红外光谱分析习题解答

红外光谱分析习题解答
解:影响红外吸收峰强度的主要因素:红外吸收的强度主要由振动能级的跃迁概率和振动过程中偶极矩的变化决定。从基态向第一激
跃迁的概率大,因此基频吸收带一般较强。另外,基频振动过程中偶极矩的变化越大,则其对应的红外吸收越强。因此,如果化学键两
接原子的电负性差异越大,或分子的对称性越差,则伸缩振动时化学键的偶极矩变化越大,其红外吸收也越强,这就是
C=O
的强度大
=C
的原因。一般来说,反对称伸缩振动的强度大于对称收缩振动的强度,伸缩振动的强度大于变形振动的强度。
解:由量子力学可知,简单双原子分子的吸收频率可用下式表示:
μπk
c 21 (1) A
N M M M M )(212
1+ (2)
) 式中:
σ为波数(cm -1),c 为光在真空中的速度(310-10cm S -1),k 为化学键力常数(N cm -1)
) 式中:
M 1和M 2分别为两种原子的摩尔质量,N A 为阿伏加德罗常数(6.021023mol -1
) (2)式代入(1)得
2
1212121)
(1307
)(221M M M M k M M M M k c
N k c A +=+=
πμπ
教材P 153公式(10-6)系数为1370有误】
Cl 键的键力常数
1
2
2
12
12
1.0079
.13453.350079.1453.35130729931307-?+??
???+??? ??cm N M M M M σ
解:依照上题的计算公式
2
1212121)
(1307
)(221M M M M k M M M M k c
N k c A +=+=
πμπ
=9 N cm -1,M H =1.0079,M F =18.998代入可计算得到HF 的振动吸收峰频率为4023cm -1
。
解:2-戊酮的最强吸收带是羰基的伸缩振动(
C=O
),分别在极性溶剂95%乙醇和非极性溶剂正己烷中,其吸收带出现的频率在正己
位于较高处。原因是乙醇中的醇羟基可以与戊酮的羰基形成分子间氢键,导致羰基的伸缩振动频率向低波数方向移动。而正己烷不能与
形成分子间氢键。
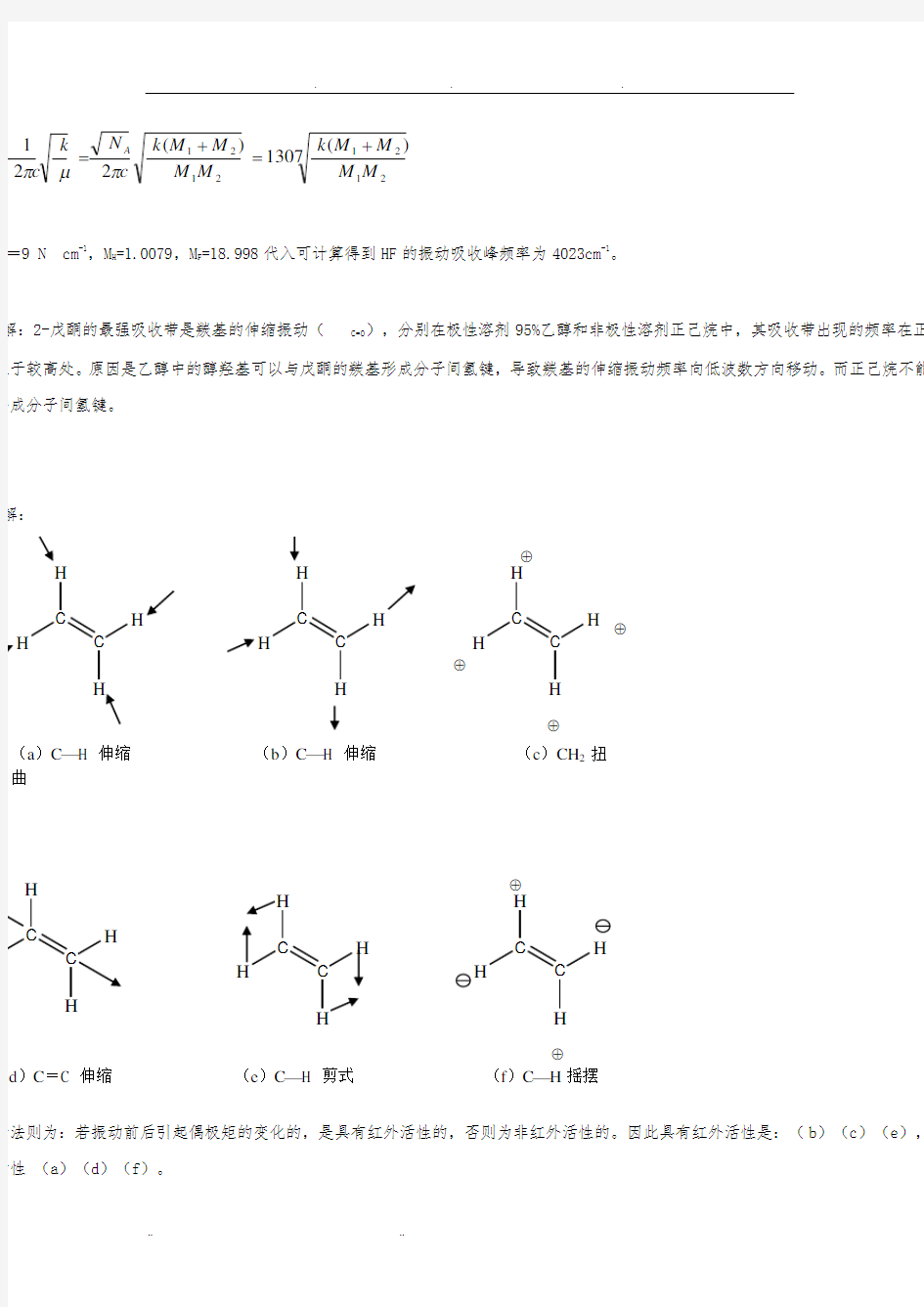
解:
断法则为:若振动前后引起偶极矩的变化的,是具有红外活性的,否则为非红外活性的。因此具有红外活性是:(b )(c )(e ),非
活性 (a )(d )(f )。
⊕
C
C
H
H
H
H
⊕
C
C
H
H
H
C
C
H
H
