生物信息学软件使用
生物信息学软件的使用(以MC4R基因为例)
第一章从NCBI上查找DNA、mRNA、蛋白质序列
一、以猪的黑素皮质素受体4(MC4R, melanocortin-4 re-ceptor)基因为例,介绍如何从NCBI 上查找DNA、mRNA、氨基酸序列。
1.首先查找MC4R的DNA序列。
在百度里输入NCBI,打开后得到的结果如下网页:
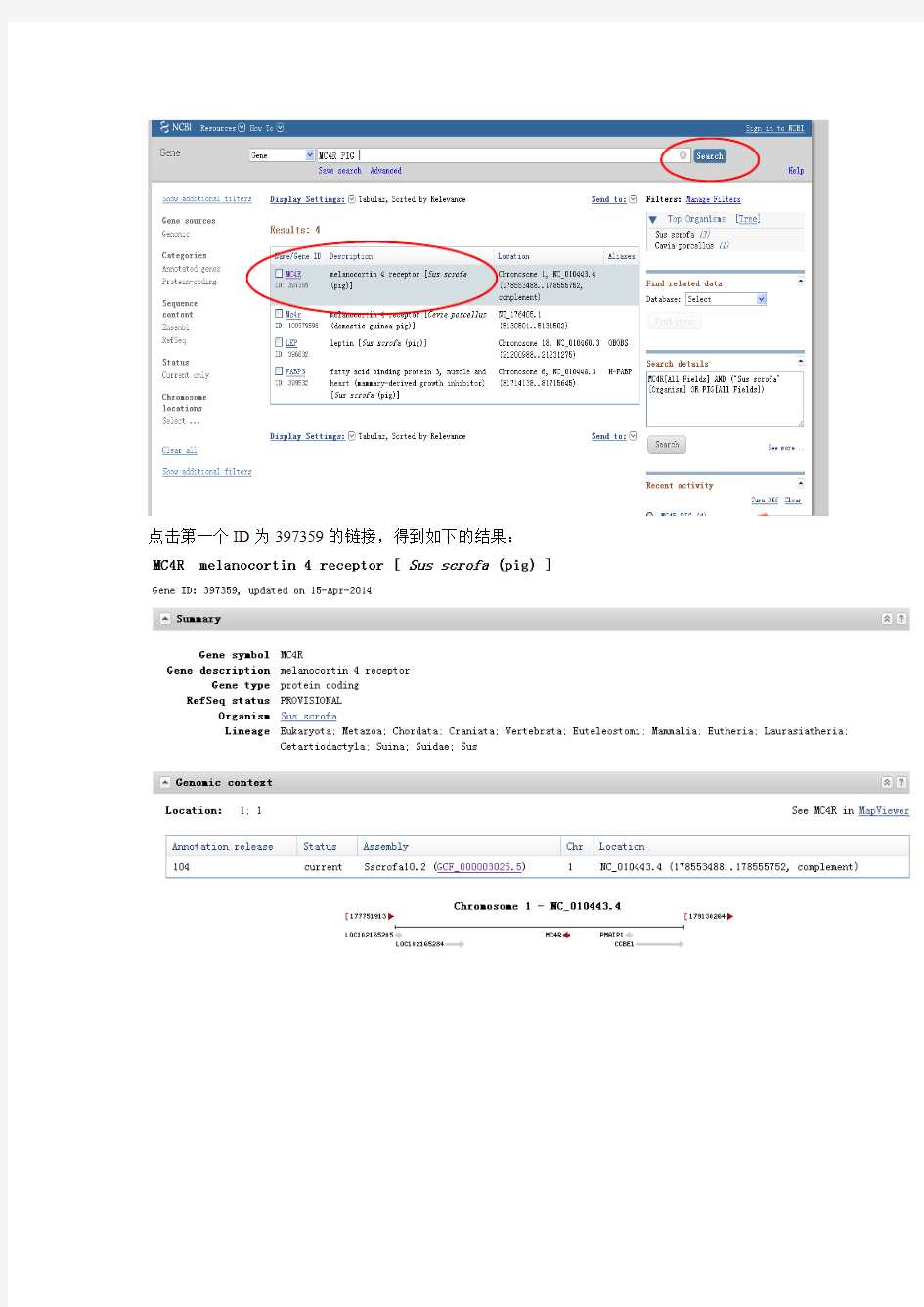
在Search 栏输入“MC4R pig”,在下拉菜单里选择Gene,然后点击Search,得到如下结果:
点击第一个ID为397359的链接,得到如下的结果:
可以看到该基因位于猪的1号染色体上,在右下方有个“Go to nucleotide”即进入核酸序列,有三种格式(用红圈标记的),经常用的是“FASTA”和“GenBank”,“FASTA”格式的比较简洁,不包含任何的数字,就全部是碱基,序列的对比和分析是就要用到这种格式;而“GenBank”格式就比较详细,可以查看到很多信息,比如碱基数、mRNA序列、内含子、外显子、CDS,以及氨基酸序列等等之类的。点击GenBank后得到如下结果:
Sus scrofa breed mixed chromosome 1,
Sscrofa10.2 DNA
LOCUS NC_010443 2265 bp DNA linear CON 29-SEP-2013 DEFINITION Sus scrofa breed mixed chromosome 1, Sscrofa10.2.
ACCESSION NC_010443 REGION: complement(178553488..178555752) GPC_000000583 VERSION NC_010443.4 GI:347618793
DBLINK BioProject: PRJNA28993
Assembly: GCF_000003025.5
KEYWORDS RefSeq.
SOURCE Sus scrofa (pig)
ORGANISM Sus scrofa
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Laurasiatheria; Cetartiodactyla; Suina; Suidae; Sus.
COMMENT REFSEQ INFORMATION: The reference sequence is identical to
CM000812.4.
On Oct 11, 2011 this sequence version replaced gi:333795951.
Assembly Name: Sscrofa10.2
The genomic sequence for this RefSeq record is from the genome
assembly released by the Swine Genome Sequencing Consortium as
Sscrofa10.2 in August 2011 (see
https://www.360docs.net/doc/549958585.html,/Projects/S_scrofa). Sscrofa10.2 is a mixed assembly of clones and contigs from the whole-genome shotgun
project AEMK00000000.1.
##Genome-Annotation-Data-START##
Annotation Provider :: NCBI
Annotation Status :: Full annotation
Annotation Version :: Sus scrofa Annotation Release 104
Annotation Pipeline :: NCBI eukaryotic genome annotation
pipeline
Annotation Software Version :: 5.1
Annotation Method :: Best-placed RefSeq; Gnomon
Features Annotated :: Gene; mRNA; CDS; ncRNA
##Genome-Annotation-Data-END##
FEATURES Location/Qualifiers
source 1..2265
/organism="Sus scrofa"
/mol_type="genomic DNA"
/db_xref="taxon:9823"
/chromosome="1"
/breed="mixed"
gene 1..2265
/gene="MC4R"
/note="melanocortin 4 receptor; Derived by automated
computational analysis using gene prediction method:
BestRefSeq."
/db_xref="GeneID:397359"
mRNA join(1..681,834..2265)
/gene="MC4R"
/product="melanocortin 4 receptor"
/inference="similar to RNA sequence, mRNA (same
species):RefSeq:NM_214173.1"
/exception="annotated by transcript or proteomic data"
/note="The RefSeq transcript has 2 indels compared to this genomic sequence; Derived by automated computational
analysis using gene prediction method: BestRefSeq."
