基因表达分析

基因表达分析
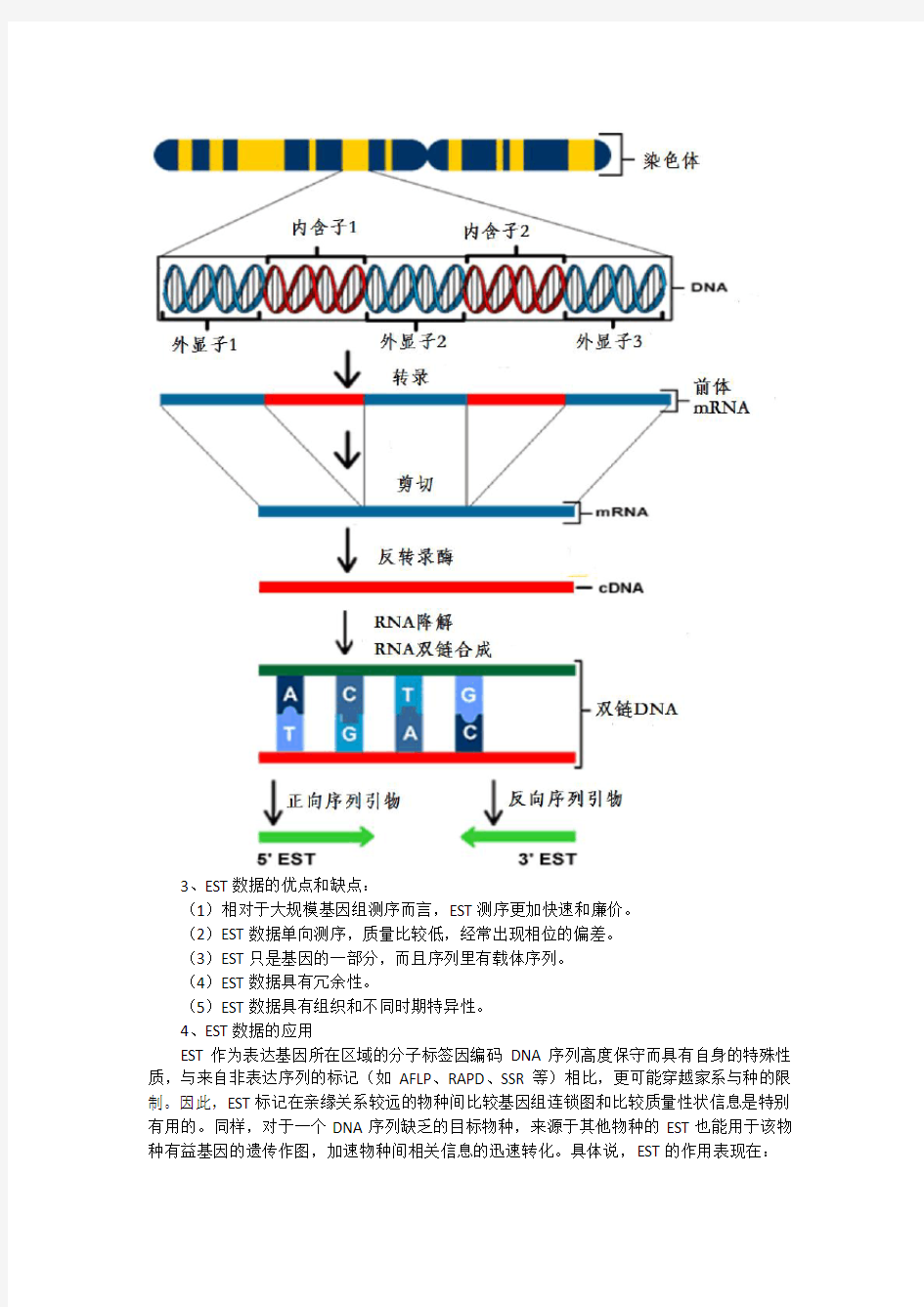
1、EST(Expressed Sequence Tag)表达序列标签(EST)分析
1、EST基本介绍
1、定义:
EST是从已建好的cDNA库中随机取出一个克隆,进行5’端或3’端进行一轮单向自动测序,获得短的cDNA部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20到7000bp不等,平均长度为400bp。
EST来源于一定环境下一个组织总mRNA所构建的cDNA文库,因此,EST也能说明该组织中各基因的表达水平。
2、技术路线:
首先从样品组织中提取mRNA,在逆转录酶的作用下用oligo(dT)作为引物进行RT-PCR 合成cDNA,再选择合适的载体构建cDNA文库,对各菌株加以整理,将每一个菌株的插入片段根据载体多克隆位点设计引物进行两端一次性自动化测序,这就是EST序列的产生过程。
3、EST数据的优点和缺点:
(1)相对于大规模基因组测序而言,EST测序更加快速和廉价。
(2)EST数据单向测序,质量比较低,经常出现相位的偏差。
(3)EST只是基因的一部分,而且序列里有载体序列。
(4)EST数据具有冗余性。
(5)EST数据具有组织和不同时期特异性。
4、EST数据的应用
EST作为表达基因所在区域的分子标签因编码DNA序列高度保守而具有自身的特殊性质,与来自非表达序列的标记(如AFLP、RAPD、SSR等)相比,更可能穿越家系与种的限制。因此,EST标记在亲缘关系较远的物种间比较基因组连锁图和比较质量性状信息是特别有用的。同样,对于一个DNA序列缺乏的目标物种,来源于其他物种的EST也能用于该物种有益基因的遗传作图,加速物种间相关信息的迅速转化。具体说,EST的作用表现在:
(1)用于构建基因组的遗传图谱与物理图谱;
(2)作为探针用于放射性杂交;
(3)用于定位克隆;
(4)借以寻找新的基因;
(5)作为分子标记;
(6)用于研究生物群体多态性;
(7)用于研究基因的功能;
(8)有助于药物的开发、品种的改良;
(9)促进基因芯片的发展等方面。
研究物种的转录组,基因组上转录表达的部分;
发现基因,一是可以为研究基因结构提供exon/intron边界,二是提供基因组上可能基因区域;
研究可变剪切;
研究基因的表达谱;
可以为制做物理图谱提供序列,为芯片提供clone数据。
正是因为EST表现出了这些巨大潜能,使其得到了充分的利用与发展。
5、常用的EST数据库
(1)NCBI dbEST
网址:https://www.360docs.net/doc/643687011.html,/dbEST/index.html
数据量:
表7-1为NCBI dbEST截至2006年8月22日的数据情况,数据库里一共有38,056,628条EST。
(2)NCBI Unigene
网址:https://www.360docs.net/doc/643687011.html,/entrez/query.fcgi?db=unigene
介绍:Unigene把dbEST的数据利用一些常规的基因数据聚在一起。对于一个cluster而言,提供了许多相关信息。Unigene经常重新构建,所以cluster标识不识固定的。
(3)The TIGR Gene Indices
The Gene Indices 更多的基于拼接(Assembly)得到的congtigs序列,而不是聚类的结果
The Gene Indices 的基因索引比NCBI Unigene多。
TIGR包括EGAD(The Expressed Gene Anatomy Database),EGAD的索引被包括在Human Gene Indices
(4)其他的一些常用数据库
SANBI,南非,收集人的EST contigs
MIPS,慕尼黑,SBI收集.Unigene的BIAST可搜集conlig
TIGEM,意大利,EST搜索及组装工具,包括本地及远程的
CBIL,宾西法尼亚州大学,DOTS组装数据库
2、EST 分析流程介绍
图7-2 EST分析流程图
(1)测序
EST数据可以从5’和3’两个方向进行测序,可以根据不同的实验目的选择测序方向
图7-3 测序方向的选择
不同方向测序的优点:
5’端测序:更有利于得到全长的cDNA序列,有助于研究基因表达的多样性。
3’短测序:有助于得到基因的特异性区域,为STS、SAGE、Microarray提供序列资源。(2)EST数据预处理过程
①Basecalling将序列的峰图从测序仪中提取出来。常见的峰图文件有SCF和ABI格式,可以在Windows用Chromas下打开。
图7-4 Chromas在Windows下打开峰图文件
②将峰图文件转化成phd、fasta文件,并去除序列中的低质量区域。
A、峰图文件转化成phd文件,并去除序列中的低质量区域。
软件:phred
基本用法:phred –id峰图文件夹–pd输出的phd文件夹–trim_phd –trim_alt “”–trim_cutoff 0.05
参数说明:
–trim_phd:将峰图文件转化成phd文件
–trim_alt:清理序列,去除低质量的区域,用–trim_cutoff的标准。如果从特定的酶切为点开始处理序列,可应用参数-trim_alt酶的序列,如果从头开始处理,用参数-trim_alt “”–trim_cutoff:去除低质量发生错误的几率,默认是0.05,意思是允许100个碱基里有5可能错误。
测序的质个量的评估公式:Q = -10 log10 (P)
公式中的Q代表了碱基的测序质量值,P代表了每个碱基出错的概率。
例如:如果每100个碱基有一个错误,那么P=0.01,这样Q就为20(我们通常说的Q20标准);如果P=0.001,Q就为30(Q30)。
注意:当P为错误阈值(cutoff,默认为0.05)时,Q近似为13,所以13就可用作背景来估计总体的质量值。
PHD文件格式介绍:
BEGIN_SEQUENCE
BEGIN_COMMENT
[信息注释]
END_COMMENT
BEGIN_DNA
[峰图序列格式是:碱基、质量值、在峰图上的位置]
END_DNA
END_SEQUENCE
例子:
BEGIN_SEQUENCE BGI.scf
BEGIN_COMMENT
CHROMAT_FILE: BGI.scf
ABI_THUMBPRINT: 0
PHRED_VERSION: 0.000925.d
CALL_METHOD: phred
QUALITY_LEVELS: 99
TIME: Wed Dec 20 07:00:52 2006
TRACE_ARRAY_MIN_INDEX: 0
TRACE_ARRAY_MAX_INDEX: 11108
TRIM: 0 630 -1.00
CHEM: unknown
DYE: unknown
END_COMMENT
BEGIN_DNA
t 15 750
g 19 766
c 25 782
a 18 793
g 18 804
g 17 819
.........
a 32 10595
t 32 10611
g 32 10635
g 32 10651
t 24 10669
c 15 10689
a 12 10707
t 12 10722
a 12 10751
c 14 10771
c 9 10785
t 19 10801
g 20 10824
t 15 10838
t 14 10854
t 14 10878
c 21 10891
c 24 10913
t 20 10933
g 22 10952
END_DNA
END_SEQUENCE
B、将phd 文件转化成fasta 文件。
软件:phd2fatsa
基本用法:phred
–id phd:文件夹
–os:输出的fasta文件
–oq:输出的质量文件
③屏蔽序列中的载体序列
软件:crossmatch
