轮烷类化合物的合成方法研究进展

2011年第31卷
有 机 化 学
V ol. 31, 2011 * E-mail: zllzll@https://www.360docs.net/doc/639481391.html,
Received September 27, 2010; revised November 16, 2010; accepted December 29, 2010.
·综述与进展·
轮烷类化合物的合成方法研究进展
纪奉元 朱亮亮*
(华东理工大学精细化工研究所 结构可控先进功能材料及其制备教育部重点实验室 上海 200237)
摘要 轮烷类互锁分子因其多样的结构和性质, 多年来一直是超分子化学领域的一个热点. 综述了近年来轮烷类化合物的合成及制备方法, 包括用传统的模板法(引入氢键、疏水作用、静电效应、配位和离子诱导等超分子作用)制备轮烷类化合物. 除此之外, 还介绍了利用“Click ”化学、“穿线-收缩”、“穿线-膨胀”、自排序组织和自由基识别等新的合成手段来制备这类化合物. 关键词 超分子; 轮烷; 模板法
Progress on Synthesis of Rotaxane Analogues
Ji, Fengyuan Zhu, Liangliang *
(Key Laboratory for Advanced Materials and Institute of Fine Chemicals , East China University of Science & Technology ,
Shanghai 200237)
Abstract Rotaxane-based interlocked molecular system has become a hot issue in supramolecular chemis-try for years, owing to its various structural architectures and performances. This tutorial review mainly fo-cuses on the synthetic strategies of the rotaxane analogues in recent years. The traditional template-directed strategies of rotaxane preparation including the employment of the supramolecular interaction such as hy-drogen bonding, hydrophobic effect, electrostatic interaction, coordination and ionic template are reviewed, respectively. So me no vel synthetic metho do lo gies, like “click” chemistry, “threading-fo llo wed-by-shrink- ing” and “threading-followed-by-swelling” protocol, self-sorting organization as well as radical recognition, are also introduced.
Keywords supramolecular; rotaxane; template-directed strategies
超分子化学是研究分子与分子之间通过非共价键的弱相互作用(如氢键、疏水相互作用等)缔合形成的复杂而有序的超分子体系的科学. 分子机器与分子器件[1]是基于超分子化学而设计的具有多组分功能性分子系统, 到现在为止仍旧是超分子化学领域的一个研究热点.
自从制造人工芯片成为现代电子技术的重要目标之一以来, 当前普遍采用的“自上而下”的硅光刻技术正在接近其物理尺度上的极限. 制备与发展分子机器和分子开关是从分子水平上拓展芯片制备的有效手段, 即
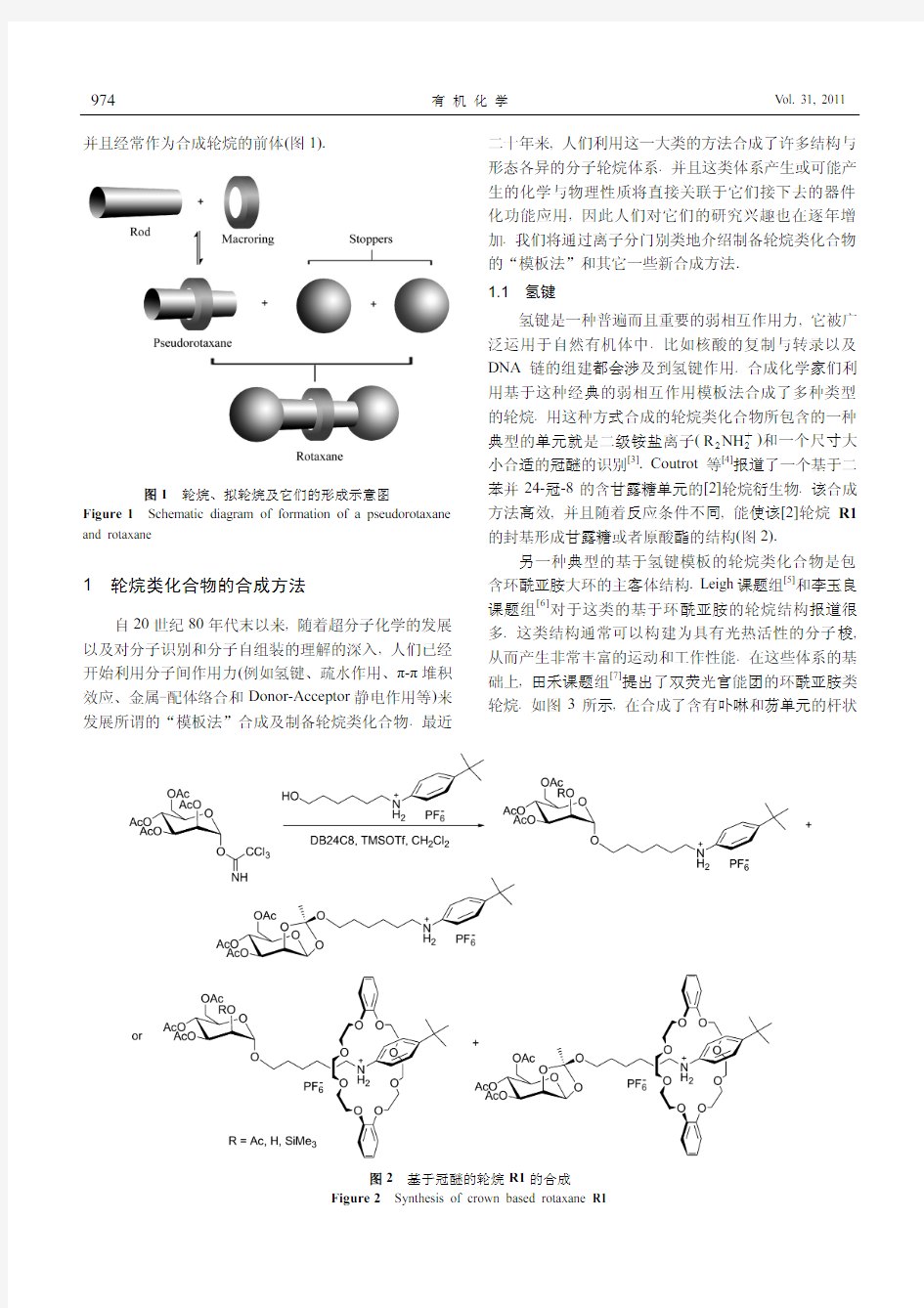
“自下而上”的技术. 轮烷类化合物[2]作为分子机器的一种主要原型, 不仅自身可以体现独特的功能性, 而且可以成为制备应用性分子器件的化学基础. 一个轮烷(rotaxane)结构通常包含一个杆状的分子组分嵌套于一个或者多个环状分子组分, 杆状分子的两侧是大的封基(stopper)以防止环的滑脱. 轮烷的化学性质稳定, 功能比较鲜明. 与之相对的是拟轮烷(pseudorotaxane)结构, 它的杆状分子的两侧没有封基, 因此它的大环可以在一定条件下发生组装-解离运动. 拟轮烷的化学性质活泼, 功能丰富多样. 拟轮烷本身也具有和轮烷相似的性能,
974
有 机 化 学
V ol. 31, 2011
并且经常作为合成轮烷的前体(图
1).
图1 轮烷、拟轮烷及它们的形成示意图
Figure 1 Schematic diagram of formation of a pseudorotaxane and rotaxane
1 轮烷类化合物的合成方法
自20世纪80年代末以来, 随着超分子化学的发展以及对分子识别和分子自组装的理解的深入, 人们已经开始利用分子间作用力(例如氢键、疏水作用、π-π堆积效应、金属-配体络合和Donor-Acceptor 静电作用等)来发展所谓的“模板法”合成及制备轮烷类化合物. 最近
二十年来, 人们利用这一大类的方法合成了许多结构与形态各异的分子轮烷体系. 并且这类体系产生或可能产生的化学与物理性质将直接关联于它们接下去的器件化功能应用, 因此人们对它们的研究兴趣也在逐年增加. 我们将通过离子分门别类地介绍制备轮烷类化合物的“模板法”和其它一些新合成方法. 1.1 氢键
氢键是一种普遍而且重要的弱相互作用力, 它被广泛运用于自然有机体中. 比如核酸的复制与转录以及DNA 链的组建都会涉及到氢键作用. 合成化学家们利用基于这种经典的弱相互作用模板法合成了多种类型的轮烷. 用这种方式合成的轮烷类化合物所包含的一种典型的单元就是二级铵盐离子(22R NH +)和一个尺寸大
小合适的冠醚的识别[3]. Coutrot 等[4]报道了一个基于二苯并24-冠-8的含甘露糖单元的[2]轮烷衍生物. 该合成方法高效, 并且随着反应条件不同, 能使该[2]轮烷R1的封基形成甘露糖或者原酸酯的结构(图2).