H
H (d )C =C 伸缩 (e )C ?H 剪式 (f )C ?H 摇摆
C
C
H
H H
H C
C
H
H H
H ⊕
⊕
C
C
H
H
H H
⊕
⊕
(a )C ?H 伸缩 (b )C ?H 伸缩 (c )CH 2扭曲
解:线性分子CS 2,理论上计算其基本振动数为:3N -5=4。其具体振动形式如下:
有红外活性的有:(b )(c )(d ),其中(c )(d )振动吸收的频率一样。
解:根据分子式计算该化合物的不饱和度:
红外吸收峰分别说明:
0 cm -1
=C -H
不饱和化合物,含有双键
0cm -1
C -H
饱和烷基
0cm -1
C=O
共轭的羰基,占有一个不饱和度 0cm -1
C=C
共轭双键,占有一个不饱和度
紫外吸收
max =104
说明,此跃迁是由
产生的。因此可能有如下结构:
(a )对称伸缩振动 (b )反对称伸缩振动
S C S S C S
S
C
S
S C S
(c )面内弯曲振动 (d )面外弯曲振动
2
2
8
015211
34=-++=-+
+=Ωn n n 2C
C C
O
H C
O
或者
(1) (2)
oodward 规则计算:
):母体基数 215nm (2): 母体基数 215nm
-烷基取代0 0nm -烷基取代 1 12nm 计算值 215nm 计算值 227nm
此该化合物为 (2)
解:在影响基团频率的因素中,由于取代基具有不同的电负性,通过静电诱导效应I ,引起分子中电子分布的变化,改变了键的力常
得键或基团的特征频率发生位移。当有电负性较强的元素与羰基上的碳原子相连时,由于诱导效应,就会发生从氧上的电子转移:
Cl 导致C=O 键的力常数变大,因而使的吸收向高波数方向移动。元素的电负性越强,诱导效应越强。吸收峰向高波数移动的程度越显
此,
C O C
CH
C H
C
CH 3
O
R
C
O
个中C =O 伸缩振动频率最高的是:
解:在酸、醛、酯、酰卤和酰胺类化合物中,都有与C =O 相连的含孤对电子基团,它们对C=O 的影响主要是通过诱导和中介这两个
的效应实现的。
当有电负性较强的元素与羰基上的碳原子相连时,由于静电诱导效应I ,使C=O 中氧原子上的电子向碳原子转移,导致C=O 键的力常
大,从而使C=O 吸收向高波数方向移动,并且元素的电负性越强,诱导效应越强,C=O 吸收峰向高波数移动的程度越显著。 中介效应M 源于含孤对电子基团上的孤对电子与C=O 上
电子发生重叠,使它们的电子云密度平均化,造成C=O 键力常数下降,使