/transcript_id="NM_214173.1"
/db_xref="GI:55741558"
/db_xref="GeneID:397359"
CDS join(534..681,834..1685)
/gene="MC4R"
/inference="similar to AA sequence (same
species):RefSeq:NP_999338.1"
/exception="annotated by transcript or proteomic data"
/note="The RefSeq protein has 1 indel compared to this
genomic sequence; Derived by automated computational
analysis using gene prediction method: BestRefSeq."
/codon_start=1
/product="melanocortin receptor 4"
/protein_id="NP_999338.1"
/db_xref="GI:55741559"
/db_xref="GeneID:397359"
/translation="MNSTHHHGMHTSLHFWNRSTYGLHSNASEPLGKGYSEGGCYEQL FVSPEVFVTLGVISLLENILVIVAIAKNKNLHSPMYFFICSLAVADMLVSVSNGSETI VITLLNSTDTDAQSFTVNIDNVIDSVICSSLLASICSLLSIAVDRYFTIFYALQYHNI MTVKRVGIIISCIWAVCTVSGVLFIIYSDSSAVIICLITVFFTMLALMASLYVHMFLM ARLHIKRIAVLPGTGTIRQGANMKGAITLTILIGVFVVCWAPFFLHLIFYISCPQNPY CVCFMSHFNLYLILIMCNSIIDPLIYALRSQELRKTFKEIICCYPLGGLCDLSSRY" ORIGIN
1 tcacagactc cccaggactt ggattggtca gaaagaagca gaggaggagc cactgtgcac
61 attttttttt ccccttcaca caccataaaa atcacagagg caactaacac tcacagcaaa
121 gcttcaggtt gggaactgat tctctctgcg aggcagctga tctgagcatg cgcacacaga
181 ttcattcttc tcccaatagc acagcagccg ctaggaaaat tattttgaaa agacctgaat
241 gcattaagac taaagttaaa gtggaagtga gaacaaaata tcaaacagca gactcgacag
301 agaatgagcg tcttgaagcc taagatttca aagtgatgct aatcagagcc ctacctgaaa
361 gagactaaaa actccatttc aagcttcgga gcatgtgata tttattcaca acaggcattc
421 caatttcagc ctcataactt tcagacagat aaagacttgg agaaaatcgc tgaggctacc
481 tgacccagga gcttaaatca ggtcagaggg gatctcaacc cacctggcgc aggatgaact
541 caacccatca ccatggaatg catacttctc tccacttctg gaaccgcagc acctacggac
601 tgcacagcaa tgccagtgag ccccttggaa aagagctact ctgaaggagg atgctacgag
661 caactttttg tctctcctga ggtgtttgtg actctgggtg tcataagcct gt
[gap 100 bp] Expand Ns
813 aaacgacg gcgtctctct gaggtgtttg
841 tgactctggg tgtcataagc ctgttggaga acattctggt gattgtggcc atagccaaga
901 acaagaatct gcattcaccc atgtactttt tcatctgtag cctggctgtg gctgatatgc
961 tggtgagcgt ttccaatggg tcagaaacca ttgtcatcac cctattaaac agcacggaca
1021 cggacgcaca gagtttcaca gtgaatattg ataatgtcat tgactcagtg atctgtagct
1081 ccttactcgc ctcaatttgc agcctgcttt cgattgcagt ggacaggtat tttactatct
1141 tttatgctct ccagtaccat aacattatga cagttaagcg ggttggaatc atcatcagtt
1201 gtatctgggc agtctgcacg gtgtcgggtg ttttgttcat catttactca gatagcagtg
1261 ctgttattat ctgcctcata accgtgttct tcaccatgct ggctctcatg gcttctctct
1321 atgtccacat gttcctcatg gccagactcc acattaagag gatcgccgtc ctcccaggca
1381 ctggcaccat ccgccaaggt gccaacatga agggggcaat taccctgacc atcttgattg
1441 gggtctttgt ggtctgctgg gcccccttct tcctccactt aatattctat atctcctgcc
1501 cccagaatcc atactgtgtg tgcttcatgt ctcactttaa tttgtatctc atcctgatca
1561 tgtgtaattc catcatcgat cccctgattt atgcactccg gagccaagaa ctgaggaaaa
1621 ccttcaaaga gatcatctgt tgctatcccc tgggtggcct ctgtgatttg tctagcagat
1681 attaaatggg gacagaggag acttataaat gcaagcataa gagactttct ccttacacag
1741 tctggacaat atgcttcaac aacagcattt tcttgtaagg catcagttga gacattctat
1801 tgtataaatt taagttcgtg attctgctca gtctctgtgt atttttaagg tcttgctacc
1861 ttttggctgt aaaatgttta tctatactac aggttatagg cacaatggat ttataaaaaa 1921 gaaaaaagtc cttatgaaaa gttaattaat gtatcttgtc attcgaaagg atttgacaca 1981 ttgcttgttt tagtaaaatg gaaatcacag tttcattaaa tatatcctaa taaatggttg 2041 ctaatattac actatacaac gctgaagtgt agaggtttga ttctagcatt gaggggagaa 2101 atactgaaac aagtgtttaa tcattaaaaa ataagctgaa atttcaacta atttaataaa 2161 acatgctcat tctccctgtg cagaaggaga aatgaagctt ctactgggag aaaaacagtt 2221 actaaaaaaa agtgggggga tattttgagt ttgaaaacta tgttt
//
2.查找mRNA和氨基酸序列
第一步和查DNA序列的一样,先打开NCBI,得到如下主页。
2.1 点击主页面的“Nucleotide”,得到下面的网页:
2.2 在“Search”栏里,输入“MC4R pig”,然后点击“Search”,得到如下结果:
出现了很多的搜索结果,可以按照自己的需要点击不同的链接,比如我想要查找mRAN 的完全编码序列,我就点击第一个“Sus scrofa MC4R mRNA, complete cds”,得到如下结果:
Sus scrofa MC4R mRNA, complete cds
GenBank: DQ388767.1
FASTA Graphics
Go to:
LOCUS DQ388767 999 bp mRNA linear MAM 15-FEB-2006 DEFINITION Sus scrofa MC4R mRNA, complete cds.
ACCESSION DQ388767
VERSION DQ388767.1 GI:87137928
KEYWORDS .
SOURCE Sus scrofa (pig)
ORGANISM Sus scrofa
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Laurasiatheria; Cetartiodactyla; Suina; Suidae;
Sus.
REFERENCE 1 (bases 1 to 999)
AUTHORS Yang,X.Q., Yu,H. and Liu,D.
TITLE The comparative analysis on MC4R gene of wild boar, domestic pig
and their crossbred
JOURNAL Unpublished
REFERENCE 2 (bases 1 to 999)
AUTHORS Yang,X.Q., Yu,H. and Liu,D.