另一种典型的基于氢键模板的轮烷类化合物是包含环酰亚胺大环的主客体结构. Leigh 课题组[5]和李玉良课题组[6]对于这类的基于环酰亚胺的轮烷结构报道很多. 这类结构通常可以构建为具有光热活性的分子梭, 从而产生非常丰富的运动和工作性能. 在这些体系的基础上, 田禾课题组[7]提出了双荧光官能团的环酰亚胺类轮烷. 如图3所示, 在合成了含有卟啉和芴单元
的杆状
图2 基于冠醚的轮烷R1的合成
Figure 2 Synthesis of crown based rotaxane R1
N o. 7
纪奉元等:轮烷类化合物的合成方法研究进展
975
Reagents and conditions: (i) 1,1,2,2-tetra-chloride ethylene, 400 K, 50%; (ii) CH 2Cl 2, 254 nm, 30%; (iii) para-xylylene diamine, Et 3N/CHCl 3, 0 ℃, 8%
图3 合成双稳态轮烷Z-R2和E-R2
Figure 3 Synthesis of bistable rotaxane Z-R2 and E-R2
分子之后, 将间苯酰氯和对二苄胺等量匀速地滴入含杆状分子的体系中, 即能在氢键模板诱导下在马来酰亚胺站点上成环. 该轮烷大环上留有炔基, 为分子的进一步修饰提供了条件. 并且它的光热双活性能使大环发生梭动, 从而调节封基上的双荧光[7a]. 1.2 疏水作用
众所周知, 疏水的客体分子在水溶液中容易进入主体大环分子的疏水空腔内部. 环糊精和葫芦脲就是这么一种大环分子, 能够和大量的匹配性化合物通过主客体识别形成包结物. Anderson [8]和Harada 等[9]就利用这种疏水作用的模板法制备了一系列的轮烷和拟轮烷. 偶氮苯和二苯乙烯在水中能够以一个相对较大的结合常数被环糊精包结, 并且它们的光活性能够调节环糊精的选择性包结. 田禾课题组[10,11]制备了许多基于环糊精的光
驱动的轮烷类化合物. 其中的一个例子见图4, 环糊精首先在水中通过疏水作用进入包结中间体A , 再和中间体B 进行Suzuki 偶合反应制备了[2]轮烷R3.[10a] R3的两侧封基是引入两个不同的萘酰亚胺衍生物, 它们具有不同波长的荧光发射性质. 因此这是一类具有双荧光波长输出的轮烷化合物, 适合于分子存储器或分子逻辑门的应用.
图5所示另一个通过环糊精疏水包结作用形成的拟轮烷R4, 它包含一个不对称的具有二茂铁单元和偶氮苯单元的杆状分子[12]. 该杆状分子能够在原位被环糊精快速的包结, 形成[3]拟轮烷R4. 通过可逆的氧化还原和光异构作用, 该主客体复合物的包结计量比和包结站点能够被选择性得控制调节. 由于不同拟轮烷组装体转换态等价于它的诱导圆二色(ICD)信号峰的数目, 因此该系统有望转换为纳米级的计数器元件.
976
有 机 化 学
V ol. 31, 2011
图4 通过疏水作用制备的基于环糊精的[2]轮烷R3
Figure 4 A cyclodextrin (CD)-based [2]rotaxane R3
prepared via hydrophobic effect
图5 [3]拟轮烷R4的制备和双模式(氧化还原和光驱动)组装体的转换
Figure 5 Synthesis of pseudo[3]rotaxane R4 and the interconversion of the dual-driven (redox- and light-driven) ensembles
葫芦脲能够和带正电荷的客体形成高稳定性的包结物[13], 这些客体的正电部分能够通过离子-偶极效应与葫芦脲外围的羰基紧密地识别. 同时葫芦脲的腔体同样具有疏水特性, 能够包结尺度合适的客体. 一个包含二甲氨基苯乙烯和对苯胺氧基链的V 字形分子的合成如图6所示[14]. V 字形的设计可以有效地避免葫芦脲的过量包结, 因此便可高效地形成新颖的[2]拟轮烷R5. 通过pH 值的调节可以控制葫芦脲在两条“分枝”之间
N o. 7 纪奉元等:轮烷类化合物的合成方法研究进展977
的运动转换. 并且, 该运动转换会引起体系吸收波长的较大位移, 以至于可用肉眼观察和区分这种“二进制”转换状态.
1.3 静电作用
Stoddart等[15]开发了一类通过给体-受体静电作用的模板法合成的轮烷类体系. 这类体系的杆状部分通常含有二萘酚(DNP)或者四硫代富瓦烯(TTF)这类给体单元, 以及含有联吡啶鎓环(CBPQT4+)这样的大环受体. 通过给体-受体静电作用结合能够使该主客体结构稳定存在. 图7所示的就是包含了这些单元的[2]轮烷R6的合成[16], 通过给体TTF和联吡啶受体的优先作用成环. 通过TTF的氧化还原能够控制大环在DNP和
TTF 图6基于葫芦脲的拟轮烷R5的形成和构型转换
Figure6Formation and the switching behaviors of the cucurbituril-based pseudorotaxane R5
图7通过给体-受体静电作用合成的[2]轮烷R6
Figure7 Synthesis of [2]rotaxanes R6 donor-acceptor electrostatic interaction
978
有 机 化 学
V ol. 31, 2011
之间梭动. 杆状分子的封基用胆固醇单元代替, 使得这个轮烷分子能够自身堆积形成凝胶状态. 同时杆状分子自身也能形成凝胶. 但有趣的是, 引入大环部分形成轮烷之后可以调节分子本身的堆积状态, 使得整个轮烷结构的堆积螺旋方向恰好和杆状分子的堆积螺旋方向相反. 1.4 配位
Sauvage 等[17]利用过渡金属模板对基元砌块的组织性将配位手段引入到制备轮烷类化合物的方法中来. 他们利用金属配位中心的高度定向性, 提出了金属轮烷的概念. 如图8所示, 杆状分子中包含了一个1,10-菲洛啉和一个三联吡啶的单元, 大环中含有2,2'-联吡啶单元, 能够在加入亚铜离子后形成四元螯合物. 将亚铜离子氧化到铜离子之后, 大环发生梭动与杆上的三联吡啶的单元形成五元螯合物. 这是电化学驱动金属轮烷运动的典型例子.
刘育等[18]设计了在客体分子上引入配位点来完成
分子间组分的连接. 如图9所示, 首先在水环境中将环糊精和4,4'-联吡啶包结形成主客体复合物. 由于吡啶氮和金属离子良好的配位作用, 当加入1 equiv.的Ni(II)之后, 复合物单元能够通过Ni(II)和吡啶氮的配位进行分子间的连接, 从而形成超分子轮烷聚合物R8. 该体系的制备实现了超分子的简单高效放大过程. 1.5 离子模板
化学模板也已经被广泛应用于这类互锁分子系统的合成中. 通过该手段合成的结构有助于发展将来的化学传感器的设计. Leigh 及其合作者[19]通过有效的金属模板合成法制备了很多[2]轮烷结构. 例如, 他们通过钯催化活性金属模板制备的轮烷R9, 如图10所示[20]. 在这里选择了一个合适的钯的配体作为大环分子. 金属钯在这里起到了双重作用: 配位引入杆分子的单元和催化合成共价键. 配合的中间体在二异丙胺中通过合适的炔的衍生物和碘化亚铜的作用下, 以相对较高的产率转化成轮烷R9
.
图8 通过配位合成的金属[2]轮烷R7
Figure 8 A metallo[2]rotaxane R7 synthesized by coordination
N o. 7
纪奉元等:轮烷类化合物的合成方法研究进展
979
图9 通过Ni(II)配位形成的超分子聚合物R8
Figure 9 A supramolecular polymer R8
formed by Ni(II) coordination
图10 钯催化活性金属模板合成轮烷R9
Figure 10 Pd(II)-catalyzed active-metal template synthesis of [2]rotaxane R9
最近, Beer 等[21]综述了通过负电荷的化学模板来构建主客体结构. 他们通过阴离子模板组装制备了一系列机械互锁型轮烷类化合物. 如图11所示, Beer 等[22]首先
合成了一个含有三唑盐的杆状分子. 它能够和卤素离子(Cl -或Br -
)以及异钛酰胺衍生物形成稳定的三元识别体. 再进一步发生环合反应. 离子交换去除阴离子模板后, 形成[2]轮烷R10还可以作为这些阴离子的探针功能使用.
980
有 机 化 学
V ol. 31, 2011
图11 通过阴离子模板组装合成的[2]轮烷R10
Figure 11 Synthesis of [2]rotaxane R10 via the anion template assembly
1.6 其它新颖合成手段
近些年来有关轮烷类化合物的合成方法学研究有了更加深入的进展, 除了传统的“模板法”之外, 又涌现了诸多新型的合成方法. “Click ”反应已经被广泛应用于高效连接形成共价键, 同时也开始被运用于有效制备诸多轮烷类化合物[23]. 裴坚等[24]报道了通过Cu(I)催化的叠氮和炔的环合反应(“Click ”反应)来高效合成[3]轮烷的例子. 图12所示的[3]轮烷R11的大环分子含有三茚并苯作为“天线”基团, 杆上的联苯乙烯作为能量收集核心, 整个结构就形成了互锁的光富集系统. 该化合物的合成是采用“一锅法”, 通过将端基分别带有叠氮和炔的中间体混合, 组装和封头都在一步完成. 这便是运用“Click ”反应有效合成功能性轮烷的例子. 即使在很稀的溶液里也能发生从三茚并苯到联苯乙烯的能量传递, 这也是因为该轮烷独特的拓扑设计能够有效地促进分子内的能量传递过程同时避免分子间的堆积.