基本用法:cross_match 序列文件载体序列–screen >screen.out
④去除嵌合(chimeric)的克隆序列
软件:perl Chimeric_Check.pl
–s:序列文件
–q:质量文件
–ns:新的序列文件
–nq:新的质量文件
说明:嵌合(chimeric)的克隆是在文库构建过程的反应中产生的,其序列特征表现为,序列的中间有很长的polyA序列,或载体序列,其形式如下:
>Back-to-back poly(A)+ tails AAAGCTGGAGCTCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCCCCCGGGCTGCAGGAATTCGAATT CGAATTCCGACAATGTCTAAGAGAGGACGTGGTGGGTCATCGGGAGCCAAGTTCCGTATTTCACTGGGTCT CCCAGTGGGAGCGGTCATCAACTGCGCTGACAACACAGGTGCCAAGAACCTGTACATCATATCCGTCAAAG GCATCAAGGGTCGTCTGAACAGGCTCCCTGCCGCTGGTGTGGGCGACATGGTTATGGCCACAGTGAAGAA AGGCAAGCCAGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAAGTCGTATCGGC GAAAAGATGGCGTGTTTCTTTACTTCGAAGACAATGCAGGGGTCATAGTAAATAACAAAGGAGAAATGAAA GGATCAGCCATCACAGGACCCGTAGCCAAGGAATGCGCAGACCTGTGGCCCAGGATTGCTTCCAATGCTGG TAGCATTGCCTGAGCGCAAATGTGGCTTGTCGTTTTCAATAAAATACTCAAAGTTTCTCGAGGGGGGGCCCG GTAACCAATTCGCCCTATAGTGAGTCGTATTA
>Linker-to-linker in middle of the sequence AAAGCTGGAGCTCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCCCCCGGGCTGCAGGAATTCGAATT CGAATTCCGACAATGTCTAAGAGAGGACGTGGTGGGTCATCGGGAGCCAAGTTCCGTATTTCACTGGGTCT CCCAGTGGGAGCGGTCATCAACTGCGCTGACAACACAGGTGCCAAGAACCTGTACATCATATCCGTCAAAG GCATCAAGGGTCGTCTGAACAGGCTCCCTGCCGCTGGTGTGGGCGACATGGTTATGGXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX GCGTGTTTCTTTACTTCGAAGACAATGCAGGGGTCATAGTAAATAACAAAGGAGAAATGAAAGGATCAGCC ATCACAGGACCCGTAGCCAAGGAATGCGCAGACCTGTGGCCCAGGATTGCTTCCAATGCTGGTAGCATTGC CTGAGCGCAAATGTGGCTTGTCGTTTTCAATAAAATACTCAAAGTTTCTCGAGGGGGGGCCCGGTAACCAAT TCGCCCTATAGTGAGTCGTATTA
⑤去除序列中的污染序列,如大肠杆菌等
软件:blast
说明:把EST数据与已知的可能污染序列数据库进行比对,去除污染。通常用e值衡量是否为污染(e<1e-100)。
⑥屏蔽序列中的重复序列
软件:repeatemasker
说明:如果EST的数据量比较大,在拼接之前要进行聚类分析,可以先屏蔽一下序列中的载体,可以提高聚类的准确性。
⑦去除序列中的polyA
软件:PolyA_Check.pl
基本用法:perl PolyA_Check.pl
–s:序列文件
–q:质量文件
–ns:新的序列文件
–nq:新的质量文件
–drop 1
参数说明:
–drop:如果只想在新生成的序列文件中标记出是否有polyA(T),请设定此参数为“-drop 0”。
⑧去除过短的序列
说明:我们通常把那些<100bp的序列去除掉,不参加后续的聚类拼接和注释分析。
经过上面的处理,我们得到了干净高质量的EST数据集,用于后续分析。
下面是一条EST数据经过预处理的过程:
屏蔽载体之前
>BGI.scf AAAGCTGGAGCTCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCCCCCGGGCTGCAGGAATTCGAATT CGAATTCCGACAATGTCTAAGAGAGGACGTGGTGGGTCATCGGGAGCCAAGTTCCGTATTTCACTGGGTCT CCCAGTGGGAGCGGTCATCAACTGCGCTGACAACACAGGTGCCAAGAACCTGTACATCATATCCGTCAAAG GCATCAAGGGTCGTCTGAACAGGCTCCCTGCCGCTGGTGTGGGCGACATGGTTATGGCCACAGTGAAGAA AGGCAAGCCAGAGCTCAGGAAAAAGGTGCATCCTGCGGTGGTGATACGACAGCGGAAGTCGTATCGGCGA AAAGATGGCGTGTTTCTTTACTTCGAAGACAATGCAGGGGTCATAGTAAATAACAAAGGAGAAATGAAAGG ATCAGCCATCACAGGACCCGTAGCCAAGGAATGCGCAGACCTGTGGCCCAGGATTGCTTCCAATGCTGGTA GCATTGCCTGAGCGCAAATGTGGCTTGTCGTTTTCAATAAAATACTCAAAGTTTAAAAAAAAAAAAAAAAAA AAAACTCGAGGGGGGGCCCGGTAACCAATTCGCCCTATAGTGAGTCGTATTA
屏蔽载体后
> BGI.scf vector EcoR ⅠAAAGC XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX ATTC GAA TTCCGACAATGTCTAAGAGAGGACGTGGTGGGTCATCGGGAGCCAAGTTCCGTATTTCACTGGGTCTCCCA GTGGGAGCGGTCATCAACTGCGCTGACAACACAGGTGCCAAGAACCTGTACATCATATCCGTCAAAGGCAT CAAGGGTCGTCTGAACAGGCTCCCTGCCGCTGGTGTGGGCGACATGGTTATGGCCACAGTGAAGAAAGGC AAGCCAGAGCTCAGGAAAAAGGTGCATCCTGCGGTGGTGATACGACAGCGGAAGTCGTATCGGCGAAAAG ATGGCGTGTTTCTTTACTTCGAAGACAATGCAGGGGTCATAGTAAATAACAAAGGAGAAATGAAAGGATCA GCCATCACAGGACCCGTAGCCAAGGAATGCGCAGACCTGTGGCCCAGGATTGCTTCCAATGCTGGTAGCAT TGCCTGAGCGCAAATGTGGCTTGTCGTTTTC AATAA AATACTCAAAGTTT AAAAAAAAAAAAAAAAAAAAA A CTCGAG GGGGGGGCCCGGTAA XXXXXXXXXXXXXXXXXXXXXXXXXXXX
Xho ⅠpolyA signal polyA tail
预处理后得到的干净的EST 数据
