISMEAR = -5),此方法也可
以得到合理的电子态密度(DOS)。不过唯一缺点是对于局域电子的占据情况不使用变分,因此对于金属体系来说,得到的力和应力张量可能会存在5%到10%的误差。对于半导体和绝缘体来说,此方法可以得到正确的力,因为这两种体系局域电子占据只有0和1两种可能。
计算金属体系的力和声子频率时,推荐使用Methfessel-Paxton方法(ISMEAR > 0),此方法也可以精确的得到体系的总能,不过需要设置合理的展宽值(SIGMA),展宽太大总能精度会降低,太小则需要使用稠密的K点网格。因此在SIGMA值尽可能大的情况下,要确保OUTCAR 文件输出的自由能(free energy)与总能(total energy)之差可以忽略,即“entropy T*S”的值要小于1 meV/atom(需要注意的是,OUTCAR文件中能量的单位为eV,需要转化为meV,并且平均到每个原子或原子对)。对于半导体和绝缘体,一定要避免使用ISMEAR > 0,否则将会得到错误的结算结果(会出现某些电子态的占据小于0或大于1的情况),导致一些性质(如声子频率)的误差超过20%,如果不仔细检查输出,将很难发现并定位这种错误。对于绝缘体推荐使用ISMEAR = 0或ISMEAR = -5。
高斯展开(ISMEAR = 0)在大多数情况下可以得到合理的结果。在这种情况下,需要通过设置的SIGMA值去推测SIGMA = 0时的结果,VASP的输出文件OUTCAR直接给出了SIGMA = 0的值,即“energy( SIGMA→0 )”对应的能量。此方法中如果使用的SIGMA值较大,也会产生类似四面体方法中的误差,不同的是,高斯展开并不能通过控制SIGMA值来控制误差的大小,因此相比之下,使用四面体方法更加灵活。另外需要注意的是,高斯展开得到的力和应力张量是与自由能(free energy)对应的,而不是SIGMA→0的能量。四面体方法(ISMEAR = -5)对于计算金属体系来说更加容易使用。
总结:
如果对自己的计算体系不了解,不能确定体系是金属,半导体,还是绝缘体的话,推荐使用ISMEAR = 0,同时设置SIGMA的值在0.03到0.05范围内进行计算。
对于半导体或绝缘体,使用四面体方法(ISMEAR = -5),如果体系晶胞很大(或计算时只有一个或两个K点),使用ISMEAR = 0,同时设置SIGMA的值在0.03到0.05范围。
金属体系的弛豫使用ISMEAR = 1或ISMEAR = 2,同时需要设置SIGMA(使“entropy T*S”的值小于1 meV/atom),对于金属体系,SIGMA值为0.2时基本就可以得到合理结果。
半导体和绝缘体避免使用ISMEAR>0。
计算电子带密度(DOS)以及精确计算体系的总能,使用四面体方法(ISMEAR = -5),如果体系为金属,在计算时不能进行弛豫(可以先使用ISMEAR = 1或ISMEAR = 2进行弛豫,再用
ISMEAR = -5计算DOS或总能)。
拓展阅读中提到有些情况需要测试并设置SIGMA(展宽)值,本计算体系没有进行设置,即使用默认的SIGMA = 0.2。
3、KPOINTS
对于原子或者分子体系,使用一个K点即可满足计算需求,内容如下:
----------------------------------------------------------------------------------------------------------------------
1 Gamma-point only
2 0
3 Monkhorst Pack
4 1 1 1
5 0 0 0
---------------------------------------------------------------------------------------------------------------------- 其中,第1行的“Gamma-point only”同样可依据个人习惯命名,第2行的“0”表示自动产生K点,第3行“Monkhorst Pack”表示按照Monkhorst 网格进行K点选取,第4行“1 1 1”表示在晶胞的xyz方向上K点的个数均为1,第5行“0 0 0”是K点相对于原网格点的位移。
4、POTCAR
POTCAR是V ASP的赝势文件,此文件包含了计算某原子所需要的赝势信息,直接从赝势库复制使用即可。本计算只包含氧元素,因此只需要给出氧元素的POTCAR,其基本内容如下:
----------------------------------------------------------------------------------------------------------------------
1 PAW_PBE O 08Apr2002
2 6.00000000000000
......