“Click ”反应形成的三唑环很多时候也被用来当作大环识别的站点[25,26], 这样就能够进一步丰富轮烷类化合物的运动和功能性. 李玉良课题组[26]合成了一个包含异钛酰胺和环醚结构大环的[2]轮烷R12 (图13). 该轮烷也是通过“Click ”反应完成杆状部分的连接. 这里形成的三唑环能够跟大环的异钛酰胺和环醚部分形成氢
N o. 7
纪奉元等:轮烷类化合物的合成方法研究进展
981
图12 通过Click 反应合成的[3]轮烷R11 Figure 12 [3]rotaxane R11
synthesized by click reaction
图13 以三唑环作为识别站点的[2]轮烷R12
Figure 13 [2]rotaxane R12 based on a 1,2,3-triazole ring as a novel recognition station
982有机化学V ol. 31, 2011
键, 因此可以作为大环识别的有效站点. 通过酸碱控制可以驱动该[2]轮烷的梭动.
传统的方法制备轮烷通常是包含了一个“穿线”过程将杆穿过大环被其包结, 接着再通过“封头”的手段合成或引入封基使其亚单元互锁而防止滑脱. 最近报道了更多新颖地制备轮烷的手段. Asakawa等[27]开发了一种“穿线-收缩”手段, 即包括首先将杆状分子穿入到大环中, 再接着通过金属配位作用减小大环空间(图14). 穿线过程是实现二苄亚胺盐和含萨罗酚的冠醚环的组装, 接着引入醋酸钯和萨罗酚单元发生配位使大环空间收缩. 这样就使得大环和杆互锁, 同时将一个拟轮烷结构转化为轮烷R13.
Chiu等[28]开发了“穿线-膨胀”的手段制备了[2]轮烷R14(图15). 该方法也是先制备拟轮烷, 接着通过使杆上的端基(顺式-1-烷基烯-2-乙烯-环丙烷)发生“膨胀”生成轮烷. 在常温下该端基稳定, 使得拟轮烷结构也能得以分离. 在加热条件下该端基能够通过Cope重排反应迅速生成大的环庚二烯基团, 即可防止大环的滑脱.
自排序组织是选择性地将不同的分子亚单元分别引入超分子结构中不同位置的过程. 近些年人们已将自排序组织方法引入到轮烷类化合物的构建中来[29,30]. 黄飞鹤等[30]运用自排序法将两个不同的AB型二元单体组装成超分子交替轮烷共聚物. 如图16所示, 单体DB24C8是由二苯并24-冠-8和对草快衍生物相连, 而单体BPP34C10是由二苯并34-冠-10和二苄亚胺盐组成. 二苯并24-冠-8对于二苄亚胺盐能够很好
的识别而图14通过“穿线-收缩”手段合成轮烷R13
Figure14 Synthesis of R13 through the threading-followed-by-shrinking protocol
N o. 7
纪奉元等:轮烷类化合物的合成方法研究进展
983
图15 通过“穿线-膨胀”手段合成轮烷R14
Figure 15 Synthesis of R14
through the threading-followed-by-swelling protocol
图16 将二元单体BPP34C10和DB24C8通过自排序组织形成的超分子交替共聚物R15
Figure 16 Formation of supramolecular alternating copolymers R15 form self-sorting organization of heteroditopic monomers BPP34C10 and DB24C8
二苯并34-冠-10和对草快能够形成稳定的包结物. 因此这两个单体能够发生交替的自排序组织, 形成线性的超分子共聚物R15.
Stoddart 等[31]最近创造了一种合成轮烷类化合物的新方法, 即通过含自由基的物种的相互作用来完成主客体的组装(图17). 他们在体系中加入联吡啶鎓环和紫精衍生物, 这两种含正电的化合物原本是彼此排斥的. 当把它们均还原形成带自由基的物种时, 便能够相互吸引地组装在一起. 接着再通过化学反应引入二萘酚和四硫代富瓦烯等单元形成[2]轮烷R16, 该轮烷可以通过电化学控制联吡啶鎓环在二萘酚、四硫代富瓦烯和紫精三个
站点间的梭动. 3 总结与展望
本文描述了目前常用的几种制备轮烷类化合物的模板法, 包括氢键、疏水作用、静电作用、金属配位以及离子模板诱导等. 同时也介绍了几种制备轮烷类化合物的新合成手段, 诸如通过“Click ”反应、“穿线-收缩”、“穿线-膨胀”、自排序组织和自由基识别等. 虽然基于轮烷的合成分子机器经历了二十年的发展, 但它依旧还是超分子化学领域的一个新兴分支, 依然吸引着更多的杰出化学家、物理学家以及工程师投身到其中来, 也不断有更多有关于新类型的报道涌现. 在接下去一段时间里, 人们关于分子机器的研究或会集中在结构合成
984
有 机 化 学
V ol. 31, 2011
图17 通过自由基识别合成轮烷R16
Figure 17 Synthesis of R16 via the radical recognition
方法学和多模式复杂体系的开发上. 超分子化学家和工程师的最终目标是要利用这些合成的轮烷类化合物构建功能性和实用性的分子器件, 服务于人们的日常生活. 相信总会有一天, 基于分子机器开发的高端的科技产品会遍布人们生活的各个角落并发挥着巨大的作用.
References
1 Balzani, V.; Credi, A.; Venturi, M. Molecular Devices and
Machines : Concepts and Perspectives f or the Nanoworld , 2nd ed., Wiley-VCH, Weinheim, Germany, 2008.
2 (a) Tian, H.; Wang, Q.-C. Chem. Soc. Rev. 2006, 35, 361.
(b) Ma, X.; Tian, H. Chem. Soc. Rev . 2010, 39, 70.
3 Horn, M.; Ihringer, J.; Glink, P. T.; Stoddart, J. F. Chem .
Eur. J . 2003, 9, 4046.
4 Coutrot, F.; Busseron, E.; Montero, J.-L. Org. Lett . 2008,
10, 753.
5 Crowley, J. D.; Goldup, S. M.; Lee, A.-L.; Leigh, D. A.;
McBurney, R. T. Chem. Soc. Rev . 2009, 38, 1530.
6 Li, Y.; Li, H.; Li, Y.-L.; Liu, H.-B.; Wang, S.; He, X.-R.;
Wang, N.; Zhu, D.-B. Org. Lett . 2005, 7, 4835.
7 (a) Ji, F.-Y.; Zhu, L.-L.; Ma, X.; Wang, Q.-C.; Tian, H.
Tetrahedron Lett. 2009, 50, 597.
(b) Ji, F.-Y.; Zhu, L.-L.; Zhang, D.; Chen, Z.-F.; Tian, H. Tetrahedron 2009, 65, 9081.
8 Oddy, F. E.; Brovelli, S.; Stone, M. T.; Klotz, E. J. F.; Ca-cialli, F.; Anderson, H. L . J. Mater. Chem. 2009, 19, 2846. 9 Harada, A. J. Org. Chem . 2010, 75, 1040.
10 (a) Qu, D.-H.; Wang, Q.-C; Ren, J.; Tian, H. Org. Lett .
2004, 6, 2085.
(b) Wang, Q.-C.; Qu, D.-H.; Tian, H. Angew. Chem ., Int. Ed . 2004, 43, 2661.
(c) Qu, D.-H.; Wang, Q.-C.; Tian, H. Angew. Chem ., Int. Ed . 2005, 44, 5296.
(d) Zhu, L.-L.; Ma, X.; Ji, F.-Y.; Wang, Q.-C.; Tian, H. Chem. Eur. J . 2007, 13, 9216.
11 (a) Wang, Q.-C.; Ma, X.; Qu, D.-H.; Tian, H. Chem. Eur. J .
2006, 12, 1088.
(b) Zhu, L.-L.; Li, X.; Ji, F.-Y.; Ma, X.; Wang, Q.-C.; Tian, H. Langmuir 2009, 25, 3482.
(c) Zhu, L.-L.; Qu, D.-H.; Zhang, D.; Chen, Z.-F.; Wang, Q.-C.; Tian, H. Tetrahedron 2010, 66, 1254.
12 Zhu, L.-L.; Zhang, D.; Qu, D.-H.; Wang, Q.-C.; Ma, X.;
Tian, H. Chem. Commun . 2009, 46, 2587.
13 (a) Wooklee, J.; Samal, S.; Selvapalam, N .; Kim, H.-J.;
Kim, K. Acc. Chem. Res . 2003, 36, 621.
(b) Lagona, J.; Mukhopadhyay, P.; Chakrabarti, S.; Isaacs, L. Angew. Chem., Int. Ed . 2005, 44, 4844.
(c) Ma, X.; Wang, Q.-C.; Qu, D.-H.; Xu, Y.; Ji, F.-Y.; Tian, H. Adv. Funct. Mater . 2007, 17, 829.
14 Zhang, H.-Y.; Wang, Q.-C.; Liu, M.-H.; Ma, X.; Tian, H.
Org. Lett . 2009, 11, 3234.
15 Fang, L.; Olson, M. A.; Benítez, D.; Tkatchouk, E.;
Goddard III, W. A.; Stoddart, J. F. Chem. Soc. Rev . 2010, 39, 17.
16 Zhao, Y.-L.; Aprahamian, I.; Trabolsi, A.; Erina, N .;
Stoddart, J. F. J. Am. Chem. Soc . 2008, 130, 6348.
17 Collin, J.-P.; Durola, F.; Lux, J.; Sauvage, J.-P. Angew.
Chem., Int. Ed . 2009, 48, 8684.