> BGI.scf ATTCGAATTCCGACAATGTCTAAGAGAGGACGTGGTGGGTCATCGGGAGCCAAGTTCCGTATTTCACTGGG TCTCCCAGTGGGAGCGGTCATCAACTGCGCTGACAACACAGGTGCCAAGAACCTGTACATCATATCCGTCAA AGGCATCAAGGGTCGTCTGAACAGGCTCCCTGCCGCTGGTGTGGGCGACATGGTTATGGCCACAGTGAAG AAAGGCAAGCCAGAGCTCAGGAAAAAGGTGCATCCTGCGGTGGTGATACGACAGCGGAAGTCGTATCGGC GAAAAGATGGCGTGTTTCTTTACTTCGAAGACAATGCAGGGGTCATAGTAAATAACAAAGGAGAAATGAAA GGATCAGCCATCACAGGACCCGTAGCCAAGGAATGCGCAGACCTGTGGCCCAGGATTGCTTCCAATGCTGG TAGCATTGCCTGAGCGCAAATGTGGCTTGTCGTTTTCAATAAAATACTCAAAGTTT
3、EST数据的聚类(Clustering)
聚类的目的:把同属于一个基因的EST数据聚在一起。
聚类的作用:有助于产生更长的一致性序列,可以降低数据的冗余性,更正数据的错误,有助发现同一基因的不同剪切形式。
聚类方法:
有指导的聚类:利用物种或邻近物种的基因/蛋白质数据做指导,将EST数据比对到参考序列上。
无指导的聚类:利用序列自身的相似性。
常用EST 聚类软件介绍:
BLASTclust
介绍:利用单链法的聚类方法,通过序列间的两两比对,建立距离矩阵,它有两个聚类标准(i)序列的相似性水平,如匹配的同一性水平(ii)匹配区域的长度,一般来说,两个序列要聚在一起,匹配区域至少要覆盖每个序列的70%。NCBI Unigene数据库就利用了BLASTclust。BLASTclust的速度较快,适合中等规模的EST数据。
网址:https://www.360docs.net/doc/643687011.html,/apps/blast/doc/blastclust.html
CLOBB(Cluster on the basis of BLAST similarity)
介绍:它以Blast作为搜索引擎,所以速度较慢,适合中小规模的EST数据。它能记录类的合并过程,鉴别那些大的类,定义了不同类型的重叠区域(overlap),最大程度的避免错误的聚类,提高了准确性。它要求的最小重叠区域是≥30bp,相似度≥95%。
网址:https://www.360docs.net/doc/643687011.html,/CLOBB/
D2cluster
介绍:它用了字符串的搜索方法,最小的字符串是6bp,用了最小单连接的聚类方法,快速准确地把EST聚类,它允许的最小重叠区域≥100bp,相似度≥90%。
Unicluster
介绍:这个软件运用了并行的处理机制和一些启发式算法,使聚类更加快速,适合大规模的EST数据。
网址:https://www.360docs.net/doc/643687011.html,/pubsoft/software.html
4、EST数据的拼接(Assembly)
拼接是把同属于的一个转录本的EST序列,联结起来,得到一个一致性(consensus sequences)序列,降低数据的冗余性。
4.1 常用的拼接软件:
(1)phrap
网址:https://www.360docs.net/doc/643687011.html,/phredphrap/phrap.html
基本用法:phrap要拼接的序列-new_ace -minamtch 30 -minscore 30 -repeate_stringency 0.95 >phrap.out
参数说明:
-new_ace:生成ace文件,便于后面有consed查看拼接的结果。
-minmatch:序列最小的匹配长度,默认是30bp。
-minscore:序列匹配的最小分值,默认是30。
-repeate_stringency:匹配的相似度,默认是0.95。
使用提示:
当EST数据比较大,有几千条时,在拼接之前并没有聚类,直接用phrap聚类拼接了,可以适当提高拼接的标准,一般经验的参数是:-minamtch 42 –minscore 40 –repeate_stringency 0.99
(2)cap3
网址:https://www.360docs.net/doc/643687011.html,/
http://pbil.univ-lyon1.fr/cap3.php
基本用法:cap3要拼接的序列-o 30 -p 90
参数说明:
-o:序列最小的匹配长度,默认是30bp。
-s:序列匹配的最小分值,默认是500。
-p:匹配的相似度,默认是75。
使用提示:
一般说来,cap3要比phrap运行速度要慢,phrap牺牲了一些匹配的敏感性,phrap拼接的序列要比cap3长,cap3拼接得比phrap准确。
(3)d2_cluster
网址:http://www.sanbi.ac.za/
4.2 拼接结果的检测
软件:consed
基本用法:在拼接的目录下运行(目录下面有ace文件)cosned –nophd
参数说明:
-nophd:如果你只有序列文件,没有峰图和phd文件,请加上这个参数。
使用提示:
(1)cosned需要图形页面的支持,所以你登录所用的终端可选用xwin32。
(2)主要检查那些cluster比较大的contig就可以,如果发现有问题,可以把这个congtig 的序列提出来,用更加严格的参数再次拼接一下,下面有两个例子。
图7-6 有问题的拼接
图7-7 正常的拼接结果
5、Unigene开放阅读框(ORF)的预测1)ORF的预测
软件:getorf(EMBOSS软件包里的程序)
基本用法:getorf要预测的序列预测的ORF序列-minsize 100 –find 3 –reverse 1
参数说明:
-minsize:预测的ORF最小长度,基本上此参数设定的越大,预测的越准确。
-find:要生成核酸序列,从翻译起始为点到翻译终止位点,请设定为3。
-reverse:如果要在互补链上找ORF,设定此参数为1,否则设定此参数为0。
使用说明:一般一个unigene预测出很多可能的ORF,我们一般选取最长的ORF,作为这条基因的开放阅读框。
2)cDNA 是否为全长的判断方法:
(1)直接从序列上评价:
5'端:如果有同源全长基因的比较,可以通过与其它生物已知的对应基因5'末端进行比较来判断。如果无同源基因的新基因,则首先判断编码框架是否完整,即在开放阅读框(ORF)的第1个ATG上游有无同框架的终止密码子;其次,判断是否有转录起始点,一般加在5'帽结构后有一段富含嘧啶的区域,或者是cDNA 5'序列与基因组序列中经过酶切保护的部分相同,则可以确定得到的cDNA的5'端是完整的。
3'端:同样可以用其它生物已知的对应基因3'末端进行比较来判断,或编码框架的下游有终止密码子,或有1个以上的PolyA加尾信号,或无明显加尾信号的则也有PolyA尾。(2)用实验方法证实:
可以通过引物延伸法确定5'端和3'端的长度,如:5'端RACE,3'端RACE,或者通过Northern Blot证实大小是否一致。
6、基因的特异表达分析
我们构建没有均一化的cDNA文库,进行EST测序的一个重要目的就是,它可以帮助我们了解基因在不同组织,不同的发育时期的表达情况。我在上面已经将EST聚类拼接成Unigene,这样我们就可以统计同一个Unigene在不同的文库里的表达情况,即EST在这个文库的数目。进而我们可以从统计学上,对基因的表达情况做出评估,通常用p值来衡量在不同库间表达是否显著。p≤0.05为一般的显著,p≤0.01为非常显著。
软件:IDEG.6
网址:http://telethon.bio.unipd.it/bioinfo/IDEG6/
介绍:IDEG.6集合多种目前常用的统计方法,如Audic and Claverie两个样品的检验,Fish精准检验,卡方检验,R检验等。