8 TITEL = PAW_PBE O 08Apr2002
……
---------------------------------------------------------------------------------------------------------------------- 其中,第1行为POTCAR的基本信息,表明本文件是氧元素的PAW_PBE泛函,此赝势文件是2002年4月8日发表的;第2行为氧原子的最外层电子数。一般而言,POTCAR可简单地通过第8行的“TITEL”来判断。
以上即为V ASP的四个基本的输入文件。服务器提交V ASP任务需要提交脚本,每个课
题组都会有不同的提交脚本,把相应的提交脚本放入输入文件的目录即可提交计算任务。
二、V ASP的输出文件(lecture1-02)
计算完成后,V ASP将有若干输出,本节主要关注OUTCAR和OSZICAR两个文件。
1、OUTCAR
OUTCAR文件会给出V ASP详细的计算过程,其中包括了计算的参数、电子迭代过程、KS方程的本征值、应力张量、原子受力、局域电荷分布、磁矩、介电性质等。
一般而言,当V ASP计算完成时,首先需要检查是否正常结束。打开OUTCAR文件,并检查文件结尾,其内容如下,则说明V ASP正常完成计算:
---------------------------------------------------------------------------------------------------------------------- 892 General timing and accounting informations for this job:
893 ========================================================
895
896 Total CPU time used (sec): 2.648
897 User time (sec): 2.261
898 System time (sec): 0.387
899 Elapsed time (sec): 5.095
900
901 Maximum memory used (kb): 157240.
902 Average memory used (kb): 0.
903
904 Minor page faults: 30651
905 Major page faults: 0
906 Voluntary context switches: 680
---------------------------------------------------------------------------------------------------------------------- 此结尾包含了计算所使用的各种时间以及内存等信息。
向上查找OUTCAR文件的内容,找到如下信息(这一部分信息会在OUTCAR中多次出现,我们一定要从文件的尾部向上寻找,找到最后一次输出的信息):
----------------------------------------------------------------------------------------------------------------------
726 Free energy of the ion-electron system (eV)
727 ---------------------------------------------------
728 alpha Z PSCEN = 0.27139542
729 Ewald energy TEWEN = -91.92708002
730 -Hartree energ DENC = -281.59389770
731 -exchange EXHF = 0.00000000
732 -V(xc)+E(xc) XCENC = 26.05297763
733 PAW double counting = 356.05703734 -357.82340294
734 entropy T*S EENTRO = -0.30852464
735 eigenvalues EBANDS = -83.30627042
736 atomic energy EATOM = 432.26319604
737 Solvation Ediel_sol = 0.00000000
738 ---------------------------------------------------
739 free energy TOTEN = -0.31456930 eV
740
741 energy without entropy = -0.00604466 energy(sigma->0) = -0.16030698 ---------------------------------------------------------------------------------------------------------------------- 这一部分给出了体系的各种能量信息。结合本节的拓展阅读,我们可以看到第734行“entropy T*S”的值为-0.309 eV,大于1 meV/atom(本结果为309 meV/atom),因此本计算结果的精度较低,这是因为我们没有设置SIGMA值,即使用了默认的0.2(不加参数设置时即使用默认值)。第739行为自由能(free energy),其值为-0.315 eV,通过“free energy”与“entropy T*S”之差:-0.315 - (-0.309) = -0.006 eV即可得到本体系的总能。体系的总能也可以通过第741行查看,其值与我们计算的差值一致,为-0.006 eV。不过由于本计算的SIGMA 值太大造成了“entropy T*S”较大,所以得到的“energy(sigma->0)”值与总能有较大差别,为-0.160 eV。一般我们在提取体系总能时便使用“energy(sigma->0)”的值。
2、OSZICAR
OSZICAR只包括了体系电子迭代过程的简化信息,对于本计算,其内容如下:
----------------------------------------------------------------------------------------------------------------------