18 Liu, Y.; Zhao, Y.-L.; Zhang, H.-Y.; Song, H.-B. Angew.
Chem., Int. Ed . 2003, 42, 3260.
19 Crowley, J. D.; Goldup, S. M.; Lee, A.-L.; Leigh, D. A.;
McBurney, R. T. Chem. Soc. Rev. 2009, 38, 1530.
20 Berná, J.; Crowley, J. D.; Goldup, S. M.; H?nni, K. D.; Lee,
A.-L.; Leigh, D. A. Angew. Chem., Int. Ed . 2007, 46, 5709. 21 (a) Lankshear, M. D.; Beer, P. D. Coord. Chem. Rev . 2006,
250, 3142.
(b) Vickers, M. S.; Beer, P. D. Chem. Soc. Rev . 2007, 36, 211.
22 Mullen, K. M.; Mercurio, J.; Serpell, C. J.; Beer, P. D.
Angew. Chem., Int. Ed . 2009, 48, 4875.
23 Zhou, W.; Zheng, H.; Li, Y.; Liu, H.; Li, Y. Org. Lett . 2010,
12, 4078.
24 Wang, J.-Y.; Han, J.-M.; Yan, J.; Ma, Y.-H.; Pei, J. Chem.
N o. 7 纪奉元等:轮烷类化合物的合成方法研究进展985
Eur. J.2009, 15, 3585.
25 Zhao, Y.-L.; Dichtel, W. R.; Trabolsi, A.; Saha, S.; Apra-
hamian, I.; Stoddart, J. F. J. Am. Chem. Soc.2008, 130, 11294.
26 Zheng, H.; Zhou, W.; Lv, J.; Yin, X.; Li, Y.; Liu, H.; Li, Y.
Chem. Eur. J.2009, 15, 13253.
27 Narita, Y. M.; Shimizu, T.; Asakawa, M. J. Am. Chem. Soc.
2004, 126, 16740.
28 Chiu, C.-W.; Lai, C.-C.; Chiu, S.-H. J. Am. Chem. Soc.
2007, 129, 3500. 29 Jiang, W.; Winkler, H. D. F.; Schalley, C. A. J. Am. Chem.
Soc.2008, 130, 13852.
30 Wang, F.; Han, C.-Y.; He, C.-L.; Zhou, Q.-Z.; Zhang, J.-Q.;
Wang, C.; Li, N.; Huang, F.-H. J. Am. Chem. Soc.2008, 130, 11254.
31 Trabolsi, A.; Khashab, N.; Fahrenbach, A. C.; Friedman, D.
C.; Colvin, M. T.; Cotí, K. K.; Benítez,
D.; Tkatchouk,
E.;
Olsen, J.-C.; Belowich, M. E.; Carmielli, R.; Khatib, H. A.;
Goddard III, W. A.; Wasielewski, M. R.; Stoddart, J. F. Nat.
Chem. 2010, 2, 42.
(Y1009271 Ding, W.)
多芳基脒类化合物的合成
多芳基脒类化合物骨架的合成 郭小玲,王继涛,孟凡超,蒋继军 (西北农林科技大学植物保护学院,陕西杨凌712100 ) [摘要]【目的】合成多芳基脒类化合物无骨架;【方法】三头脒骨架:由均苯三酚在氮气保护下以及碳酸钾存在的条件下与对氰基苄溴进行偶联得到;四头脒骨架:由季戊四醇四对甲苯磺酸酯与对氰基苯酚偶联得到;【结果】合成了以苯环为核的三头芳基脒骨架以及以季戊四基为核的四头芳基脒骨架,并且利用重结晶方法纯化了产物,数据表明,所采用的合成方法产率高、纯度好,为下一步的成脒反应以及活性测定奠定了基础。 [关键词] 季戊四醇四对甲苯磺酸酯,1,3,5-三(4-氰基苯甲氧基)苯,季戊四醇四对氰基苯基醚,合成 近年来,人们在活性寡糖的研究中发现,将母体活性寡糖小分子(α-Gal monomer)制成寡聚物(α-Gal polymers)后,其活性有时可大大增加[10,11],但活性提高程度会有所不同。聚合结构中母体寡糖小分子数目的比例越高,其活性提高程度越大。生命科学基础研究结果表明:蛋白质(受体)与配体(ligand,内源性活性物质或者外源性小分子药物)[12]由于几何互补性而靠近,并主要在几何互补作用下,蛋白质构象受到诱导,以匹配性更好的亚稳态构象进行初步结合。继而在调整结合期,氢键、疏水作用等短程精细作用开始表现并最终使二者微扰调整到合适的构象结合,这就是所谓的“诱导契合”[13]。根据多效价效应原理,多效价能够引起一些蛋白受体的聚合,受体聚合后与配体也将产生诱导契合的效果,从而使它们之间结合力增强,表现出多效价效应。多效价配体可和受体上的主要结合点作用,也可和结合亚点作用结合靶标酶的结构信息,利用多效价效应来发现高效、安全的新型农药分子,可看作是一种基于结构的合理设计。 在生物体系中,多位点结合比单位点结合更具优势。首先,多位点结合更牢固,高亲和性的多效价化合物作为一些防治对象靶标部位的抑制剂具有很大的潜在价值。其次,多效价作用能提高受体的选择性。 农药活性的提高很大程度上取决于其对靶标结合程度的增强。有的防治对象可能存在多个作用位点,如一些杀虫剂既有第一作用位点乙酰胆碱酯酶,也有第二作用位点腺苷酸三磷酸酶(A TPase)。在设计聚合物时也可充分考虑这些靶标的特点,从而进行合理的设计,开发出能与多靶标结合的化合物,以提高目标化合物的生物活性[14]。近几年,日本Kagabu Shinzo等报道了以不同链长的亚甲基、烯基、炔基等连接体合成对称的烟碱类化合物的二聚体,并表现出一定的生物活性[15]。 Pang等针对乙酰胆碱酯酶的结构设计了一系列不同链长的二效价他克林聚合物,旨在目标化合物能同时结合乙酰胆碱酯酶的催化位点和外周位点。结果发现当聚合物的链长为7个亚甲基时活性最好,是其母体的1475倍。 芳基二脒类化合物能够实现与DNA小沟的紧密结合而芳基单脒化合物则不能,这也可以利用多效价效应来解释。这容易使我们想到,更多头的芳基脒类是不是同样具有多效价作用而有可能具有更高的生物活性?于是本文在此假设的基础上,决定设计并合成以不同化学结构为核的具有多个芳基脒头的化合物,并研究其生物活性,目标化合物的合成路线见图1。
茚酮类化合物的研究进展
2010年第30卷 有 机 化 学 V ol. 30, 2010 * E-mail: jlliu@https://www.360docs.net/doc/639481391.html, Received November 19, 2009; revised December 25, 3009; accepted February 1, 2010. ·综述与进展· 茚酮类化合物的研究进展 段义杰 刘建利* 王翠玲 (西北大学生命科学学院 西部资源生物与现代生物技术教育部重点实验室 西安 710069) 摘要 茚环结构存在于天然产物、合成药物、农药等分子中. 茚酮作为原料用于生物活性化合物的合成具有很强的工业应用前景. 同时在有机发光材料、染料合成方面也有应用, 还作为可光除去的有机保护基团. 对此类化合物的合成、应用进行了总结, 以促进相关的研究进展. 关键词 茚酮; 合成; 应用 Progress in the Studies of Indanones Duan, Yijie Liu, Jianli * Wang, Cuiling (Key Laboratory of Resource Biology and Biotechnology in Western China , Ministry of Education , School of Life Science , Northwest University , Xi'an 710069) Abstract Indan ring frameworks are ubiquitous in a large number of natural products, bioactive and phar-maceutically interesting molecules. Indanones therefore are very useful molecules as starting building blocks for the synthesis of biologically active compounds and thus are of tremendous industrial interest. It is also very useful in organic light-emitting devices, dyes and photoremovable protecting groups. The synthetic methods and application of this kind of molecules are reviewed in this paper. Keywords indanone; synthesis; application 茚酮的基本结构有1-茚酮、2-茚酮、1,2-茚二酮、1,3-茚二酮、茚三酮(Scheme 1). 