TITLE Direct Submission
JOURNAL Submitted (05-FEB-2006) Northease Agricultural University, Animal Science & Technology, Xiangfang Block Gongbin Road Mucai Street,
Harbin, Heilongjiang 150030, China
FEATURES Location/Qualifiers
source 1..999
/organism="Sus scrofa"
/mol_type="mRNA"
/db_xref="taxon:9823"
CDS 1..999
/codon_start=1
/product="MC4R"
/protein_id="ABD28176.1"
/db_xref="GI:87137929"
/translation="MNSTHHHGMHTSLHFWNRSTYGLHSNASEPLGKGYSEGGCYEQL FVSPEVFVTLGVISLLENILVIVAIAKNKNLHSPMYFFICSLAVADMLVSVSNGSETI VITLLNSTDTDAQSFTVNIDNVIDSVICSSLLASICSLLSIAVDRYFTIFYALQYHNI MTVKRVGIIISCIWAVCTVSGVLFIIYSDSSAVIICLITVFFTMLALMASLYVHMFLM ARLHIKRIAVLPGTGTIRQGANMKGAITLTILIGVFVVCWAPFFLHLIFYISCPQNPY CVCFMSHFNLYLILIMCNSIIDPLIYALRSQELRKTFKEIICCYPLGGLCDLSSRY"
variation 171
/replace="g"
variation 175
/replace="c"
variation 551
/replace="c"
variation 758
/replace="c"
variation 892
/replace="a"
ORIGIN
1 atgaactcaa cccatcacca tggaatgcat acttctctcc acttctggaa ccgcagcacc
61 tacggactgc acagcaatgc cagtgagccc cttggaaaag gctactctga aggaggatgc
121 tacgagcaac tttttgtctc tcctgaggtg tttgtgactc tgggtgtcat aagcttgttg
181 gagaacattc tggtgattgt ggccatagcc aagaacaaga atctgcattc acccatgtac
241 tttttcatct gtagcctggc tgtggctgat atgctggtga gcgtttccaa tgggtcagaa
301 accattgtca tcaccctatt aaacagcacg gacacggacg cacagagttt cacagtgaat 361 attgataatg tcattgactc agtgatctgt agctccttac tcgcctcaat ttgcagcctg 421 ctttcgattg cagtggacag gtattttact atcttttatg ctctccagta ccataacatt 481 atgacagtta agcgggttgg aatcatcatc agttgtatct gggcagtctg cacggtgtcg 541 ggtgttttgt tcatcattta ctcagatagc agtgctgtta ttatctgcct cataaccgtg 601 ttcttcacca tgctggctct catggcttct ctctatgtcc acatgttcct catggccaga 661 ctccacatta agaggatcgc cgtcctccca ggcactggca ccatccgcca aggtgccaac 721 atgaaggggg caattaccct gaccatcttg attggggtct ttgtggtctg ctgggccccc 781 ttcttcctcc acttaatatt ctatatctcc tgcccccaga atccatactg tgtgtgcttc 841 atgtctcact ttaatttgta tctcatcctg atcatgtgta attccatcat cgatcccctg 901 atttatgcac tccggagcca agaactgagg aaaaccttca aagagatcat ctgttgctat 961 cccctgggtg gcctctgtga tttgtctagc agatattaa
//
由此,就可以得到我们想要的mRNA序列和氨基酸序列。
第二章 PCR引物的设计
1.用NCBI设计PCR引物
打开NCBI的首页,然后点击BLAST,得到如下结果:
点击“Specialized BLAST”中的“Primer-BLAST”,得到如下界面:
如果我要设计扩增mRNA的引物,那么就把mRNA序列输入进去,其他参数可以根据自己
需要进行设定,一般默认就好,然后点击“Get Primers”,得到如下界面:
第三章利用生物软件对RNA的二级结构进行预测
1. 利用生物软件(http://www.genebee.msu.su/services/rna2_reduced.html)对mRNA的二级结构进行预测,打开网址后得到如下网页:
交”,得到如下结果:
第四章 DNA序列、蛋白质序列的BLAST对比分析
1.我就以MC4R的蛋白序列为例,首先打开NCBI,得到如下界面:
点击右边的“BLAST”,得到如下界面:
一般使用“nucleotide blast”和“protein blast”,前者是核酸的对比,后者是蛋白质的对比,我查找的是蛋白质对比,所以点击“protein blast”,得到如下结果:
输入猪MC4R基因编码的蛋白质序列,然后点击BLAST,得到如下界面:
第五章预测分析蛋白质的一级结构、二级结构以及三级结构
一,以猪的以猪的黑素皮质素受体4(MC4R,melanocortin-4 re-ceptor)基因的氨基酸序列为例,通过不同的在线生物分析软件对MC4R编码的蛋白质的一级结构进行分析。
1. 通过ExPASy中的protparam(https://www.360docs.net/doc/549958585.html,/tools/protparam.html)对蛋白质的分子量、等电点进行预测,输入网址后结果如下:
输入氨基酸序列后,点击“Compare parameters”,得到如下结果:
ProtParam
User-provided sequence:
10 20 30 40 50 60 MNSTHHHGMH TSLHFWNRST YGLHSNASEP LGKGYSEGGC YEQLFVSPEV FVTLGVISLL
70 80 90 100 110 120
ENILVIVAIA KNKNLHSPMY FFICSLAVAD MLVSVSNGSE TIVITLLNST DTDAQSFTVN
130 140 150 160 170 180
IDNVIDSVIC SSLLASICSL LSIAVDRYFT IFYALQYHNI MTVKRVGIII SCIWAVCTVS
190 200 210 220 230 240
GVLFIIYSDS SAVIICLITV FFTMLALMAS LYVHMFLMAR LHIKRIAVLP GTGTIRQGAN
250 260 270 280 290 300
MKGAITLTIL IGVFVVCWAP FFLHLIFYIS CPQNPYCVCF MSHFNLYLIL IMCNSIIDPL
310 320 330
IYALRSQELR KTFKEIICCY PLGGLCDLSS RY
Number of amino acids: 332 (氨基酸数目)
Molecular weight: 36946.7 (分子量)
Theoretical pI: 7.13 (等电点)
Amino acid composition:(氨基酸组成)
Ala (A) 19 5.7% Arg (R) 9 2.7% Asn (N) 15 4.5% Asp (D) 9 2.7% Cys (C) 15 4.5% Gln (Q) 6 1.8% Glu (E) 8 2.4% Gly (G) 17 5.1% His (H) 12 3.6% Ile (I) 38 11.4% Leu (L) 39 11.7% Lys (K) 8 2.4% Met (M) 12 3.6% Phe (F) 19 5.7% Pro (P) 9 2.7% Ser (S) 33 9.9% Thr (T) 19 5.7% Trp (W) 3 0.9% Tyr (Y) 15 4.5% Val (V) 27 8.1% Pyl (O) 0 0.0% Sec (U) 0 0.0% (B) 0 0.0% (Z) 0 0.0% (X) 0 0.0%
Total number of negatively charged residues (Asp + Glu): 17
Total number of positively charged residues (Arg + Lys): 17
Atomic composition: (原子组成)
Carbon C 1692
Hydrogen H 2645
Nitrogen N 415
Oxygen O 455
Sulfur S 27
Formula: C1692H2645N415O455S27
Total number of atoms: 5234 (总原子数)
Extinction coefficients: (消光系数)
Extinction coefficients are in units of M-1 cm-1, at 280 nm measured in water. Ext. coefficient 39725
Abs 0.1% (=1 g/l) 1.075, assuming all pairs of Cys residues form cystines Ext. coefficient 38850
Abs 0.1% (=1 g/l) 1.052, assuming all Cys residues are reduced
Estimated half-life: (半衰期)
The N-terminal of the sequence considered is M (Met).
The estimated half-life is: 30 hours (mammalian reticulocytes, in vitro).
>20 hours (yeast, in vivo).
>10 hours (Escherichia coli, in vivo).
Instability index: (不稳定系数)
The instability index (II) is computed to be 46.15
This classifies the protein as unstable.