7、基因的注释和功能分类
1)NCBI NT数据库
介绍:这是一个非冗余的核酸数据库,包括了GenBank、RefSeq、PDB的数据。
网址:https://www.360docs.net/doc/643687011.html,
软件:blastn
一般标准:le-5
2)NCBI NR数据库
介绍:这是一个非冗余的蛋白质数据库,包括了SwissProt、PIR(Protein Information Resource)、PRF(Protein Research Foundation)、PDB(Protein Data Bank)蛋白质数据库非冗余的数据以及从GenBank 和RefSeq的CDS数据翻译来的蛋白质数据。
网址:https://www.360docs.net/doc/643687011.html,
软件:blastp(blastx)
一般标准:1e-5
3)SwissProt数据库
介绍:SWISS-PROT是经过注释的蛋白质序列数据库,由欧洲生物信息学研究所(EBI)维护。数据库由蛋白质序列条目构成,每个条目包含蛋白质序列、引用文献信息、分类学信息、注释等,注释中包括蛋白质的功能、转录后修饰、特殊位点和区域、二级结构、四级结构、与其它序列的相似性、序列残缺与疾病的关系、序列变异体和冲突等信息。Swiss-Prot中尽可能减少了冗余序列,并与其它30多个数据建立了交叉引用,其中包括核酸序列库、蛋白质序列库和蛋白质结构库等。
网址:https://www.360docs.net/doc/643687011.html,/sprot/
软件:blastp(blastx)
一般标准:1e-5
4)KEGG数据库
介绍:KEGG(Kyoto Encyclopedia of Genes and Genomes)京都基因和基因组百科全书是系统分析基因功能,联系基因组信息和功能信息的知识库。基因组信息存储在GENES数据库里,包括完整和部分测序的基因组序列;更高级的功能信息存储在PATHWAY数据库里,包括图解的细胞生化过程如代谢、膜转运、信号传递、细胞周期,还包括同系保守的子通路等信息;KEGG的另一个数据库是LIGAND,包含关于化学物质、酶分子、酶反应等信息。KEGG提供了Java的图形工具来访问基因组图谱,比较基因组图谱和操作表达图谱,以及其它序列比较、图形比较和通路计算的工具,可以免费获取。
网址:http://www.genome.jp/kegg/
软件:blastp(blastx)
一般标准:1e-5
使用提示:通过与KEGG数据库进行比对,我们可以了解基因可能参与的代谢途径。
图7-8 KEGG的代谢途径
5)COG数据库
介绍:COGs(Clusters of Orthologous Groups of proteins)蛋白质直系同源簇数据库是对细菌、藻类和真核生物的21个完整基因组的编码蛋白,根据系统进化关系分类构建而成。COG 库
对于预测单个蛋白质的功能和整个新基因组中蛋白质的功能都很有用。通过某个蛋白质与所有COGs中的蛋白质进行比对,可以把它归入适当的COG家族。
网址:https://www.360docs.net/doc/643687011.html,/COG
软件:blastp(blastx)
一般标准:1e-5
使用说明:做完全部基因与COG数据库比对后,我可以对基因在功能上进行一下分类,如图7-9所示。
图7-9 COG功能分类图
6)Interpro
介绍:Interpro是一个关于蛋白家族、功能保守区域、和功能位点的数据库,它整合了已知功能蛋白的特点,并应用于功能未知的蛋白进行注释。
网址:https://www.360docs.net/doc/643687011.html,/interpro/
软件:interproscan
(7)GO
介绍:GO(Gene Ontology)是用一套具有动态形式的控制字汇来解释真核生物的基因或蛋白质在细胞内所扮演的角色及生医学方面的知识,同时这些字汇随着生命科学研究的进步,一直不断的累积与改变。一个本体会被一个控制字汇来描述并给予统一的名称,到目前为止,在Gene Ontology下有三大独立的本体被建立:biological process,molecular function及cellular component。一个基因或蛋白质可从3个层面进行注解,首先是构成在细胞内的特定组件cellular 过程(biologicalprocess),因此科学家试着收集各真核生物(如SGD、MGI、FlyBase、..)的基因或蛋白质,利用已知component,其次是此组件在分子功能上所扮演的角色,最后是基因或蛋白质参与的生物的文献资料及序列比较资讯为基础,将所有的真核生物的基因或蛋白质都基于在此系统下作注解与分类。
网址:https://www.360docs.net/doc/643687011.html,/ or https://www.360docs.net/doc/643687011.html,/GO/index.html
软件:interproscan
提示:我么也可以通过基因与SwissProt/COG数据比对,把已知蛋白的GO信息转加给你的基因,比对的标准,可以设定为1e-10或更高一点。图7-10是一张GO的功能分类图,可以到https://www.360docs.net/doc/643687011.html,/cgi-bin/wego/index.pl画GO的分类图。
图7-10 GO功能分类图
7.1.3 BGI EST Pipeline(BEP)介绍
北京华大基因研究中心(BGI)总结了多年EST分析的经验,开发了一套EST分析的软件包,它整合了上面介绍的各项分析,使得EST分析简易化、流程化,以适应高通量的EST 分析要求。使用该软件包进行EST分析,可以大大提高分析的速度和准确性,进而提高了科学研究的速度。下面介绍一下该软件包的用法,如需获得该软件包请与BGI EST分析小组联系。
1、硬件和软件配置
操作系统:BEP是Linux下开发并运行的。
程序语言:BEP后台程序使用Perl和C SHELL编写。
软件和数据库需求:
BEP需要下列第三方软件和公共数据库:
Perl5.0或更高版本,可以到下面网址下载https://www.360docs.net/doc/643687011.html,(特别要注意安装Perl的GD 模块,分析中要用到GD模块画图)。
Linux平台的blastall和formatdb,可以到下面网址下载:
ftp://https://www.360docs.net/doc/643687011.html,/blast/executables/
Phred,请与swxfr@https://www.360docs.net/doc/643687011.html,联系索要软件。
Cross_match,请与phg@https://www.360docs.net/doc/643687011.html,联系索要软件。
uicluster2-1.1可以到下面网址下载:https://www.360docs.net/doc/643687011.html,/pubsoft/software.html Getorf,可以到下面网址下载:https://www.360docs.net/doc/643687011.html,
InterProScan,可以到下面网址下载:ftp://https://www.360docs.net/doc/643687011.html,/pub/software/unix/iprscan/t Non-Redundant Protein Database(NR):可以到下面网址下载:ftp://https://www.360docs.net/doc/643687011.html, Non-Redundant Nucleotide Database(NT):可以到下面网址下载:ftp://https://www.360docs.net/doc/643687011.html, SWISSPROT数据库:可以到下面网址下载:http://www.ebi.ac.ck/swissprot