1 N E dE d eps ncg rms rms(c)
2 DA V: 1 0.391538037185E+02 0.39154E+02 -0.95952E+02 14 0.335E+02
3 DA V: 2 0.395443442858E+01 -0.35199E+02 -0.34375E+02 28 0.480E+01
4 DA V: 3 -0.157563036656E+00 -0.41120E+01 -0.39089E+01 14 0.376E+01
5 DA V: 4 -0.310473026346E+00 -0.15291E+00 -0.13905E+00 14 0.660E+00
6 DA V: 5 -0.313452365096E+00 -0.29793E-02 -0.29751E-02 28 0.911E-01 0.307E-01
7 DA V: 6 -0.314469929004E+00 -0.10176E-02 -0.17937E-03 14 0.380E-01 0.155E-01
8 DA V: 7 -0.314569298818E+00 -0.99370E-04 -0.24636E-04 14 0.157E-01
9 1 F= -.31456930E+00 E0= -.16030698E+00 d E =-.308525E+00
---------------------------------------------------------------------------------------------------------------------- 此文件的第1行给出了每列所代表的含义,其中N为电子迭代的步数,E为每个电子步体系的总能,dE为前后两个电子步对应体系的总能的差,d eps为本征值的变化,rms为残差向量,rms(c)为电荷密度的残差向量。对于本计算,可见从第1个电子步到第4个电子步,rms(c)一列均无数据,表明在前4步计算过程中,电荷是保持固定的,从第5步开始,电荷开始更新。第9行为体系的能量信息,此行与上述OUTCAR的第739和741行对应,这里的F即为“free energy TOTEN”对应的能量,E0即为“ energy(sigma->0)”所对应的能量,单位同样均为eV,在取体系能量时我们同样取E0对应的值。
课后练习:
1.仿照孤立氧原子体系,计算孤立氢原子的能量。
2.仿照孤立氧原子体系,计算孤立碳原子的能量。
课程拓展:
1.把VSAP输出的W A VECAR文件复制到输入文件夹中(即输入文件包括POSCAR、
INCAR、KPOINT、POTCAR、以及W A VECAR),重新提交计算,查看OSZICAR有什么不同。
2.把原子放在晶胞的中央,比较所得到的体系能量。
课后练习解析:
1、(输入文件详见lecture1-03)同孤立氧原子类似,计算氢原子需要构建一个相应的POSCAR 结构文件。我们同样把一个氢原子放入边长为8.00 ?的正交晶胞中,文件内容如下:
----------------------------------------------------------------------------------------------------------------------
1 H atom
2 1.0
3 8.00 0.00 0.00
4 0.00 8.00 0.00
5 0.00 0.00 8.00
6 H
7 1
8 Cartesian
9 0.00 0.00 0.00
----------------------------------------------------------------------------------------------------------------------
对于赝势文件,我们也需要选择对应氢元素的POTCAR,直接通过从赝势库中复制使用:
----------------------------------------------------------------------------------------------------------------------
1 PAW_PBE H 15Jun2001
2 1.00000000000000
......
8 TITEL = PAW_PBE H 15Jun2001
……
---------------------------------------------------------------------------------------------------------------------- 注意这里“TITEL”为氢元素的PAW_PBE泛函。
KPOINTS文件和INCAR文件同计算氧原子的一致即可。经提交计算完成后(输出文件详见lecture1-04),查看OUTCAR文件。从OUTCAR文件的最后:
---------------------------------------------------------------------------------------------------------------------- 924 Total CPU time used (sec): 1.404
925 User time (sec): 1.162
926 System time (sec): 0.242
927 Elapsed time (sec): 1.690
928
929 Maximum memory used (kb): 99120.
930 Average memory used (kb): 0.
931
932 Minor page faults: 26949
933 Major page faults: 0
934 V oluntary context switches: 204
---------------------------------------------------------------------------------------------------------------------- 我们可以确认计算正常结束。通过OSZICAR文件,我们可快速查看相应的能量:
---------------------------------------------------------------------------------------------------------------------- ......