其中茚三酮(Ninhydrine)非常有名, 又称水合茚三酮、水合茚满三酮. 茚酮结构广泛存在于天然产物、药物、农药等生物活性分子中, 也是有机发光、光致变色、染料等材料中的结构单元. 因此此类化合物具有广泛的应用前景[1]. 1 天然存在的茚酮及其衍生物 天然存在的茚酮化合物有 100多个, 其中重要的化合物有pterosin P (1), mukagolactone (2)和monachosorin A (3). 这些及相关结构的分子显示出多种生物活性, 例如平滑肌松弛活性、环氧化酶抑制活性等. 从海洋藻青菌中分离的化合物4显示抑制人血管内皮因子生长的 Scheme 1 活性, 在肿瘤血管生成调节方面具有应用前景[2] (Scheme 2). 一个新的茚酮类化合物2,6-dimethyl-1-oxo-4-indan- ecarboxylic acid (5)最近被从植物中分离出来, 虽然它的结构中有一个手性碳, 但该化合物不显示旋光性, 可能
金刚烷胺的合成
金刚烷胺 摘要:金刚烷胺具有药理活性,抗病毒那么,本文以两种方法合成,分别以溴代金刚烷胺为中间产物合成和以金刚醇硝酸酯为中间产物合成。然后,反应生成盐酸金刚烷胺。 关键词:金刚烷胺,盐酸金刚烷胺,合成 金刚烷胺又称金刚胺、三环癸胺,首先是作为抗病毒药使用,后来人们发现其有抗帕金森病作用。 【理化性质】为白色结晶性粉末,味苦,易溶于水或乙醇。 【作用与用途】金刚烷胺能阻断甲型流感病毒脱壳及其核酸释放至呼吸道上皮细胞中;此外,本品尚可能影响已进入细胞的病毒的早期复制。本品作用并无宿主特异性。能阻止病毒进入宿主细胞,影响病毒的脱氢,抑制病毒的复制。其抗病毒谱较窄,对A型流感病毒有明显抑制作用。主要用于禽流感治疗,用药后可明显降低死亡率。 盐酸金刚烷胺为一种对称的三环状胺,可以抑制病毒穿入宿主细胞,并影响病毒的脱壳,抑制其繁殖,起治疗和预防病毒性感染作用。盐酸金刚烷胺为白色结晶性粉末,无臭,味苦。在水或乙醇中易溶,在三氯甲烷中溶解,在丙酮中微溶,同时具有酸溶碱不溶的性质,金刚烷胺抗病毒谱较窄,主要是用于亚洲A型流感的预防,对于B型流感病毒与风疹病毒、麻疹病毒、流行性腮腺炎病毒及单纯疱疹病毒感染均无效。 金刚烷中含有两种碳原予即叔碳原子和仲碳原子,决定了金刚烷化学的反应特性主要通过与这两种碳原予相连的氢原子而体现出来。桥碳原子上氢的反应活性比单纯的叔碳原子上氢的反应活性低,不容易发生取代反应,原因在于由三个不同空间角度的环结构组成的化合物,而金刚烷高度对称的结构使得桥碳原子与相连的三个原子能够形成独特的半平面结构,令sp2杂化成为可能,从而在一定的条件下可以发生取代反应。
合成金刚烷胺的中间产物的合成 溴代金刚烷的合成 卤代反应:在FeS04的H2s04溶液中,金刚烷和溴反应加热到30-40得到85%溴代产物.其中含有97.5%的1.溴代金刚烷。 金刚烷醇硝酸酯的合成 ①向已加入5.4 g(O.04 m01)金刚烷和O.27 g十二烷基苯磺酸钠圆底烧瓶中加入100mL二氯甲烷,搅拌均匀,冰水浴条件下用恒压滴液漏斗向烧瓶中缓慢滴加混酸(将20 mL 98%的浓硫酸加入9.2 mL发烟硝酸中混合而成),约20 min滴加完毕。撤冰水浴,5min之后,改为25℃恒温水浴,反应23 h。 ②停止反应后,在冰浴条件下缓慢向烧瓶中加入尽可能少的冰水并缓慢搅拌,直到没有红棕色气体产生而且底层的深绿色消失,放罨1 h以确定没有酸性气体。旋转蒸发,蒸出二氯甲烷溶剂。 ③将混合物倒入冰水后,直到冰全部融化,过滤得浅黄色固体。或者直接将混合物中加入少量氯化钠和乙酸乙酯溶剂(反应过程中有表面活性剂,加入乙酸乙酯以降低表面张力,从而容易进行分离萃取。待氯化钠溶解完后进行萃取,合并多次萃取的有机层,旋转蒸发后得浅黄色固体物质。 金刚烷胺的合成 1.由溴代金刚烷合成金刚烷胺标准物(工业应用生产金刚烷胺的最主要途径) ①直接固体加热 将109溴代金刚烷与4.5 g尿素混合,直接加热。到180℃时,反应开始,有明显的反应现象,就是突然膨胀,温度猛升至230.240℃。反应结束后,自然降温,加入浓盐酸过量使充分溶解,后加入过量的氢氧化钠,使呈碱性,移入蒸馏罐,进行水蒸气蒸馏,后滤干得金刚烷胺4.0 g。 ②以甲苯为溶剂的氨解反应 为使反应进行的够完全充分,考虑加入甲苯作溶剂,使反应物于液相中进行反应。 于温度182℃条件下回流反应6 h,但反应结果并不理想,转化率只有40%。加入甲苯作溶剂,对反应产生不利的影响,因为甲苯的沸点相对偏低,没有使反应达到所要求的240℃,因此换沸点更高的溶剂对反应将产生什么样的影响有待于作进一步探讨。
酚类化合物
酚类化合物主要来源于石油加工产品,煤焦油,煤液化油,三者中酚类化合物的组成具有很大的相似性。煤焦油,煤液化油中主要的含氧酸性物质即为酚类化合物,其含量受煤种,工艺条件影响很大,低温馏分段中的酚含量较高,质量分数可达30%以上,如此高的酚含量会显著增加后续过程的氢耗量,导致生产成本的增加;此外,酚类化合物的不稳定性不利于油品的存储与运输;酚类化合物作为一种重要的有机中间体和生产原料而被广泛应用到各大领域,因而具有相当大的市场需求和应用价值。然而,我国市场每年的酚类供应都存在较大缺口,随着国家对煤炭资源利用的愈发重视,从煤焦油和煤液化油产品中提取酚类化合物不仅符合国家能源战略的需求,也是挖掘煤焦油和煤液化油的潜在价值。 一、目前获得酚类的方法 酚类物质最初发现于蔬菜,水果,谷物等植物中,如生育酚,儿茶素,白黎芦醇,芝麻林酚,大豆黄素等等,这些天然的酚类化合物大多具有抗氧化性,可以延缓衰老,对于癌症也有一定的抵制作用,所以其医药上的应用潜力越来越得到人们的重视。 煤液化油中提取酚类化合物的原因有一下几点: 1)人们在煤焦油和液化油产品的加工过程中发现,酚类化合物由于其具有特殊的结构特点,会影响油品的安定性[3, 4]、煤液化工艺中的循环溶剂性能[5],因此分离出煤焦油或液化油中的酚类物质将有助于油品的存储,运输,及优化工艺结构。 2)酚类化合物具有弱酸性,是煤焦油液化油中含氧化合物[6]的主要组成部分。在后续加工过程中,高的酚含量将显著增加氢耗量,氢气在合成工业中是一种贵重的原料,这无疑会提升生产的成本。 3)酚类化合物是一种高附加值产品,表1-5 为典型酚类化合物的用途[1],可见酚类化合应用范围非常广,涉及医药、农药、有机合成等等,与人们的生活和工业生产密切相关。从油品中分离酚类化合物将大大增加煤加工产品的附加值,具有很高的经济效益。 4)随着工业的发展,石化能源的消耗带来了巨大的含酚废水排放量[7, 8],是世界上主要的污染物之一,已经严重威胁到人们的生活,健康及安全。由于现行的工艺条件限制,在油品加工过程中会产生的大量含酚废水需要处理,增加生产成本,还会污染环境,与绿色工艺的要求相差甚远,急需对其加以改进。如果能从源头萃取分离出绝大部分的酚类化合物,既不会对后续加工产生负面影响,又能简化工艺流程,
5位氟代的嘧啶化合物的合成
5位氟代的嘧啶化合物的合成 【摘要】具有特定结构和实用价值的5-氟嘧啶类化合物作为一种重要的医药中间体越来越受到人们的关注。本论文主要研究的课题是采用工业原料氟乙酸甲酯,以丙酰氯与其发生亲核取代反应制得丙酰氟乙酸甲酯,再与甲脒乙酸盐缩合,采用适当比例的混合溶剂提取得一系列的5位氟代的嘧啶化合物。用红外光谱仪,熔点测定仪等对所取得的物质进行结构鉴定。 【关键词】氟嘧啶;酰化反应;环合 引言 近年来,5氟嘧啶类化合物作为一种重要的医药中间体越来越受到人们的关注,以它们为基础的5-氟尿嘧啶、伏立康唑等化合物的合成研究也取得了很大的进展。5氟嘧啶环的合成、氟嘧啶C2、C4和C6位的烷基化及其卤代一直是合成上的难点和热点。鉴于嘧啶环的药用活性及氟原子的特殊性质,在嘧啶环上引入氟原子已成为合成新药物的重要考虑因素。本文主要进行的是5位氟代的嘧啶类化合物及其衍生物的合成方法和工艺研究,综合考察各相关反应的产率、反应操作的难易程度和中间体的纯度、性状等各方面因素。 1氟嘧啶类化合物的合成方法 目前5氟嘧啶类化合物的合成方法主要有三类:一类是从氟乙酸乙酯出发,经过一系列反应得到最终产物;另一类是直接对现有的氟嘧啶类化合物进行烷基化、a-卤代烷基化以及基团的取代反应;还有一类就是从2,3,3,3-四氟丙酸甲酯出发,经过一系列反应最终得到产物。具体的合成方法举例如下: 1.1从氟乙酸乙酯出发6-(1-溴乙基)-4-氯-5-氟嘧啶的合成[1][2]。本方法采用工业原料氟乙酸乙酯,以丙酰氯与其发生亲核取代反应制得α-氟丙酰乙酸乙酯(2),再与甲脒乙酸盐缩合,采用适当比例的混合溶剂提取得(3),再用POCl3使化合物(3)的羟基氯化、NBS溴代得目标化合物(1)。合成路线如图1所示: 该合成路线工艺简单、反应成本低、反应条件温和,适合工业化生产,并保持了较高的产率,总收率为41.7%[1]。原料氟乙酸乙酯价廉易得,除第一步反应收率较低以外,其余各步收率较为良好。其合成条件对设备要求不高,只要用脲和胍来替换第二步反应中的脒,就可以分别得到不同的反应中间体及产物。 1.2从氟嘧啶类化合物出发2,4-二氯-6-乙基-5-氟嘧啶的合成[3]。本合成方法以2,4-二氯-5-氟嘧啶为原料,经格氏反应,氧化而得。合成路线如图2所示。本实验的格氏试剂的制备中常用碘或碘钾盐作催化剂,然后采用1,2-二溴乙烷作为反应引发剂。导致本反应对溶剂要求高,使用前需经过严格的纯化,去除其中的过氧化物,水,醇,酸等杂质。有副反应产生,产物纯化较为困难。且反应中的原料2,4-二氯-5-氟嘧啶本就是5位氟代的嘧啶类化合物之一,原料难得昂