Aliphatic index: 119.76 (脂肪系数)
Grand average of hydropathicity (GRAVY): 0.765 (总平均亲水性)由以上结果可以知道猪MC4R基因所编码的蛋白质的分子量为36946.7,等电点为7.13,脂肪系数为119.76,总平均亲水性为0.765 ,以及其他的指标。
2.利用ProtScale(https://www.360docs.net/doc/549958585.html,/cgi-bin/protscale.pl)对氨基酸序列做疏水性分析;输入网址后结果如下:
输入MC4R的氨基酸序列,点击“Submit”,得到如下的结果:
ProtScale
User-provided sequence:
10 20 30 40 50 60 MNSTHHHGMH TSLHFWNRST YGLHSNASEP LGKGYSEGGC YEQLFVSPEV FVTLGVISLL
70 80 90 100 110 120
ENILVIVAIA KNKNLHSPMY FFICSLAVAD MLVSVSNGSE TIVITLLNST DTDAQSFTVN
130 140 150 160 170 180
IDNVIDSVIC SSLLASICSL LSIAVDRYFT IFYALQYHNI MTVKRVGIII SCIWAVCTVS
190 200 210 220 230 240 GVLFIIYSDS SAVIICLITV FFTMLALMAS LYVHMFLMAR LHIKRIAVLP GTGTIRQGAN
250 260 270 280 290 300
MKGAITLTIL IGVFVVCWAP FFLHLIFYIS CPQNPYCVCF MSHFNLYLIL IMCNSIIDPL
310 320 330
IYALRSQELR KTFKEIICCY PLGGLCDLSS RY
SEQUENCE LENGTH: 332
Using the scale Hphob. / Kyte & Doolittle, the individual values for the 20 amino acids are: (所用氨基酸标度信息)
Ala: 1.800 Arg: -4.500 Asn: -3.500 Asp: -3.500 Cys: 2.500 Gln: -3.500 Glu: -3.500 Gly: -0.400 His: -3.200 Ile: 4.500 Leu: 3.800 Lys: -3.900 Met: 1.900 Phe: 2.800 Pro: -1.600 Ser: -0.800 Thr: -0.700 Trp: -0.900 Tyr: -1.300 Val: 4.200 : -3.500 : -3.500 : -0.490
Weights for window positions 1,..,9, using linear weight variation model: (分析所用参数信息)
1 2 3 4 5 6 7 8 9
1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00
edge center edge
经蛋白质疏水性预测分析表明,其中间部位则表现出较强的亲水性,按分值大小划分其疏水最大值为3.367,分别位于第197和第253 AA处;最小值为-1.878,位于第311AA 处。
生物信息学软件及使用概述
生物信息学软件及使 刘吉平 liujiping@https://www.360docs.net/doc/549958585.html, 用概述 生 物秀-专心做生物! w w w .b b i o o .c o m
生物信息学是一门新兴的交叉学生物信息学的概念: 科,它将数学和计算机知识应用于生物学,以获取、加工、存储、分类、检索与分析生物大分子的信息,从而理解这些信息的生物学意义。 生 物秀-专心做生物! w w w .b b i o o .c o m
分析和处理实验数据和公共数据,生物信息学软件主要功能 1.2.提示、指导、替代实验操作,利用对实验数据的分析所得的结论设计下一阶段的实验 3.实验数据的自动化管理 4.寻找、预测新基因及其结构、功能 5.蛋白质高级结构及功能预测(三维建模,目前研究的焦点和难点) 生 物秀-专心做生物! w w w .b b i o o .c o m
功能1. 分析和处理实验数据和公共数据,加快研究进度,缩短科研时间 ?核酸:序列同源性比较,分子进化树构建,结构信息分析,包括基元(Motif)、酶切点、重复片断、碱基组成和分布、开放阅读框(ORF ),蛋白编码区(CDS )及外显子预测、RNA 二级结构预测、DNA 片段的拼接; ?蛋白:序列同源性比较,结构信息分析(包括Motif ,限制酶切点,内部重复序列的查找,氨基酸残基组成及其亲水性及疏水性分析),等电点及二级结构预测等等; ?本地序列与公共序列的联接,成果扩大。 生 物秀-专心做生物! w w w .b b i o o .c o m
Antheprot 5.0 Dot Plot 点阵图 Dot plot 点阵图能够揭示多个局部相似性的复杂关系 生 物秀-专心做生物! w w w .b b i o o .c o m
生物信息学数据库或软件
一、搜索生物信息学数据库或者软件 数据库是生物信息学的主要内容,各种数据库几乎覆盖了生命科学的各个领域。 核酸序列数据库有GenBank,EMBL,DDB等,核酸序列是了解生物体结构、功能、发育和进化的出发点。国际上权威的核酸序列数据库有三个,分别是美国生物技术信息中心(NCBI)的GenBank ,欧洲分子生物学实验室的EMBL-Bank(简称EMBL),日本遗传研究所的DDBJ 蛋白质序列数据库有SWISS-PROT,PIR,OWL,NRL3D,TrEMBL等, 蛋白质片段数据库有PROSITE,BLOCKS,PRINTS等, 三维结构数据库有PDB,NDB,BioMagResBank,CCSD等, 与蛋白质结构有关的数据库还有SCOP,CATH,FSSP,3D-ALI,DSSP等, 与基因组有关的数据库还有ESTdb,OMIM,GDB,GSDB等, 文献数据库有Medline,Uncover等。 另外一些公司还开发了商业数据库,如MDL等。
生物信息学数据库覆盖面广,分布分散且格式不统一, 因此一些生物计算中心将多个数据库整合在一起提供综合服务,如EBI的SRS(Sequence Retrieval System)包含了核酸序列库、蛋白质序列库,三维结构库等30多个数据库及CLUSTALW、PROSITESEARCH等强有力的搜索工具,用户可以进行多个数据库的多种查询。 二、搜索生物信息学软件 生物信息学软件的主要功能有: 分析和处理实验数据和公共数据,加快研究进度,缩短科研时间; 提示、指导、替代实验操作,利用对实验数据的分析所得的结论设计下一阶段的实验;寻找、预测新基因及预测其结构、功能; 蛋白高级结构预测。 如:核酸序列分析软件BioEdit、DNAClub等;序列相似性搜索BLAST;多重系列比对软件Clustalx;系统进化树的构建软件Phylip、MEGA等;PCR 引物设计软件Primer premier6.0、oligo6.0等;蛋白质二级、三级结构预测及三维分子浏览工具等等。 NCBI的网址是:https://www.360docs.net/doc/549958585.html,。 Entrez的网址是:https://www.360docs.net/doc/549958585.html,/entrez/。 BankIt的网址是:https://www.360docs.net/doc/549958585.html,/BankIt。 Sequin的相关网址是:https://www.360docs.net/doc/549958585.html,/Sequin/。 数据库网址是:https://www.360docs.net/doc/549958585.html,/embl/。
生物信息学分析方法