Cluster of Othologues Groups Proteins Database(COG),可以到下面网址下载:https://www.360docs.net/doc/643687011.html,/COG
Kyoto Encyclopedia of Genes and Genomes Database(KEGG),可以到下面网址下载:http://www.genome.ad.jp/kegg
2、程序的安装:
(1)解压文件BEP_Backend_Programs.tar.gz
$ gzip - d BEP_Backend_Programs.tar.gz
$ tar - xvf BEP_Backend_Programs.tar.gz
(2)在解压后的目录里有一个BEP.Config文件,请修改第三方软件和数据库的位置,下面是个例子:
binpath = /disk2/prj0317/Est_Pipeline/bin
perl = /usr/local/bin/perl
phred = /usr/local/genome/bin/phred
phd2fasta = /usr/local/genome/bin/phd2fasta
cross_match = /usr/local/genome/bin/cross_match
blastall = /usr/local/genome/bin/blastall
phrap_manyreads = /usr/local/genome/bin/phrap
phrap = /usr/local/genome/bin/phrap
getorf = /ust/local/genome/EMBOSS -2.6.0/bin/getorf
InterProScan = /disk16/prj0317/interpro_bin/iprscan/interProScan.pl
nr = /disk2/database/public.3800A/NCBI/https://www.360docs.net/doc/643687011.html,/BLAST/nr/2006 - 07 - 11/nr
nt = /disk2/database/public.3800A/NCBI/https://www.360docs.net/doc/643687011.html,/BLAST/nr/2006 - 07 - 11/nt swissprot = /disk2/prj0317/Database/swissprot/uniprot_sprot_2006_06_27.fasta
kegg = /disk2/prj0317/Est_Pipeline/Database/kegg/kegg_35.fa
cog = /disk2/prj0317/Est_Pipeline/Database/cog/myva
uicluster = /disk2/prj0317/Est_Pipeline/Bio_Soft/uicluster2 - 1.1/bin/uicluster2 请确保上面第三方软件和数据库已经在你系统中安装并测试完毕。
(3)运行.prel BEP_Byte.pl适当调整参数就可以进行EST分析了。
3、程序使用说明及结果介绍
1)项目信息参数
参数:
-project:EST项目的名称,给一个项目说明性标识符号,如WTEA。(必需参数)
-outdir:结果的输出目录,会在该目录下面生一个Output目录(各项分析的结果会在这个目录下面,还有一个Script目录,生成程序运行的过程日志文件。(必需参数)
-system:程序默认为直接投放任务,同时支持SGE qsub投放任务,需要将此参数设定为“-system sge”。
2)输入选项参数
参数:
-input_chromato:如果输入的是峰图文件,请选择“-input_chromato峰图文件”。可以输入峰图的文件夹,或文件夹的压缩文件,目前支持zip和tar.gz压缩格式。
-input_sequence:如果输入的是序列文件,请选择“-input_sequence序列文件”。
-input_qual:如果有输入序列文件的质量文件,请选择“-input_qual质量文件”。
-input_vector:要屏蔽的载体序列,如果你选择了下面的“-crossmatch”要屏蔽载体,那么这个参数是必须的。
注意:
(1)请输入你要做分析的EST序列的峰图文件或序列文件,目前只支持fasta格式的序
列文件,要屏蔽载体,不要忘记输入载体序列。
(2)下面介绍各项分析的参数,如果你要做哪些分析,请选上该项分析的标识参数,例如:如果你要处理峰图生成序列文件,请选择-phred参数,要做nt库注释,请选择-nt参数。
3)EST基本的分析选各项
(1)处理峰图,生成序列文件。
参数:
-phred:要处理峰图,请选上这个参数。
-phred_trim_cutoff:设定去除低质量碱基时,碱基可能出现错误的概率。默认是0.05。
结果示范与说明:
Phred:(Output目录下面)
FASTA:Phred结果中fasta结果文件夹
PHD:Phred结果中phd结果文件夹
WTEA.seq:序列文件
WTEA.seq.qual:序列的质量文件
(2)提纯序列文件(去除载体,polyA,短序列)。
参数:
-crossmatch:要屏蔽序列中载体序列,请选上这个参数。
-wipe_polya:要去除载体中的polyA尾巴,请选上这个参数。如果不想去除polyA尾,请将这个参数设定为“-wipe_polya 0”,流程默认去除polyA尾巴序列。
-filter_short:过滤短的序列,默认过滤小于100bp的序列,可以自由设定过滤的标准。如果不想过滤,请设定此参数为“-filter_short 0”。
结果示范与说明:
Raw:
WTEA.seq:序列文件
WTEA.seq.qual:序列的质量文件
WTEA.seq.screen:去除载体序列后的文件
WTEA.seq.screen.checked:去除载体序列后的文件进一步检测
WTEA.seq.screen.qual:去除载体序列后的文件的质量文件
WTEA_Raw_EST_Info.xls:为提纯前的EST序列信息(长度、GC等)
nopolyA.seq:去除polyA后的序列
nopolyA.seq.qual:去除polyA后的序列的质量文件
(3)序列聚类(也可以不做这步,直接用下步的phrap聚类拼接)
参数:
-clustering:要聚类EST,请选上这个参数
-clu_matchlen:两个EST聚类在一起的最小比配长度,默认是40bp
-clu_errlimit:在clu_matchlen匹配长度中最大的错误匹配碱基数,默认是2
(4)序列拼接
参数:
-phrap:要拼接EST,得到一致序列,请选上这个参数
-phrap_minmatch:两个EST拼接在一起的最小比配长度,默认是30bp
-phrap_minscore:匹配的最小分值,默认是30
-phrap_stringency:匹配区域的identiry,默认是0.95
结果示范与说明:
a . 先聚类后拼接的结果(有两部分)
Clustering_Assembly:
WTEA.CleanEST.seq.clus:聚类的结果
WTEA_Cluster_Assembly.fasta:类大于等于2个EST的拼接结果
WTEA_Cluster_Assembly.list:每一类拼接后得到一致序列与EST的对应关系列表WTEA_Sinlets.fasta:单独得EST为一类的序列
WTEA_Sinlets.list:单独得EST为一类的序列与EST的对应关系列表Unigene:
WTEA.Unigene2EST.xls:拼接后每一个一致序列(unigene)与原来的EST的对应关系WTEA.Unigene.seq:拼接后一致序列(unigene)文件
b . 直接用phrap拼接的结果
Unigene:
Contigs_EST.xls:拼接后每条contigs对应的ESTs信息
WTEA.CleanEST.seq.ace:拼接的ACE文件
WTEA.CleanEST.seq.contigs:拼接的congtigs文件
WTEA.CleanEST.seq.contigs.qual:拼接的congtigs文件的质量文件
WTEA.CleanEST.seq.singlets:拼接的singlets文件
WTEA.Unigene.seq:拼接的congtigs+singlets文件
phrap.list:Phrap的输出结果简化信息
phrap.out:Phrap的输出结果
(5)开放阅读框(ORF)的预测:
参数:
-orf:要预测unigene的开放阅读框,请选上这个参数
-orf_minsize:开放阅读框的最小长度,默认是100bp
结果示范与说明:
Orf:
https://www.360docs.net/doc/643687011.html,age.list:Codon Usage的统计信息
WTEA.Unigene.seq_3_20.orf:预测的ORF
WTEA.Unigene.seq_3_20.orf.longest:预测的ORF每条基因选取了一条最长的WTEA.Unigene.seq_3_20.orf.longest.pro:预测的ORF每条基因选取了一条最长的蛋白序列(6)基因的功能注释和分类:
a . NCBI NT Database
参数:
-nt:用NT数据库对unigene进行注释
-nt_e:BLAST E VALUE,经验参数le-5
-nt_group:并行的处理数据,将输入的序列分成分去注释,默认是3
-nt_blast_type:BLAST的类型,默认是blastn
结果示范与说明:
Nt:
WTEA.Unigene.Nt.B:与NT库比对的BLASTN原始结果
WTEA.Unigene.Nt.B.O:与NT库比对的BLASTN提取结果
WTEA.Unigene.Nt.B.O.Best:与NT库比对的BLASTN提取结果,选取一个最好的
b . NCBI NR Database
参数:
-nr:用NR数据库对unigene进行注释
-nr_e:BLAST E VALUE,经验参数1e-5
-nr_group:并行的处理数据,将输入的序列分成分去注释,默认是3
-nr_blast_type:BLAST的类型,默认是blastx
结果示范与说明:
Nr:
WTEA.Unigene.Nr.B:与NR库比对的BLASTX原始结果
WTEA.Unigene.Nr.B.O:与NR库比对的BLASTX提取结果
WTEA.Unigene.Nr.B.O.Best:与NR库比对的BLASTX提取结果,选取一个最好的
c . SwissProt Database
参数:
-swissprot:用NR数据库对unigene进行注释
-swissprot_e:BLAST E VALUE,经验参数1e-5
-swissprot_group:并行的处理数据,将输入的序列分成分去注释,默认是3
-swissprot_blast_type:BLAST的类型,默认是blastx
结果示范与说明:
Swissprot:
WTEA.Unigene.Swissprot.B:与Swissprot库比对的BLASTX原始结果
WTEA.Unigene.Swissprot.B.O:与Swissprot库比对的BLASTX提取结果
WTEA.Unigene.Swissprot.B.O.Best:与Swissprot库比对的BLASTX提取结果,选取一个最好的d . COG Database(Clusters of Orthologous Groups of proteins)
参数:
-cog:用COG数据库对unigene进行注释
-cog_e:BLAST E VALUE,经验参数le-5
-cog_group:并行的处理数据,将输入的序列分成分去注释,默认是3
-cog_blast_type:BLAST的类型,默认是blastx
结果示范与说明:
COG:
WTEA.Unigene.COG.B:与COG库比对的BLASTX原始结果
WTEA.Unigene.COG.B.O:与COG库比对的BLASTX提取结果
WTEA_COG_Class.txt:COG注释信息
WTEA_COG_Class.txt_Statistic.xls:COG分类的统计信息
WTEA_COG_Map.png:COG分类的图
e . KEGG Pathway Database(Clusters o
f Orthologous Groups of proteins)
参数:
-kegg:用KEGG数据库对unigene进行注释
-kegg_e:BLAST E VALUE,经验参数1e-5
-kegg_group:并行的处理数据,将输入的序列分成分去注释,默认是3
-kegg_blast_type:BLAST的类型,默认是blastx
结果示范与说明:
Kegg:
WTEA.Unigene.ec:基因注释的ec号
WTEA.Unigene.ec.map:基因注释的ec号对应代谢图
WTEA.Unigene.kegg.B:与KEGG库比对的BLASTX原始结果
WTEA.Unigene.kegg.B.O:与KEGG库比对的BLASTX提取结果
WTEA.Unigene.kegg.B.O.Best:与KEGG库比对的BLASTX提取结果,选取一个最好的WTEA.Unigene.kegg.Pathway:基因对应的PANTHWAY信息
f . Interpro and GO(这套系统用interproscan去注释基因,得到基因GO注释信息)
参数:
-interpro:用interproscan对基因进行注释,并得到基因的GO信息
-interpro_type:如果序列是核酸序列参数为nt,如果序列是蛋白序列参数为aa
-interpro_group:将序列分成几份,默认是3
结果示范与说明:
Interpro:
WTEA.gene.interpro.txt:基因的intepro注释信息
WTEA.go.Biological.Process:基因Biological Process方面GO注释的信息
https://www.360docs.net/doc/643687011.html,ponent:基因Cellular Component方面GO注释的信息
WTEA.go.Molecular.Function:基因Molecular Function方面GO注释的信息
WTEA.go.seg:GO的分类图
WTEA.go.txt:基因的GO注释信息
WTEA.go.txt_gene_list.txt:每一类GO分类对应的基因
WTEA.interpro.classify.txt:基因interpro注释信息的统计
WTEA.ipr.go:基因interpro以及GO注释信息
WTEA_.GO_Anno.txt:基因的GO注释信息
merged.txt:Interproscan结果
(7)常规分析举例:
①从峰图开始分析,要做这套流程的所有分析,可以参照下面的命令行:
$ perl BEP_Byte.pl -project WTEA -outdir ./ -input_chromato test_scf.tar.gz - input_vector../../.. /bin/vector/pBluescript +. fa -phred -crossmatch -clustering -phrap - orf - swissprot - cog - kegg - interpro