恶唑类化合物的合成方法综述
2005届本科毕业(学位)论文河西学院化学系 第一章:噁唑类化合物的合成方法综述 1.引言:根据杂原子在环数目很多。的五元环体系叫唑含有两个杂原子且其中 一个杂原子为N,的化合物是噁唑类化合O3-唑。五元环中杂原子为N、中位置不同,有可分为1,2-唑和1,4)等。(物,其种类较多,有噁唑(1)、噁唑啉(2)、噁唑烷(3)、噁唑酮、苯并噁唑111OOOONNNNH4312ONnNO5 [1]。噁唑类化合物是一类重要的杂环化合物,一些具有噁唑环的化合物具有生物活性[2]。同时它们在中间体、药物合成中也具氨基噁唑具有杀真菌、抗菌、抗病毒作用例如2-[345][6],,。5)是耐高温的高聚物有广泛的用途。分子结构中含有噁唑环的聚苯并噁唑(噁唑(1)是1,3位含有O、N原子的五元环,为有像吡啶一样气味且易溶于水的液体,是非常稳定的化合物,它在热的强酸中很稳定,不发生自身氧化反应,不参与任何的正常的生物化学过程。其二氢和四氢杂环化合物叫做噁唑啉或4,5-二氢唑啉(2)和噁唑烷或四氢噁唑啉(3)。 [1]年确定的,但一向没有人作过大量深入的在1887虽然噁唑环这个名称还是Hantzsch研究,因为这个环系不常见于天然产物中,而且制备也相当困难。直到青霉素的出现,才推动了噁唑的研究。青霉素本身虽没有噁唑环,但它最初是疑为是属于这个环系的。青霉素实际含有一个噻唑环,而噁唑是噻唑的氧的类似物。因为青霉素是一个很重要的药品,研究的范围也由噻唑推广到了噁唑。下面我们就将噁唑类化合物的合成方法进行综述。 2.合成方法 噁唑类化合物可由提供N,O原子的化合物来合成。 页37共页1第 2005届本科毕业(学位)论文河西学院化学系 法合成噁唑环2.1.Cornforth[7] 1947年由。其过程如下:Cornforth等人首次合成第一个含有噁唑环的化合物NHCCHEtO222)HN=CHOCH(CHCHOH + HClHCN + (CH) 23223ClNCCEtO 2HC(OEt)3AcOH)EtOCCH-N=CHOCH(CH2232)CHOCH(CHHCKOEt23加热KO HOOCCEtO2NNN水解喹啉加热CuO, OO O[7]羧酸乙酯的路线如下据此设计合成噁唑-4-。 EtCO 2ClNH2NHEtOCCH N222PrOi Et EtOKHCO2OiPr H57%EtCOEtCO22NN AcOH OiPr OKO34%82% 2.2. 碱催化酰氨基磺酰烯关环合成法[8]-1-苯磺酰烯在碱催化下关环可得到噁 唑化合物。用3-酰氨基-2-碘HNPhSOMeNNH2Na IPhSONaOH22PhOSHCMeMe 22aq ,EtAc hv 80℃℃THF 0HOOOI94%38%
抗糖尿病药物合成进展
抗糖尿病药物合成进展 摘要:糖尿病是一种病因复杂的慢性疾病,严重威胁着人体健康,其患病率呈逐年上升趋势,治疗糖尿病已成为全球性的重大公共卫生问题。研究者不断开发研制新型、有效、安全的治疗药物,现研究的抗糖尿病化合物主要分为化学合成类药物包括胰岛素增敏剂、胰岛素促分泌素、肠促胰岛素、钠-葡萄糖共转运蛋白2抑制剂、α-葡萄糖苷酶抑制剂、胰岛淀粉样多肽类似物,以及一些对天然活性物质改性的药物。本文就目前开发的抗糖尿病化合物进行了分类,简单阐述了其作用机制与合成方法,并提出今后抗糖尿病药物研究发展趋势。 关键词:糖尿病;降糖药;合成;机理 Synthesis and Advances in Anti-diabetes Drugs Abstract:As a chronic disease, diabetes threatens human health seriously. The prevalence of diabetes is increasing annually, and the treatment of diabetes has become a major global public health problem.Researchers develop new, effective and safe therapeutic drugs constantly, the synthetic antidiabetic drugs includes insulin sensitizers, insulin secretagogues, incretins,sodium - glucose co-transporter 2 inhibitors, á-glucosidase inhibitors, amylin analogs and some of the natural active substances modified drugs. In this paper, the current study of anti-diabetic compounds were classified, and briefly described its mechanism of action and synthesis methods, put forward the development trend of the future anti-diabetic drug research . Keywords: diabetes; hypoglycemic drugs; syntheses;mechanism 1.引言 糖尿病是一种内分泌代谢性型疾病,由体内胰岛素绝对或相对不足所致。近年来,糖尿病的患病率逐年增加,2015年全球患糖尿病的人口约为4.15亿,预计2040年将会增至6.42亿[1]。如果不能积极有效地对糖尿病进行治疗,极易引发诸多威胁糖尿病病人的生命安全的并发症,因此,研制出安全、高效抗糖尿病药物来维持正常人体血糖水平对于预防糖尿病及其并发症意义重大。 1.1糖尿病分类 临床上将糖尿病划分为两类,一类是I型糖尿病,另一类Ⅱ型糖尿病。前者为胰岛素依赖型,是胰岛β细胞受到破坏,血浆胰岛素水平低于正常值从而导致高血糖的疾病类型。这类糖尿病约占所有糖尿病患者10%,其治疗只能依赖于外源性胰岛素,抗病药物研究方向是研发给药方便、安全有效的胰岛素制剂及替代品[2];后者为非胰岛素依赖型,是由于胰岛素分泌相对缺乏及胰岛素作用环节不健全所致血糖升高的疾病类型[3]。这类疾病的特征是胰岛β细胞功能恶化,肝脏、骨骼肌、脂肪组织对胰岛素的敏感性逐渐降低。目前,大部分糖尿病患者为Ⅱ型糖尿病,占全世界总糖尿病病例的90%以上[4]。对于II 型糖尿病,治疗关键在于开发促使胰岛β细胞分泌更多胰岛素,改善机体对胰岛素敏感性的化学药物。 1.2糖尿病发病机理
【CN109912606A】一种嘧啶并吲唑类化合物的合成方法【专利】
(19)中华人民共和国国家知识产权局 (12)发明专利申请 (10)申请公布号 (43)申请公布日 (21)申请号 201910303026.6 (22)申请日 2019.04.16 (71)申请人 新乡医学院 地址 453003 河南省新乡市红旗区金穗大 道601号新乡医学院 (72)发明人 高庆贺 邱培勇 刘兆敏 杨利敏 吴曼曼 (74)专利代理机构 新乡市平原智汇知识产权代 理事务所(普通合伙) 41139 代理人 路宽 (51)Int.Cl. C07D 487/04(2006.01) (54)发明名称一种嘧啶并吲唑类化合物的合成方法(57)摘要本发明公开了一种嘧啶并吲唑类化合物的合成方法,属于有机合成技术领域。本发明的技术方案要点为:一种嘧啶并吲唑类化合物的合成方法,具体步骤为:将芳香醛类化合物、3-氨基吲唑类生物和三乙胺溶于溶剂中,再加入NH 4I和氧化剂,然后于110-150℃反应制得目标产物嘧啶并吲唑类化合物。本发明合成过程简单高效,通过无过渡金属催化的一锅串联反应一步直接制得嘧啶并吲唑类化合物,避免了由于多步反应中多种试剂的使用以及对各步反应中间体的纯化处理等引起的资源浪费和环境污染,合成过程操作方便,原料简单,反应条件温和,底物适用范围广,同时以三乙胺为原料极大地降低了生产成 本。权利要求书2页 说明书18页CN 109912606 A 2019.06.21 C N 109912606 A