核酸和蛋白质序列分析 蛋白质, 核酸, 序列 关键词:核酸序列蛋白质序列分析软 件 在获得一个基因序列后,需要对其进行生物信息学分析,从中尽量发掘信息,从而指导进一步的实验研究。通过染色体定位分析、内含子/外显子分析、ORF分析、表达谱分析等,能够阐明基因的基本信息。通过启动子预测、CpG岛分析和转录因子分析等,识别调控区的顺式作用元件,可以为基因的调控研究提供基础。通过蛋白质基本性质分析,疏水性分析,跨膜区预测,信号肽预测,亚细胞定位预测,抗原性位点预测,可以对基因编码蛋白的性质作出初步判断和预测。尤其通过疏水性分析和跨膜区预测可以预测基因是否为膜蛋白,这对确定实验研究方向有重要的参考意义。此外,通过相似性搜索、功能位点分析、结构分析、查询基因表达谱聚簇数据库、基因敲除数据库、基因组上下游邻居等,尽量挖掘网络数据库中的信息,可以对基因功能作出推论。上述技术路线可为其它类似分子的生物信息学分析提供借鉴。本路线图及推荐网址已建立超级链接,放在北京大学人类疾病基因研究中心网站(https://www.360docs.net/doc/549958585.html,/science/bioinfomatics.htm),可以直接点击进入检索网站。 下面介绍其中一些基本分析。值得注意的是,在对序列进行分析时,首先应当明确序列的性质,是mRNA序列还是基因组序列?是计算机拼接得到还是经过PCR扩增测序得到?是原核生物还是真核生物?这些决定了分析方法的选择和分析结果的解释。 (一)核酸序列分析 1、双序列比对(pairwise alignment) 双序列比对是指比较两条序列的相似性和寻找相似碱基及氨基酸的对应位置,它是用计算机进行序列分析的强大工具,分为全局比对和局部比对两类,各以Needleman-Wunsch 算法和Smith-Waterman算法为代表。由于这些算法都是启发式(heuristic)的算法,因此并没有最优值。根据比对的需要,选用适当的比对工具,在比对时适当调整空格罚分(gap penalty)和空格延伸罚分(gap extension penalty),以获得更优的比对。 除了利用BLAST、FASTA等局部比对工具进行序列对数据库的搜索外,我们还推荐使用EMBOSS软件包中的Needle软件(http://bioinfo.pbi.nrc.ca:8090/EMBOSS/),和Pairwise BLAST (https://www.360docs.net/doc/549958585.html,/BLAST/)。以上介绍的这些双序列比对工具的使用都比较简单,一般输入所比较的序列即可。 (1)BLAST和FASTA FASTA(https://www.360docs.net/doc/549958585.html,/fasta33/)和BLAST (https://www.360docs.net/doc/549958585.html,/BLAST/)是目前运用较为广泛的相似性搜索工具。这两
生物信息学软件使用
生物信息学软件的使用(以MC4R基因为例) 第一章从NCBI上查找DNA、mRNA、蛋白质序列 一、以猪的黑素皮质素受体4(MC4R, melanocortin-4 re-ceptor)基因为例,介绍如何从NCBI 上查找DNA、mRNA、氨基酸序列。 1.首先查找MC4R的DNA序列。 在百度里输入NCBI,打开后得到的结果如下网页: 在Search 栏输入“MC4R pig”,在下拉菜单里选择Gene,然后点击Search,得到如下结果:
点击第一个ID为397359的链接,得到如下的结果:
可以看到该基因位于猪的1号染色体上,在右下方有个“Go to nucleotide”即进入核酸序列,有三种格式(用红圈标记的),经常用的是“FASTA”和“GenBank”,“FASTA”格式的比较简洁,不包含任何的数字,就全部是碱基,序列的对比和分析是就要用到这种格式;而“GenBank”格式就比较详细,可以查看到很多信息,比如碱基数、mRNA序列、内含子、外显子、CDS,以及氨基酸序列等等之类的。点击GenBank后得到如下结果: Sus scrofa breed mixed chromosome 1, Sscrofa10.2 DNA LOCUS NC_010443 2265 bp DNA linear CON 29-SEP-2013 DEFINITION Sus scrofa breed mixed chromosome 1, Sscrofa10.2. ACCESSION NC_010443 REGION: complement(178553488..178555752) GPC_000000583 VERSION NC_010443.4 GI:347618793 DBLINK BioProject: PRJNA28993 Assembly: GCF_000003025.5 KEYWORDS RefSeq. SOURCE Sus scrofa (pig) ORGANISM Sus scrofa Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Laurasiatheria; Cetartiodactyla; Suina; Suidae; Sus. COMMENT REFSEQ INFORMATION: The reference sequence is identical to CM000812.4. On Oct 11, 2011 this sequence version replaced gi:333795951. Assembly Name: Sscrofa10.2 The genomic sequence for this RefSeq record is from the genome assembly released by the Swine Genome Sequencing Consortium as Sscrofa10.2 in August 2011 (see https://www.360docs.net/doc/549958585.html,/Projects/S_scrofa). Sscrofa10.2 is a mixed assembly of clones and contigs from the whole-genome shotgun
启动子生物信息学分析软件
https://www.360docs.net/doc/549958585.html,/seq_tools/promoter.html 2. PlantCARE(plant cis-acting regulatory elements), a database of plant cis-acting regulatory elements http://bioinformatics.psb.ugent.be/webtoo ls/plantcare/html/ 3. promoter 2.0 prediction server http://www.cbs.dtu.dk/services/Promoter/ 4. 启动子分析网址: 1 https://www.360docs.net/doc/549958585.html,/seq_tools/promoter.html 2 http://alggen.lsi.upc.es/recerca/menu_recerca.html 3 http://www.cbs.dtu.dk/services/Promoter/ 4 https://www.360docs.net/doc/549958585.html,/~molb470/ ... s/solorz/index.html 5 https://www.360docs.net/doc/549958585.html,/molbio/proscan/ http://bip.weizmann.ac.il/toolbo ... ters.html#databases https://www.360docs.net/doc/549958585.html,/seq_tools/promoter.html https://www.360docs.net/doc/549958585.html,.sg/promoter/CGrich1_0/CGRICH.htm https://www.360docs.net/doc/549958585.html,/pub/programs.html#pmatch https://www.360docs.net/doc/549958585.html,.hk/~b400559/arraysoft_pathway.html#Promoter http://www.dna.affrc.go.jp/PLACE/signalup.html http://intra.psb.ugent.be:8080/PlantCARE/ http://www.cbs.dtu.dk/services/Promoter/ https://www.360docs.net/doc/549958585.html,/molbio/proscan/ https://www.360docs.net/doc/549958585.html,/molbio/signal/ https://www.360docs.net/doc/549958585.html,/thread-41571-1-1.htm 常用启动子分析网址: http://bip.weizmann.ac.il/toolbox/seq_analysis/promoters.html#databas es