吸收频率向低波数移动。
对同一基团来说,若诱导效应I 和中介效应M 同时存在,则振动频率最后位移的方向和程度,取决于这两种效应的净结果。因此,不
其它因素条件影响,在酸、醛、酯、酰卤和酰胺类化合物中,出现C =O 伸缩振动频率的大小顺序为:酰卤酸酯醛酰胺。
、解:
(Ⅰ) (Ⅱ)
较结构(Ⅰ) 和(Ⅱ)可知,3330cm -1
和1600 cm -1
处的锐陡带应分别源于结构(Ⅱ)中的
N -H
和
C=O
。这也可以从该材料在2300 cm
0 cm -1
处无吸收看出,因为2300 cm -1
和3600 cm -1
处的吸收分别对应-C≡N 和-OH 。因此结构(Ⅱ)更可能为该材料的结构组成。
、解:芳环C -H 面外弯曲振动位于900-650cm -1
围。出现12条强吸收带。谱带位置及数目与苯环的取代情况有关,如下表所示,利
围的吸收带可判断苯环上取代基的相对位置。
芳烃的C -H 面外弯曲振动(cm -1
)
C
O
F
F
类型
C -H
5个氢取代(单取代) 750(s ),700(s ) 4个邻接的氢(邻二取代) 770
735(s )
3个邻接的氢(间二取代) 810
750(s ),710690(s ) 2个邻接的氢(对二取代)
860
800(s )
表中s 代表单峰。
此:化合物A 吸收带在767 cm -1
和629 cm -1
处 间位
化合物B 吸收带在792 cm -1
处 对位 化合物C 吸收带在724 cm -1处 邻位
、解:根据苯环中C -H 面外弯曲振动吸收峰在900600 cm -1
区域的特征,C 7H 7Br 在801 cm -1
有一单吸收带,说明苯环为对位取代,则
合物为对溴甲苯。
、解:根据苯环中C -H 面外弯曲振动吸收峰在900600 cm -1
区域的特征,该氯苯在900cm -1
和690 cm -1
间无吸收带,说明苯环上所有
都被氯取代了,因此该化合物为六氯苯。
、解:
CH 2
NH 2
H 3C
C
O
N CH 3
CH 3
3500 cm -1
3400 cm -1
-
NH 2
as
s
叔胺无此带
3030 cm -1
=C -H 氢伸缩振动
2960 cm -1
,2870 cm -1
-CH 3饱和碳氢
as
s
1450 cm -1
,1380 cm -1
-CH 3饱和碳氢
as
s
1600 cm -1-1500 cm -1
苯环骨架伸缩振
动
1650 cm -1
C=O