10 1 F= -.12017738E+00 E0= -.63758417E-01 d E =-.112838E+00
---------------------------------------------------------------------------------------------------------------------- 2、输入文件详见lecture1-05,输出文件详见lecture1-06。
课程拓展解析:
1、在W A VECAR存在的情况下进行计算(输入文件详见lecture1-07),OSZICAR内容如下
(输出文件详见lecture1-08):
----------------------------------------------------------------------------------------------------------------------
1 N E dE d eps ncg rms rms(c)
2 DA V: 1 -0.314608546170E+00 -0.31461E+00 -0.91868E-05 14 0.676E-02 0.118E-02
3 DA V: 2 -0.314604560030E+00 0.39861E-05 -0.11779E-05 1
4 0.257E-02
4 1 F= -.31460456E+00 E0= -.16034224E+00 d E =-.308525E+00
---------------------------------------------------------------------------------------------------------------------- 一般情况下,误差小于10 meV时,我们认为V ASP达到了计算的精度要求,此结果与之前的计算相比,误差为F≈0.04 meV,E0≈0.04 meV,因此我们可以认为结果一致。但是体系的电子迭代步数变成了2步,比W A VECAR不存在的情况下更少了,与此同时,计算所花费的时间也将更少。WA VECAR包含了体系的波函数信息,使用W A VECAR进行计算可以使初始的波函数更加合理,这种方法常用于V ASP的续算过程,但需要注意的是,WA VECAR必须是同一个体系产生的,并且INCAR和KPOINTS也要保持一致。
2、把氧原子放在晶胞的中央,即改变POSCAR文件内容如下(输入文件详见lecture1-09):----------------------------------------------------------------------------------------------------------------------
1 O atom
2 1.0
3 8.00 0.00 0.00
4 0.00 8.00 0.00
5 0.00 0.00 8.00
6 O
7 1
8 Cartesian
9 4.00 4.00 4.00
---------------------------------------------------------------------------------------------------------------------- 这里我们可以简单的修改第9行,改变氧原子的笛卡尔坐标值,把原来的“0.00 0.00 0.00”(晶胞的顶点)替换为“4.00 4.00 4.00”(晶胞的中心)实现。相应的体系如下图所示:
计算完成后,同样通过OSZICAR查看其能量信息(输出文件详见lecture1-10):
---------------------------------------------------------------------------------------------------------------------- ……
10 1 F= -.31460322E+00 E0= -.16034090E+00 d E =-.308525E+00
---------------------------------------------------------------------------------------------------------------------- 可见孤立氧原子的能量与放置在晶胞中的位置无关,与实际情况吻合。
对于POSCAR而言,原子也可以使用分数坐标来定义。以氧原子在晶胞中心为例,在xyz三个方向上,氧原子均处于晶胞的12?处,因此可以由如下方式表示(输入文件详见lecture1-11):
----------------------------------------------------------------------------------------------------------------------
1 O atom
2 1.0
3 8.00 0.00 0.00
4 0.00 8.00 0.00
5 0.00 0.00 8.00
6 O
7 1
8 Direct
9 0.50 0.50 0.50
---------------------------------------------------------------------------------------------------------------------- 此方法需要修改POSCAR的第8行和第9行。其中第8行的“Direct”表示使用分数坐标来定义原子位置,而第9行则为原子在xyz三个方向上相对于晶胞的值。在以分数坐标表示的体系中,用分数坐标值乘以晶格常数,即为相应的笛卡尔坐标的值。
使用此方法书写POSCAR同样可以得到一致的计算结果(输出文件详见lecture1-12):---------------------------------------------------------------------------------------------------------------------- ......
10 1 F= -.31460322E+00 E0= -.16034090E+00 d E =-.308525E+00
----------------------------------------------------------------------------------------------------------------------
课程回顾:
1.计算孤立氧原子能量的四个输入文件(POSCAR,INCAR,KPOINTS,POTCAR)。
2.结构文件POSCAR的写法。
3.参数文件INCAR中ISMEAR的用法。
4.判断计算是否正常结束(OUTCAR),读取体系的能量(OUTCAR,OSZICAR)。
5.结构文件POSCAR中原子坐标的两种写法(笛卡尔坐标,分数坐标)。
本节课相应文件请至链接: https://https://www.360docs.net/doc/476959086.html,/s/1YrlkpjwNdS3CfKI796Esew 提取码: whtg