1.一种嘧啶并吲唑类化合物的合成方法,其特征在于具体步骤为:将芳香醛类化合物1、3-氨基吲唑类化合物2和三乙胺溶于溶剂中,再加入NH 4I和氧化剂,然后于110-150℃反应制得目标产物嘧啶并吲唑类化合物3, 该合成方法中的反应方程式为: 其中R 1为苯基、取代苯基、2-萘基、1-萘基、吡啶基或噻吩基,该取代苯基为3,4,5-三甲氧基苯基、3,4-二甲基苯基、2,4-二氯基苯基或一元取代苯基,一元取代苯基苯环上的取代基为甲基、叔丁基、甲氧基、氟、氯、溴、三氟甲基或硝基,R 2为氢、甲氧基、氟、氯、溴、碘或硝基,溶剂为氯苯、甲苯、1,4-二氧六环、乙腈、N -甲基-2-吡咯烷酮或N ,N -二甲基甲酰胺,氧化剂为二叔丁基过氧化物、过氧化二异丙苯、过氧化苯甲酰、过氧化苯甲酸叔丁酯、二甲亚砜、氧气或空气。 2.根据权利要求1所述的嘧啶并吲唑类化合物的合成方法,其特征在于:所述芳香醛类化合物1、3-氨基吲唑类化合物2、三乙胺、NH 4I与氧化剂的投料摩尔比为1:1:2.5:1.5:3,芳香醛类化合物1与溶剂的投料配比为1mmol:4mL。 3.根据权利要求1所述的嘧啶并吲唑类化合物的合成方法,其特征在于:所述氧化剂为二叔丁基过氧化物、过氧化二异丙苯、过氧化苯甲酰、过氧化苯甲酸叔丁酯、二甲亚砜或氧气时,合成过程在密封条件下进行;所述氧化剂为空气时,合成过程在密封或敞开条件下进行。 4.一种嘧啶并吲唑类化合物的合成方法,其特征在于具体步骤为:将苯甲醛1a、3-氨基吡唑并吡啶类化合物4和三乙胺溶于溶剂氯苯中,再加入NH 4I,然后于120℃反应制得目标产物吡啶并吡唑并嘧啶类化合物5, 该合成方法中的反应方程式为: 其中R 3为氢或甲基。 5.根据权利要求4所述的嘧啶并吲唑类化合物的合成方法,其特征在于:所述苯甲醛1a、3-氨基吡唑并吡啶类化合物4、三乙胺与NH 4I的投料摩尔比为1:1:2.5:1.5,苯甲醛1a与溶剂氯苯的投料配比为1mmol:4mL。 6.根据权利要求1或4所述的嘧啶并吲唑类化合物的合成方法,其特征在于:所述嘧啶并吲唑类化合物为下列化合物之一: 权 利 要 求 书1/2页2CN 109912606 A
轮烷类化合物的合成方法研究进展
2011年第31卷 有 机 化 学 V ol. 31, 2011 * E-mail: zllzll@https://www.360docs.net/doc/639481391.html, Received September 27, 2010; revised November 16, 2010; accepted December 29, 2010. ·综述与进展· 轮烷类化合物的合成方法研究进展 纪奉元 朱亮亮* (华东理工大学精细化工研究所 结构可控先进功能材料及其制备教育部重点实验室 上海 200237) 摘要 轮烷类互锁分子因其多样的结构和性质, 多年来一直是超分子化学领域的一个热点. 综述了近年来轮烷类化合物的合成及制备方法, 包括用传统的模板法(引入氢键、疏水作用、静电效应、配位和离子诱导等超分子作用)制备轮烷类化合物. 除此之外, 还介绍了利用“Click ”化学、“穿线-收缩”、“穿线-膨胀”、自排序组织和自由基识别等新的合成手段来制备这类化合物. 关键词 超分子; 轮烷; 模板法 Progress on Synthesis of Rotaxane Analogues Ji, Fengyuan Zhu, Liangliang * (Key Laboratory for Advanced Materials and Institute of Fine Chemicals , East China University of Science & Technology , Shanghai 200237) Abstract Rotaxane-based interlocked molecular system has become a hot issue in supramolecular chemis-try for years, owing to its various structural architectures and performances. This tutorial review mainly fo-cuses on the synthetic strategies of the rotaxane analogues in recent years. The traditional template-directed strategies of rotaxane preparation including the employment of the supramolecular interaction such as hy-drogen bonding, hydrophobic effect, electrostatic interaction, coordination and ionic template are reviewed, respectively. So me no vel synthetic metho do lo gies, like “click” chemistry, “threading-fo llo wed-by-shrink- ing” and “threading-followed-by-swelling” protocol, self-sorting organization as well as radical recognition, are also introduced. Keywords supramolecular; rotaxane; template-directed strategies 超分子化学是研究分子与分子之间通过非共价键的弱相互作用(如氢键、疏水相互作用等)缔合形成的复杂而有序的超分子体系的科学. 分子机器与分子器件[1]是基于超分子化学而设计的具有多组分功能性分子系统, 到现在为止仍旧是超分子化学领域的一个研究热点. 自从制造人工芯片成为现代电子技术的重要目标之一以来, 当前普遍采用的“自上而下”的硅光刻技术正在接近其物理尺度上的极限. 制备与发展分子机器和分子开关是从分子水平上拓展芯片制备的有效手段, 即 “自下而上”的技术. 轮烷类化合物[2]作为分子机器的一种主要原型, 不仅自身可以体现独特的功能性, 而且可以成为制备应用性分子器件的化学基础. 一个轮烷(rotaxane)结构通常包含一个杆状的分子组分嵌套于一个或者多个环状分子组分, 杆状分子的两侧是大的封基(stopper)以防止环的滑脱. 轮烷的化学性质稳定, 功能比较鲜明. 与之相对的是拟轮烷(pseudorotaxane)结构, 它的杆状分子的两侧没有封基, 因此它的大环可以在一定条件下发生组装-解离运动. 拟轮烷的化学性质活泼, 功能丰富多样. 拟轮烷本身也具有和轮烷相似的性能,
吡啶并嘧啶类化合物的合成研究进展_任青云
2005年第25卷有机化学V ol. 25, 2005第12期, 1530~1541 Chinese Journal of Organic Chemistry No. 12, 1530~1541 * E-mail: he1208@https://www.360docs.net/doc/639481391.html, Received November 16, 2004; revised March 3, 2005; accepted April 20, 2005. 吡啶并[2,3-d]嘧啶类化合物的合成 吡啶并[2,3-d]嘧啶及其氧代衍生物具有潜在的生物 学和药理学活性, 该类化合物是人们合成和研究得最多
No. 12 任青云等:吡啶并嘧啶类化合物的合成研究进展 1531 的一类吡啶并嘧啶类衍生物. 综合近二十年来各类文献, 其合成方法主要分为两大类: (1)从吡啶环出发关环; (2)从嘧啶环出发关环. 本文即依此作为此类化合物合成的主要分类依据. 1.1 从吡啶环出发. 1.1.1 含α,ω-二腈的吡啶环在卤化氢作用下关环 在有机合成反应中α,ω-二腈在卤化氢作用下环化反应历来是制备杂环化合物的一条有效途径[20](Eq. 1). 1995年, Victory 等[21]利用该反应成功合成了一系列吡啶并[2,3-d ]嘧啶化合物, 并发现随卤化氢酸性不同而生成不同的化合物. 如Scheme 1, 当HX 为氯化氢时生成化合物1, 当HX 为溴化氢或碘化氢时则同时生成两种异构体1和2. 后来, 发现不同溶剂对反应的选择性也有很大影响, 如采用甲苯作溶剂, 加热或室温条件下分别与氯化氢或溴化氢反应, 结果都只得到一种关环产物1, 且收率也有明显的提高, 当HX 为溴化氢时1的收率大于 75%. Scheme 1 1.1.2 由氨基烟碱腈在酸或碱作用下关环 1988年, Hosmane 等 [22] 报道由2-氨基烟碱腈与原甲 酸三甲酯在催化剂三氟乙酸作用下生成N -(3-氰基吡啶基-2)-甲脒(3), 再与稍过量的甲基肼反应, 关环生成4-β-甲基肼基吡啶并[2,3-d ]嘧啶(4), 收率为46%. 后来发现在适当的条件下, 氨和肼一样能使甲脒关环(Eq. 2). 采用原甲酸三甲酯生成脒中间体再与各种亲核小分子关环, 这是制备杂环化合物的一条重要途径, 该方法经过改进后在合成步骤与收率等方面均有很大的提高, 在 烟碱腈亦可与盐酸胍在丁醇钠催化下发生Michael 加成关环, 生成吡啶并[2,3-d ]嘧啶化合物5, 收率为61%[23] (Eq. 3). 1997年, Quintela 等[24]合成了具有抗组胺活性的吡啶并[2,3-d ]嘧啶类化合物8, 采用氨基吡啶6与富电碳原子合成子N ,N -二甲基二氯亚甲基亚胺氯(7)反应, 经分子内关环得到目标产物, 收率为80%~90% (Scheme 2). 该方法的特点是反应活性高、收率好, 反应中提到的富电合成子亦可以应用到其他相关合成反应当中 . Scheme 2 2001年, Kumar 等[25]采用硫脲与氨基烟碱腈加热反应, 得到4-氨基-5,7-二取代吡啶并[2,3-d ]嘧啶-2-(1H )-硫酮(9) (Eq. 4), 该系列化合物均具有一定的杀菌活性 .