生物信息学常用工具
常用DNA和蛋白质序列数据分析工具: ●序列比对工具: a)BLAST: ●网络比对,包括基础的Blast比对、参数、特殊Blast如PSI-Blast、Blast2 等; ●本地比对,包括程序下载、安装、数据库的下载及格式化、Blast程序的 运行等。 b)多序列比对ClustalX(Windows系统) 包括程序下载、安装、及程序的运行、结果的输入输出等。 ●真核生物基因结构的预测: a)基因可读框的识别: Genescan; CpG岛、转录终止信号和启动子区域预测; CpGPlot; POLYAH; PromoterScan; b)基因密码子偏好性: CodonW; c)采用mRNA序列预测基因: Spidey; d)ASTD数据库 ●分子进化遗传分析工具 ●MEGA;
●Phylip; ●蛋白质结构和功能预测 a)一级结构 ProtParam蛋白质序列理化参数检索; ProtScale蛋白质疏水性分析; COILS卷曲螺旋预测; b)二级结构 PredictProtein蛋白质结构预测; PSIPRED不同蛋白质结构预测方法; c)InterProScan: 模式和序列谱研究 Prosite:蛋白质结构域、家族和功能为点数据库; Pfam:蛋白质家族比对和HMM数据库; BLOCK:模块搜索数据库; SMART:简单模块架构搜索工具; TMHMM:跨膜结构预测工具; d)三级结构 Swiss-Model Workspace: 同源建模的网络综合服务器; Phyre:线串法预测蛋白质折叠; HMMSTR/Rosetta:从头预测蛋白质结构; Swiss-PdbViewer:分子建模和可视化工具; 序列模体的识别和解析; MEME程序包; ●蛋白质谱数据分析
常用生物信息学软件
常用生物信息学软件 一、基因芯片 1、基因芯片综合分析软件。 ArrayVision 7.0 一种功能强大的商业版基因芯片分析软件,不仅可以进行图像分析,还可以进行数据处理,方便protocol的管理功能强大,商业版正式版:6900美元。 Arraypro 4.0 Media Cybernetics公司的产品,该公司的gelpro, imagepro一直以精确成为同类产品中的佼佼者,相信arraypro也不会差。 phoretix? Array Nonlinear Dynamics公司的基因片综合分析软件。 J-express 挪威Bergen大学编写,是一个用JA V A语言写的应用程序,界面清晰漂亮,用来分析微矩阵(microarray)实验获得的基因表达数据,需要下载安装JA V A运行环境JRE1.2后(5.1M)后,才能运行。 2、基因芯片阅读图像分析软件 ScanAlyze 2.44 ,斯坦福的基因芯片基因芯片阅读软件,进行微矩阵荧光图像分析,包括半自动定义格栅与像素点分析。输出为分隔的文本格式,可很容易地转化为任何数据库。 3、基因芯片数据分析软件 Cluster 斯坦福的对大量微矩阵数据组进行各种簇(Cluster)分析与其它各种处理的软件。 SAM Significance Analysis of Microarrays 的缩写,微矩阵显著性分析软件,EXCEL软件的插件,由Stanford大学编制。 4.基因芯片聚类图形显示 TreeView 1.5 斯坦福开发的用来显示Cluster软件分析的图形化结果。现已和Cluster成为了基因芯片处理的标准软件。 FreeView 是基于JA V A语言的系统树生成软件,接收Cluster生成的数据,比Treeview 增强了某些功能。 5.基因芯片引物设计 Array Designer 2.00 DNA微矩阵(microarray)软件,批量设计DNA和寡核苷酸引物工具 三、序列综合分析 V ector NTI Suite 8.0 不喜欢装备各种专业性强的软件,而希望用一个综合性的软件代替的同志可以选择本软件。本阶段的大部分功能它都有。该软件具体特有良好的数据库管理(增加、修改、查找),对要操作的数据放在一个界面相同的数据库中统一管理。软件中的大部分分析可以通过在数据库中进行选定(数据)->分析->结果(显示、保存和入库)三步完成。在分析主界面,软件可以对核酸蛋白分子进行限制酶分析、结构域查找等多种分析和操作,生成重组分子策略和实验方法,进行限制酶片段的虚拟电泳,新建输入各种格式的分子数据、
生物信息学分析报告
目录 1序列信息提取 (2) 2Gene Ontology (GO)功能注释 (2) 2.1序列比对(BLAST) (2) 2.2GO功能条目提取(Mapping) (2) 2.3功能注释(Annotation) (3) 2.4补充注释(Annotation augmentation) (3) 2.5GO功能注释统计 (3) 2.6GO Slim注释与统计 (4) 3KEGG通路注释 (5) 4蛋白质相互作用网络分析 (6) References (8)
1 序列信息提取 原始数据中质谱鉴定成功的蛋白质共计695个,序列信息批量提取自UniProtKB数据库,以FASTA格式保存(2014040152BT76DF0L.fasta)。 2 Gene Ontology (GO)功能注释 基因本体(Gene Ontology) 是一个标准化的基因功能分类体系,提供了一套动态更新的标准化词汇表,并以此从三个方面描述生物体中基因和基因产物的属性:参与的生物过程(Biological Process),分子功能(Molecular Function) 和细胞组分(Cellular Component) 1。 2.1序列比对(BLAST) 我们利用本地化序列比对软件NCBI BLAST+(ncbi-blast-2.2.28+-win32.ext)将鉴定到的蛋白质与 SwissProt Mammals数据库中的蛋白质序列进行比对。根据相似性原理,所得的同源蛋白的功能信息可以用于目标蛋白的功能注释。我们仅保留排名前10条且E-value ≤1e-3的比对序列进行后续的分析(GO.xlsx表中sheet TopBlastHits)。所得的比对相似性范围为36-100% ,其中大部分目标蛋白序列的比对相似性为90% 或以上(图1)。 图1序列比对相似性分布 2.2GO功能条目提取(Mapping) BlastGO2是一个用于基因/蛋白质功能注释和数据分析的应用软件。我们利用Blast2GO(Version 2.7.1)中的Mapping功能对所有鉴定成功的蛋白的比对序列所关联的GO功能条目进行提取,共提取到与其中692个鉴定成功的蛋白序列(99.6%)相关的21,078条GO功能条目。
生物信息学分析
生物信息学分析 人类X染色体图谱(来自国家生物技术信息中心网站)。 生物信息学是一个跨学科的领域,目的是开发理解生物数据的方法和软件工具。生物信息学作为一个跨学科的科学领域,结合了生物学、计算机科学、信息工程、数学和统计学的相关知识用于分析和解释生物数据。通过数学和统计技术,生物信息学已经被用于对生物数据库进行计算机分析。 生物信息学既是生物研究主体的总称,该研究主体使用计算机编程作为其方法论的一部分;也是对重复使用的特定分析“管道”的引用,特别是在基因组学领域。生物信息学的常见用途包括候选基因的鉴定和单核苷酸多态性(SNPs)。通常,这种鉴定的目的是为了更好地理解疾病的遗传基础、独特的适应性、理想的特性(特别是农业物种)或种群间的差异。以一种不太正式的方式,生物信息学也试图理解核酸和蛋白质序列中的组织原则,称为蛋白质组学。 1 介绍 生物信息学已经成为生物学许多领域的重要组成部分。在实验分子生物学中,图像和信号处理等生物信息学技术允许从大量原始数据中提取有用的结果。在遗传学领域,它有助于对基因组及其观察到的突变进行测序和注释。它在生物文献的文本挖掘以及生物和基因本体的发展中起着组织和查询生物数据的作用。它还在基因和蛋白质表达和调
节的分析中发挥作用。生物信息学工具有助于比较遗传和基因组数据,更概括的说,有助于理解分子生物学的进化方面。在更综合的层面上,它有助于分析和编目作为系统生物学重要组成部分的生物路径和网络。在结构生物学中,它有助于对DNA、RNA、[2][3] 蛋白质[4] 以及生物分子间的相互作用进行模拟和建模。[5][6][7][8] 1.1 历史 历史上,生物信息学这个术语和它今天的意义并不一样。波利恩·霍格威和本·海茨帕在1970年创造了这个词,用来指对生物系统中信息过程的研究。[9][10][11] 这一定义将生物信息学定位为一个平行于生物化学(研究生物系统中的化学过程)的领域。[9] 序列 遗传物质序列在生物信息学中经常使用,使用计算机比手工更容易管理。 20世纪50年代初,弗雷德里克·桑格确定胰岛素序列后,蛋白质序列的获取成为可能,计算机成为分子生物学中的关键。手动比较多个序列被证明是不切实际的。这一领域的先驱是玛格丽特·奥克利·戴霍夫。[12] 她编译了第一批蛋白质序列数据库,最初作为书籍出版,[13] 并开创了序列比对和分子进化的方法。[14] 生物信息学的另一个早期贡献者是艾文·卡巴特,他在1970年开创了生物序列分析方