注解:test_ scf.tar.gz是峰图文件夹的压缩文件,也可以输入峰图文件夹的位置。
②从序列文件开始分析,要做这套流程的所有分析,可以参照下面的命令行:
$ perl BEP_Byte.pl -project WTEA -outdir ./ -input_sequence test.seq - input_qual test.seq.qual -input_vector../../.. /bin/vector/pBluescript +. fa -crossmatch -clustering -phrap - orf -nt - swissprot - cog - kegg - interpro
注解:如果输入序列,就没有必要加上-phred选项了。
③如果是用SGE qsub对列管理系统,请加上“- system sge”。例如:
$ perl BEP_Byte.pl -project WTEA -outdir ./ -system sge - input_chromato test_scf.tar.gz - input_vector../../.. /bin/vector/pBluescript +. fa -phred -crossmatch -clustering -phrap - orf - nt - nr -swissprot - cog - kegg - interpro
7.1.4 EST的应用
1 基因可变剪切的识别
在真核等高等生物中,基因在转录以后存在多种剪切形式,剪切成mRNA来翻译蛋白质序列,这体现了基因的多样性。我们通过建立cDNA文库,测得EST序列,可以了解基因的不同剪切形式。
可变剪切的可分类为:
(1)内含子残留
(2)可变的donor位点
基因表达的分析技术
第二篇细胞的遗传物质 第三章基因表达的分析技术 生物性状的表现均是通过基因表达调控实现的。对基因结构与基因表达调控进行研究,是揭示生命本质的必经之路。在基因组研究的过程中,逐步建立起一系列行之有效的技术。针对不同的研究内容,可建立不同的研究路线。 第一节PCR技术 聚合酶链反应(polymerase chain reaction,PCR)技术是一种体外核酸扩增技术,具有特异、敏感、产率高、快速、简便等突出优点。。PCR技术日斟完善,成为分子生物学和分子遗传学研究的最重要的技术。应用PCR技术可以使特定的基因或DNA片段在很短的时间内体外扩增数十万至百万倍。扩增的片段可以直接通过电泳观察,并作进一步的分析。 一、实验原理 PCR是根据DNA变性复性的原理,通过特异性引物,完成特异片段扩增。第一,按照欲检测的DNA的5'和3'端的碱基顺序各合成一段长约18~24个碱基的寡核苷酸序列作为引物(primer)。引物设计需要根据以下原则:①引物的长度保持在18~24bp之间,引物过短将影响产物的特异性,而引物过长将影响产物的合成效率;②GC含量应保持在45~60%之间;③5'和3'端的引物间不能形成互补。第二,将待检测的DNA变性后,加入四种单核苷酸(dNTP)、引物和耐热DNA聚合酶以及缓冲液。通过95℃变性,在进入较低的温度使引物与待扩增的DNA链复性结合,然后在聚合酶的作用下,体系中的脱氧核苷酸与模板DNA链互补配对,不断延伸合成新互补链,最终使一条DNA双链合成为两条双链。通过变性(92~95℃)→复性(40~60℃)→引物延伸(65~72℃)的顺序循环20至40个周期,就可以得到大量的DNA片段。理论上循环20周期可使DNA扩增100余万倍。
全基因组表达谱分析方法(DGE)
全基因组表达谱分析方法(DGE)----基于新一代测序技术的 技术路线 该方法首先从每个mRNA的3’端酶切得到一段21bp的TAG片段(特异性标记该基因);然后通过高通量测序,得到大量的TAG序列,不同的TAG序列的数量就代表了相应基因的表达量;通过生物信息学分析得到TAG代表的基因、基因表达水平、以及样品间基因表达差异等信息。技术路线如下: 1、样品准备: a) 提供浓度≥300ng/ul、总量≥6ug、OD260/280为1.8~2.2的总RNA样品; 2、样品制备(见图1-1): a) 类似SAGE技术,通过特异性酶切的方法从每个mRNA的3’末端得到一段21bp 的特异性片段,用来标记该基因,称为TAG; b) 在TAG片段两端连接上用于测序的接头引物; 3、上机测序: a) 通过高通量测序每个样品可以得到至少250万条TAG序列; 4、基本信息分析: a) 对原始数据进行基本处理,得到高质量的TAG序列; b) 通过统计每个TAG序列的数量,得到该TAG标记的基因的表达量; c) 对TAG进行注释,建立TAG和基因的对应关系; d) 基因在正义链和反义链上表达量间的关系; e) 其它统计分析; 5、高级信息分析: a) 基因在样品间差异表达分析; b) 库容量饱和度分析;
c) 其它分析; 测序优势 利用高通量测序进行表达谱研究的优势很明显,具体如下: 1.数字化信号:直接测定每个基因的特异性表达标签序列,通过计数表达标签序列的数目来确定该基因的表达量,大大提高了定量分析的准确度。整体表达差异分布符合正态分布,不会因为不同批次实验引起不必要的误差。 2.可重复性高:不同批次的表达谱度量准确,能够更准确的进行表达差异分析。 3.高灵敏度:对于表达差异不大的基因能够灵敏的检测其表达差异;能够检测出低丰度的表达基因。 4.全基因组分析,高性价比:由于该技术不用事先设计探针,而是直接测序的方式,因此无需了解物种基因信息,可以直接对任何物种进行包括未知基因在内的全基因组表达谱分析,因此性价比很高。 5.高通量测序:已有数据表明,当测序通量达到200万个表达标签时,即可得到样本中接近全部表达基因的表达量数据,而目前每个样本分析可以得到300 万~600万个表达标签。
随机信号分析习题
随机信号分析习题一 1. 设函数???≤>-=-0 , 0 ,1)(x x e x F x ,试证明)(x F 是某个随机变量ξ的分布函数。并求下列 概率:)1(<ξP ,)21(≤≤ξP 。 2. 设),(Y X 的联合密度函数为 (), 0, 0 (,)0 , other x y XY e x y f x y -+?≥≥=? ?, 求{}10,10<<< 8. 两个随机变量1X ,2X ,已知其联合概率密度为12(,)f x x ,求12X X +的概率密度? 9. 设X 是零均值,单位方差的高斯随机变量,()y g x =如图,求()y g x =的概率密度 ()Y f y \ 10. 设随机变量W 和Z 是另两个随机变量X 和Y 的函数 22 2 W X Y Z X ?=+?=? 设X ,Y 是相互独立的高斯变量。求随机变量W 和Z 的联合概率密度函数。 11. 设随机变量W 和Z 是另两个随机变量X 和Y 的函数 2() W X Y Z X Y =+?? =+? 已知(,)XY f x y ,求联合概率密度函数(,)WZ f z ω。 12. 设随机变量X 为均匀分布,其概率密度1 ,()0X a x b f x b a ?≤≤? =-???, 其它 (1)求X 的特征函数,()X ?ω。 (2)由()X ?ω,求[]E X 。 13. 用特征函数方法求两个数学期望为0,方差为1,互相独立的高斯随机变量1X 和2X 之和的概率密度。 14. 证明若n X 依均方收敛,即 l.i.m n n X X →∞ =,则n X 必依概率收敛于X 。 15. 设{}n X 和{}n Y (1,2,)n = 为两个二阶矩实随机变量序列,X 和Y 为两个二阶矩实随机变量。若l.i.m n n X X →∞ =,l.i.m n n Y Y →∞ =,求证lim {}{}m n m n E X X E XY →∞→∞ =。 