恶唑类化合物的合成方法综述
恶唑类化合物的合成方法 综述 Prepared on 22 November 2020
第一章:恶唑类化合物的合成方法综述 1.引言: 含有两个杂原子且其中一个杂原子为N的五元环体系叫唑,数目很多。根据杂原子在环中位置不同,有可分为1,2-唑和1,3-唑。五元环中杂原子为N、O的化合物是恶唑类化合物,其种类较多,有恶唑(1)、恶唑啉(2)、恶唑烷(3)、恶唑酮、苯并恶唑(4)等。 恶唑类化合物是一类重要的杂环化合物,一些具有恶唑环的化合物具有生物活性[1]。例如2-氨基恶唑具有杀真菌、抗菌、抗病毒作用[2]。同时它们在中间体、药物合成中也具有广泛的用途[3,4,5]。分子结构中含有恶唑环的聚苯并恶唑(5)是耐高温的高聚物[6]。 恶唑(1)是1,3位含有O、N原子的五元环,为有像吡啶一样气味且易溶于水的液体,是非常稳定的化合物,它在热的强酸中很稳定,不发生自身氧化反应,不参与任何的正常的生物化学过程。其二氢和四氢杂环化合物叫做恶唑啉或4,5-二氢唑啉(2)和恶唑烷或四氢恶唑啉(3)。 虽然恶唑环这个名称还是Hantzsch在1887[1]年确定的,但一向没有人作过大量深入的研究,因为这个环系不常见于天然产物中,而且制备也相当困难。直到青霉素的出现,才推动了恶唑的研究。青霉素本身虽没有恶唑环,但它最初是疑为是属于这个环系的。青霉素实际含有一个噻唑环,而恶唑是噻唑的氧的类似物。因为青霉素是一个很重要的药品,研究的范围也由噻唑推广到了恶唑。下面我们就将恶唑类化合物的合成方法进行综述。 2.合成方法 恶唑类化合物可由提供N,O原子的化合物来合成。 法合成恶唑环 1947年由Cornforth等人首次合成第一个含有恶唑环的化合物[7]。其过程如下:
酚类化合物
酚类化合物 (一)主要化合物及其食物来源 酚类化合物包括了一类有益健康的化合物,其共同特性是分子中含有酚的基团,因而具有较强的抗氧化功能。根据分子组成的不同,植物性食物中的酚类化合物分为简单酚、酚酸、羟基肉桂酸衍生物及类黄酮。常见的酚类化合物有: 1.简单酚又称一元苯酚,如水果中分离出的甲酚、芝麻酚、桔酸(gallicacid)。 2.酚酸主要有香豆酸(coumaricacid)、咖啡酸(caffeicacid)、阿魏酸(ferulicacid) 和绿原酸(chlorogenicacid)等。 3.类黄酮(flavonoids),又称黄酮类化合物,包括黄酮、槲皮素、黄酮醇、黄烷醇、黄烷酮等。 4.异黄酮异黄酮广泛存在于豆科植物中,黄豆中所含异黄酮有:染料木苷元(三羟基异黄酮,又称金雀异黄素)、大豆苷元(二羟基异黄酮)、大豆苷、染料木苷、大豆黄素苷以及上述三种苷的丙二酰化合物。 5.茶多酚主要由5种单体构成,分别是表没食子儿茶素一没食子酸酯(EGCG)、表没食子儿茶素(EGC)、表儿茶素一没食子酸酯(ECG)、儿茶素(CA)和表儿茶素(EC)。其中,EGCG的含量最高,被认为是茶多酚生物学活性的主要来源。(二)生物学作用 酚类化合物与人体健康关系的研究多集中在槲皮素、大豆异黄酮、茶多酚的生物学作用方面。现将其主要的保健功能综述如下: 1.抗氧化作用植物中所含的多酚化合物是重要的抗氧化剂,可以保护低密度脂蛋白免受过氧化,从而防止动脉粥样硬化和体内过氧化反应的致癌作用。 2.血脂调节功能大豆异黄酮可以降低胆固醇,含这种成分的大豆蛋白可使动物的低密度脂蛋白和极低密度脂蛋白以及胆固醇降低30%~40%。茶多酚可减少肠内胆固醇的吸收,降低血液胆固醇,降低体脂和肝内脂肪聚积。 3.血管保护作用红葡萄酒中的多酚化合物可抑制血小板的活性,从而抑制血栓的形成,并可使已形成的血栓血小板解聚;还可促进血管内皮细胞分泌产生舒血管因子,减轻栓塞性心血管病的发生。因此,红葡萄酒所含这类化合物成分的摄入量与冠心病、心肌梗死等的发病率呈负相关关系。
恶唑类化合物的合成方法综述
第一章:噁唑类化合物的合成方法综述 1.引言: 含有两个杂原子且其中一个杂原子为N 的五元环体系叫唑,数目很多。根据杂原子在环中位置不同,有可分为1,2-唑和1,3-唑。五元环中杂原子为N 、O 的化合物是噁唑类化合物,其种类较多,有噁唑(1)、噁唑啉(2)、噁唑烷(3)、噁唑酮、苯并噁唑(4)等。 N O N O NH O 1 111 2 34 N O N O N O n 5 噁唑类化合物是一类重要的杂环化合物,一些具有噁唑环的化合物具有生物活性[1]。例如2-氨基噁唑具有杀真菌、抗菌、抗病毒作用[2]。同时它们在中间体、药物合成中也具有广泛的用途[3,4,5]。分子结构中含有噁唑环的聚苯并噁唑(5)是耐高温的高聚物[6]。 噁唑(1)是1,3位含有O 、N 原子的五元环,为有像吡啶一样气味且易溶于水的液体,是非常稳定的化合物,它在热的强酸中很稳定,不发生自身氧化反应,不参与任何的正常的生物化学过程。其二氢和四氢杂环化合物叫做噁唑啉或4,5-二氢唑啉(2)和噁唑烷或四氢噁唑啉(3)。 虽然噁唑环这个名称还是Hantzsch 在1887[1]年确定的,但一向没有人作过大量深入的研究,因为这个环系不常见于天然产物中,而且制备也相当困难。直到青霉素的出现,才推动了噁唑的研究。青霉素本身虽没有噁唑环,但它最初是疑为是属于这个环系的。青霉素实际含有一个噻唑环,而噁唑是噻唑的氧的类似物。因为青霉素是一个很重要的药品,研究的范围也由噻唑推广到了噁唑。下面我们就将噁唑类化合物的合成方法进行综述。 2.合成方法 噁唑类化合物可由提供N ,O 原子的化合物来合成。
2.1.Cornforth 法合成噁唑环 1947年由Cornforth 等人首次合成第一个含有噁唑环的化合物[7]。其过程如下: HCN + (CH 3)2CHOH + HCl H 2N=CHOCH(CH 3)2 222 EtO 2CCH 2-N=CHOCH(CH 3)2 3KOEt AcOH 水解 Cl C N HC CHOCH(CH 3)2K EtO 2C N O EtO 2C N O HOOC N O 据此设计合成噁唑-4-羧酸乙酯的路线如下[7]。 H 2 Oi Pr OiPr N CO 2Et N CO 2Et OiPr N O CO 2Et 222 AcOH 57% 82% 34% HCO 2Et EtOK 2.2. 碱催化酰氨基磺酰烯关环合成法 用3-酰氨基-2-碘-1-苯磺酰烯在碱催化下关环可得到噁唑化合物[8]。 H N O Me O NH I Me PhSO 2 H NaOH N O PhO 2SH 2C Me aq ,EtAc hv 80℃ THF 0℃ 38% 94%PhSO 2Na I 2 2.3.由西佛碱氧化法合成 在温和的反应条件下,用二醋酸碘苯作氧化剂可以以良好产率将西佛碱氧化生成2-芳基-5-甲氧基噁唑化合物[9]。 2methanol , 1h O CH 3 O N Ar N O Ar OCH 3
Pinner脒合成的反应机理及应用进展
Pinner脒合成的反应机理及应用进展 王阳阳 (西北农林科技大学理学院陕西杨凌712100) 摘要:脒类化合物在农药、医药以及其他领域上都具有很广泛的用途。合成脒类化合物的方法主要为:Pinner脒合成法。本文重点介绍了Pinner脒合成方法的机理和副反应机理,并对其在有机合成中的应用进行了探讨。 关键词:Pinner脒合成;机理;改进;应用 The reaction mechanism and application of Pinner amidine synthesis Wang Yangyang (College of science, Northwest A&F University, Yangling, 712100, China) Abstract:The amidine compounds have a very wide range of functions in the pesticide, medicine and other fields. The primary method of synthesis of amidine compounds is Pinner amidine synthesis. This article focuses on the reaction mechanism of Pinner amidine synthesis and the side reactions mechanism Its application in organic synthesis is also discussed. Key words: Pinner amidine synthesis; mechanism; improvement; application 1.前言 脒类化合物在农药和医药上具有很广泛的用途。早年发现某些脒盐可以治疗血吸虫病,但毒性较大,一些长链烷氧基取代的苯甲脒盐具有表面活性剂的作用,被称为杀虫脒[1]。现在,脒类化合物的主要用途是合成含氮的杂环化合物,如:咪唑、噻唑、嘧啶环等,在含氮杂环的合成中起着重要作用。研究发现,脒盐还可以作为水溶性偶氮类引发剂,在水溶液聚合与乳液聚合中得到广泛应用[2]。 脒类化合物的合成方法一般采用酸催化法即Pinner 脒合成法。反应式如scheme 1: Pinner脒合成: cheme 1