生物信息学工具介绍
生物信息学工具介绍 1、FASTA[10](https://www.360docs.net/doc/549958585.html,/fasta33/)和BLAST[11](http://www.nc https://www.360docs.net/doc/549958585.html,/BLAST/)是目前运用较为广泛的相似性搜索工具。比较和确定某一数据库中的序列与某一给定序列的相似性是生物信息学中最频繁使用和最有价值的操作。本质上这与两条序列的比较没有什么两样,只是要重复成千上万次。但是要严格地进行一次比较必定需要一定的耗时,所以必需考虑在一个合理的时间内完成搜索比较操作。FASTA使用的是Wilbur-Lipman 算法的改进算法,进行整体联配,重点查找那些可能达到匹配显著的联配。虽然FASTA不会错过那些匹配极好的序列,但有时会漏过一些匹配程度不高但达显著水平的序列。使用FASTA和BLAST,进行数据库搜索,找到与查询序列有一定相似性的序列。一般认为,如果蛋白的序列一致性为25-30%,则可认为序列同源。BLAST(Basic Loc al Alignment Search Tool,基本局部联配搜索工具)是基于匹配短序列片段,用一种强有力的统计模型来确定未知序列与数据库序列的最佳局部联配。BLAST 是现在应用最广泛的序列相似性搜索工具,相比FASTA 有更多改进,速度更快,并建立在严格的统计学基础之上。这两个工具都采用局部比对的方法,选择计分矩阵对序列计分,通过分值的大小和统计学显著性分析确定有意义的局部比对。BLAST根据搜索序列和数据库的不同类型分为5种:1、BLASTP是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。 2、BLASTX是核酸序列到蛋白库中的一种查询。先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。 3、BLASTN是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。 4、TBLASTN是蛋白序列到核酸库中的一种查询。与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。 5、TBLASTX是核酸序列到核酸库中的一种查询。此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。另外PSI-BLAST通过迭代搜索,可以搜索到与查询序列相似性较低的序列。其中BLASTN、BLASTP在实践中最为常用,TBLASTN在搜索相似序列
常用生物信息学软件介绍
常用生物学软件简介 1. Oligo 6是目前使用最为广泛的一款引物设计软件,除了可以简单快捷地完成各种引物和探针的设计与分析外,还具有很多其他同类软件所不具有的高级功能: a) 已知一个PCR引物的序列,搜寻和设计另一个引物的序列。b) 按照不同的物种对MM子的偏好性设计简并引物。 c) 对环型DNA片段,设计反向PCR引物。d) 设计多重PCR引物。e) 为LCR反应设计探针,以检测某个突变是否出现。f) 分析和评价用其他途径设计的引物是否合理。 g) 同源序列查找,并根据同源区设计引物。 h) 增强了的引物/探针搜寻手段。设计引物过程中,可以“Lock”每个参数,如Tm 值范围和引物3’端的稳定性等。 i) 以多种形式存储结果;支持多用户,每个用 户可保存自己的特殊设置。 网址: https://www.360docs.net/doc/549958585.html,/ 2. Vector NTI Suite是一套功能最全,而且界面最美观,最友好的分子生物学应用软件包。主要包括四个大型软件,它们分别可以对DNA、RNA、蛋白质分子进行各种分析和操作。Vector⑴ NTI:作为Vector NTI Suite的核心组成部分,它可以在生物研究的全过程中提供数据组织和序列编辑的软件支持。Vector NTI 是以一种窗口形式,且支持项目组织的数据库来完成这一功能的;通过这个数据库,可以保存和组织大部分的实验数据,比如:基因结构、载体、序列片断、引物、蛋白质、多肽、电泳Markers和限制性内切酶等。实际上,该数据库还支持对Vector NTI Suite 中各种小型的绘图和结果展示工具的管理。Vector NTI 可以按照用户要求设计克隆策略。用户只需提供克隆载体,外源片断序列,明确载体克隆的大致位置或酶切位点,其它工作由软件完成。设计结果以图文形式输出到屏幕;最后根据客户定制的条件进行模拟电泳。Vector NTI 还具有强大的设计和评估PCR引物、测序引物和杂交探针功能。BioPlot⑵:BioPlot是一个对蛋白质和核酸序列进行各种理化特性分析的综合性工具,它是一种方便的桌面程序。和其他程序不同的是,BioPlot可以绘制50种以上预定制的蛋白质特征图谱,如疏水性和抗原性;并将序列与特征图谱和活性序列区域一一对应。BioPlot还可以对核酸序列进行8种不同类型的分析,如:退火温度、自由能和GC含量等。AlignX⑶:AlignX可以对多个蛋白质或核酸序列进行同源比较,以寻找不同序列之间的同源区域或相似性很高序列中的不同碱基,并绘制进化树;为下一步设计PCR引物、探针及研究系统发育提供基础。AlignX 可以识别所有标准TXT格式,如FASTA、GeneBank、EMBL、SWISS-PROT、GenPept 和ASCII Text。ContigExpress⑷:Contig Express是用来对多个小核酸片段进行拼接而形成连续的长序列。这些小片段可以是Text序列,也可以是直
生物信息学分析工具
为了使NCBI的资料库发挥更大的进阶应用价值,NCBI研究团队发展许多可以做生物医学资料採矿与资料分析的检索与分析工具。在此依工具的使用目的将其分为六大类,每大类下分别包含工具的名称与简介,作为研究人员在选择工具时的参考。 1.资料检索--文章词语搜寻 ?Entrez一提供核酸、蛋白质、蛋白质3D结构Entrez:提供核酸、蛋白质、蛋白质3D结构、基因体图谱资讯、PubMed MEDLINE 文献等整合式查询。序列资料的来源包括GenBank、EMBL、DDBJ、RefSeq、PIR-International、PRF、Swiss-Prot与PDB(网址:https://www.360docs.net/doc/549958585.html,/Entrez/)。 特性: (1)对每一个资料库纪录做预先的相似性搜寻计算,以鉴别该资料的相关纪录。 (2)提供整合性跨资料库服务,可从一个资料库的纪录连结至其他资料库的相关纪录。 ?Batch Entrez一使使用者可在背景执行,从Entrez取得大量核酸与蛋白质序列资讯,而使用者只需输入含GI或Accession Number 的名单即可。查询结果可直接储存在使用者的电脑中(网址:https://www.360docs.net/doc/549958585.html,/entrez/batchentrez.cgi?db=Nucleotide)。 ?LinkOut一在Entrez的文章、期刊或生物资料建立连结到外部网页连结之注册服务。欲建立连结者可提供网址、资源名称、简短的网页描述与想建立的NCBI资料规格书即可(网址:https://www.360docs.net/doc/549958585.html,/entrez/linkout/doc/linkoutoverview.html)。 ?Cubby一使Entrez使用者储存与更新搜寻,并且订做他们的LinkOut设定。需填写注册申请书申请使用权限(网址 https://www.360docs.net/doc/549958585.html,/entrez/login.fcgi?call=so.SignOn..Login)。 ?Citation Matcher一可查询PubMed 资料库的PubMed ID或MEDLINE UID,提供文献的目录资讯(网址: https://www.360docs.net/doc/549958585.html,/entrez/query/static/overview.html#Citation%20Matcher)。 ?Taxonomy Browser一用来查询生物分类资料库的查询工具,可由生物学名、俗名或较高层级分类查询生物与分类血缘,同时可获得核酸、蛋白质、结构与基因体资讯,并且可向上或向下查询分类树(Taxonomic tree)(网址:https://www.360docs.net/doc/549958585.html,/Taxonomy/)。 2.序列相似度搜寻 ?BLAST一Basic Local Alignment Search Tool一核酸与蛋白质序列比对工具。BLAST网页提供提供BLAST(Basic Local Alignment Search Tool)程式、概述、使用说明与常见问题解答(网址:https://www.360docs.net/doc/549958585.html,/BLAST/)。BLAST程式包括: (1) 核酸BLAST: ?blastn程式一核酸序列比对。 ?MegaBLAST一可搜寻一批EST序列、长序列cDNA或基因体序列。 (2) 蛋白质BLAST: ?blastp程式一蛋白质序列比对。 ?PHI-BLAST程式一Pattern Hit Initiated BLAST(Zhang, et al., 1998) 一输入蛋白质序列查询蛋白质资料库,搜寻是否存在某种特定序列形式的BLAST程式。 ?PSI-BLAST程式一Position-Specific Iterated BLAST(Altschul, et al., 1997) 一输入蛋白质序列查询蛋白质资料库,搜寻是否属于某个蛋白质家族的BLAST程式。 (3)转译BLAST搜寻: ?blastx程式一核酸序列与蛋白质资料库比对。
生物信息学论文完结版
生物信息学论文 学院:生命科学技术学院 专业:生物科学 班级:2013级 老师:高亚梅 学生:蔡欣月 学号:20134083003
链孢霉GH5-1及GH6-3基因生物信息学分析蔡欣月(黑龙江八一农垦大学,生命科学技术学院,2013级生物科学专业,黑龙江省,大庆市) 【摘要】目的:分析和预测链孢霉菌GH5-1和GH6-3基因及其编码蛋白质的结构和特征。方法:利用NCBI、CBS和ExPASy网站中的各种信息分析工具,并结合VectorNTIsuite8.0生物信息分析软件包,分析预测链孢霉菌GH5-1和GH6-3基因并预测该基因编码蛋白结构的特征和功能。结果:GH5-1基因全长2006bp,编码区具有390个氨基酸,在GenBank同源序列中,其与endoglucanase 3 [Neurospora crassa OR74A]基因氨基酸序列一致性达到100%,且有GH5-1保守域。GH5-1蛋白相对分子量预测为41907.4,理论等电点为5.14。预测GH5-1编码蛋白α螺旋(H ) 、β折叠(E )、无规则卷(L )的比例分别是16.92%、33.85%、49.23%,2个GTPase结构域。GH5-1蛋白为亲水蛋白,无跨膜区,有信号肽。GH6-3基因全长1914bp,编码区具有419个氨基酸,在GenBank同源序列中,其与exoglucanase 3 [Neurospora crassa OR74A]基因氨基酸序列一致性达到100%,且有GH6-3保守域。GH6-3蛋白相对分子量预测为44839.3,理论等电点为6.51。预测GH6-3编码蛋白α螺旋(H ) 、β折叠(E )、无规则卷(L )的比例分别是29.59%、16.71%、53.75%,1个GTPase结构域。GH6-3蛋白为亲水蛋白,有跨膜区,无信号肽。结论:成功预测GH5-1和GH6-3基因及其编码蛋白生化及其结构特征,为下一步对其进行克隆和表达奠定基础。 【关键词】链孢霉菌;糖基水解酶家族5(GH5-1);糖基水解酶家族6(GH6-3)生物信息学 链孢霉菌又称脉孢菌、串珠菌、红色面包菌,俗称红霉菌,是食用菌生产中重要的竞争性杂菌之一。其广泛分布在自然界土壤中和和禾本科植物上,尤其在玉米芯上极易发生[1]。通过空气、土壤、腐烂植物、谷物等进行传播、在食用菌生产中,链孢菌和绿菌是生产中最常见的病原菌。链孢霉在高温高湿条件下最易发生,是夏季食用菌生产中危害严重的病原菌,该病原菌生活力强、生长迅速、繁殖快、分生孢子多、易传播,几乎会感染所有熟料栽培的食用菌,并且一旦感染很难彻底消灭,给生产造成较大的经济损失,严重危害所有食用菌的母种、原种、栽培种,以及香菇、木耳、银耳、银耳、灵芝等熟料菌简[2]。目前链孢霉菌的全基因组序列已经获得,但有关其蛋白和基因的各类研究仍为数较少,本文通过对链孢霉GH5-1和GH6-3基因及编码蛋白质进行生物信息学分析,分析其基本生化及结构特征,为下一步对其进行克隆表达和应用奠定基础。 一、材料与方法 1.1材料 通过ExPASy 数据库的UniProtKB(https://www.360docs.net/doc/549958585.html,或https://www.360docs.net/doc/549958585.html,/uniprot)获得链孢霉菌的GH5-1与GH6-3基因序列。GH5-1基因编号为NCU00762,NCBI的登录号为XM_959066.2,其他物种的GH5-1的氨基酸序列均来自Genbank,登录号见表1。GH6-3基因编号为NCU09680,NCBI的登录号为XM_952322.2,其他物种的GH6-3的氨基酸序列均来自Genbank,登录号见表2。 1.2方法 利用美国国家生物技术信息中心(NCBI,https://www.360docs.net/doc/549958585.html,)的基本局部比对搜索工具(BLAST,https://www.360docs.net/doc/549958585.html,/blast/),运用Blastx完成基因同源性分析。 应用ORF finder(https://www.360docs.net/doc/549958585.html,/gorf/orfig.cgi)寻找其开放读码框,并推导出可编码蛋白序列。 利用保守结构域(https://www.360docs.net/doc/549958585.html,/Structure/cdd/wrpsb.cgi)分析预测其保守域。 通过瑞士生物信息学研究所的蛋白分析专家系统(ExPASy,https://www.360docs.net/doc/549958585.html,)所提供的蛋白组学和分析工具:Protparam、Proscale程序分析GH5-1及GH6-3蛋白氨基酸组成、相对分子质量、等电点等基本理化性质;TMHMM程序预测GH5-1及GH6-3的跨膜区;SignalP程序预测GH5-1及GH6-3蛋白的信号肽,
常用的生物信息学软件的介绍和文献依据
常用的生物信息学软件的介绍和文献依据
名称简介参考文献备注 ALINE 一个产生出版质量比对的“所见 即所得”蛋白质-序列比对编辑器 19390156 AMDA 用于自动微阵列数据分析的一个 R包 16824223 AmiGO 访问本体论和注释数据19033274 AnnotationSketch 基因组注释绘图库,基因组特征可 视化 19106120 Arcadia 代谢通路的一个可视化工具,翻译 文本的生物学网络描述为图示 20453003 ArchTEx 下一代测序数据片段的最佳延长 及准确提取和可视化 22302569 ArrayExpress 将ArrayExpress数据集导入到 R/Bioconductor中 19505942 ArrayExpressHTS 用于RNA-seq数据处理和质量 评估的一个流程 21233166 arrayMagic 双色cDNA微阵列质控和预处理15454413 arrayQCplot 用图形分析和统计分析检查微阵 列数据质量的软件 16864592 BALL 生物化学算法库20973958
BALLView 用于分子建模研究和教育的一个 工具 16332707 BamTools 分析和管理BAM文件的一个 C++应用程序接口和工具包 21493652 Batch Blast Extractor 批量Blast提取器:一个自动的 blastx剖析器应用程序 18831775 BayesPeak 分析ChIP-seq数据的一个R包, 峰识别 21245054 BEDTools 比较基因组特征的一套灵活的实 用程序,支持BED,BAM, GFF格式文件 20110278 BEST 结合位点评估工具套件,整合了4 种普遍使用的motif发现程序 15814553 BIGpre 一个下一代测序数据质量评估程 序包 22289480 BiNGO 一个评估基因本体论类别在生物 网络中过代表的Cytoscape插件 15972284 Bio++ 用于序列分析、系统发生学、分子 进化和群体遗传学的一套C++库 16594991 BioCoder 一种标准化及自动化生物学实验 方案的编程语言 21059251