基因表达谱芯片的数据分析(2012-03-13 15:25:58)转载▼ 标签:杂谈分类:生物信息 摘要 基因芯片数据分析的目的就是从看似杂乱无序的数据中找出它固有的规律, 本文根据数据分析的目的, 从差异基因表达分析、聚类分析、判别分析以及其它分析等角度对芯片数据分析进行综述, 并对每一种方法的优缺点进行评述, 为正确选用基因芯片数据分析方法提供参考. 关键词: 基因芯片; 数据分析; 差异基因表达; 聚类分析; 判别分析 吴斌, 沈自尹. 基因表达谱芯片的数据分析. 世界华人消化杂志2006;14(1):68-74 https://www.360docs.net/doc/643687011.html,/1009-3079/14/68.asp 0 引言 基因芯片数据分析就是对从基因芯片高密度杂交点阵图中提取的杂交点荧光强度信号进行的定量分析, 通过有效数据的筛选和相关基因表达谱的聚类, 最终整合杂交点的生物学信息, 发现基因的表达谱与功能可能存在的联系. 然而每次实验都产生海量数据, 如何解读芯片上成千上万个基因点的杂交信息, 将无机的信息数据与有机的生命活动联系起来, 阐释生命特征和规律以及基因的功能, 是生物信息学研究的重要课题[1]. 基因芯片的数据分析方法从机器学习的角度可分为监督分析和非监督分析, 假如分类还没有形成, 非监督分析和聚类方法是恰当的分析方法; 假如分类已经存在, 则监督分析和判别方法就比非监督分析和聚类方法更有效率。根据研究目的的不同[2,3], 我们对基因芯片数据分析方法分类如下: (1)差异基因表达分析: 基因芯片可用于监测基因在不同组织样品中的表达差异, 例如在正常细胞和肿瘤细胞中; (2)聚类分析: 分析基因或样本之间的相互关系, 使用的统计方法主要是聚类分析; (3)判别分析: 以某些在不同样品中表达差异显著的基因作为模版, 通过判别分析就可建立有效的疾病诊断方法. 1 差异基因表达分析(difference expression, DE) 对于使用参照实验设计进行的重复实验, 可以对2样本的基因表达数据进行差异基因表达分 基因表达检测的最终技术目标是能确定所关注的任何组织、细胞的 RNA的绝对表达量。可以先从样本中抽提RNA,再标记RNA, 然后将这些标记物作探针与芯片杂交,就可得出原始样本中不同 RNA的量。然而用于杂交的某个特定基因的RNA的量与在一个 相应杂交反应中的信号强度之间的关系十分复杂,它取决于多种 因素,包括标记方法、杂交条件、目的基因的特征和序列。所以 芯片的方法最好用于检验两个或多个样本中的某种RNA的相对 表达量。样本之间某个基因表达的差异性(包括表达的时间、空 间特性及受干扰时的改变)是基因表达最重要的,而了解RNA 的绝对表达丰度只为进一步的应用或多或少地起一些作用。 基因表达的检测有几种方法。经典的方法(仍然重要)是根据在 细胞或生物体中所观察到的生物化学或表型的变化来决定某一 特定基因是否表达。随着大分子分离技术的进步使得特异的基因 产物或蛋白分子的识别和分离成为可能。随着重组DNA技术的 运用,现在有可能检测.分析任何基因的转录产物。目前有好几 种方法广泛应用于于研究特定RNA分子。这些方法包括原位杂交.NORTHERN凝胶分析.打点或印迹打点.S-1核酸酶分 析和RNA酶保护研究。这里描述RT-PCR从RNA水平上检查 基因表达的应用。8 f3 f- |2 L) K) b7 ]- ~- | RT-PCR检测基因表达的问题讨论 关于RT-PCR技术方法的描述参见PCR技术应用进展,在此主要讨论它在应用中的问题。理论上1μL细胞质总RNA对稀有mRNA扩增是足够了(每个细胞有1个或几个拷贝)。1μL差不多相当于50-100,000个典型哺乳动物细胞的细胞质中所含RNA的数量,靶分子的数量通常大于50,000,因此扩增是很容易的。该方法所能检测的最低靶分子的数量可能与通常的DNAPCR相同;例如它能检测出单个RNA分子。当已知量的转录RNA(用T7RNA聚合酶体外合成)经一系列稀释,实验结果表明通过PCR的方法可检测出10个分子或低于10个分子,这是反映其灵敏度的一个实例。用此技术现已从不到1个philadelphia染色体阳性细胞株K562中检测到了白血病特异的MRNA的转录子。因此没必要分离polyA+RNA,RNA/PCR法有足够的灵敏度来满足绝大多数实验条件的需要。 7 H+ F& _* S6 W( a8 p: [, @- d, { 将PCR缓冲液同时用于反转录酶反应和PCR反应,可简化实验步骤。我们发现整个反应过程皆用PCR缓冲液的结果相当于或优于先用反转录缓冲液合成CDNA,然后PCR缓冲液进行PCR扩增循环。当然,值得注意的是PCR缓冲液并不最适合第一条DNA链的合成。我们对不同的缓冲液用于大片段DNA 合成是否成功还没有进行过严格的研究。 随机信号分析常用函数及示例 1、熟悉练习使用下列MATLAB函数,给出各个函数的功能说明和内部参数的意 义,并给出至少一个使用例子和运行结果。 rand(): 函数功能:生成均匀分布的伪随机数 使用方法: r = rand(n) 生成n*n的包含标准均匀分布的随机矩阵,其元素在(0,1)内。 rand(m,n)或rand([m,n]) 生成的m*n随机矩阵。 rand(m,n,p,...)或rand([m,n,p,...]) 生成的m*n*p随机矩数组。 rand () 产生一个随机数。 rand(size(A)) 生成与数组A大小相同的随机数组。 r = rand(..., 'double')或r = rand(..., 'single') 返回指定类型的标准随机数,其中double指随机数为双精度浮点数,single 指随机数为单精度浮点数。 例:r=rand(3,4); 运行结果: r= 0.4235 0.4329 0.7604 0.2091 0.5155 0.2259 0.5298 0.3798 0.3340 0.5798 0.6405 0.7833 randn(): 函数功能:生成正态分布伪随机数 使用方法: r = randn(n) 生成n*n的包含标准正态分布的随机矩阵。 randn(m,n)或randn([m,n]) 生成的m*n随机矩阵。 randn(m,n,p,...)或randn([m,n,p,...]) 生成的m*n*p随机矩数组。 randn () 产生一个随机数。 randn(size(A)) 生成与数组A大小相同的随机数组。 r = randn(..., 'double')或r = randn(..., 'single') 返回指定类型的标准随机数,其中double指随机数为双精度浮点数,single 指随机数为单精度浮点数。 例: 万方数据 万方数据 万方数据 基因表达系列分析技术及其应用 作者:党冬梅, 魏晓萍, 惠起源, 符兆英 作者单位:延安大学医学院,陕西,延安,716000 刊名: 延安大学学报(医学科学版) 英文刊名:JOURNAL OF YANAN UNIVERSITY(MEDICAL SCIENCE EDITION) 年,卷(期):2005,3(1) 被引用次数:0次 参考文献(8条) 1.Velculescu E查看详情 1995 2.Menssen A.Hermeking H Characterization of the c-MYC regulated transcriptome by SAGE:Identification and analysis of target genes 2002(09) 3.Levens D Disentangling the MYC web 2002(09) 4.Matsumura H.Nirasawa S.Terachi R Transcript profiling in rice (Oryzn sation L.) seedlings using serial analysis of gene expression 1999(06) 5.Margulies E H.Kardia S L R.Innis J W查看详情 2001 6.Du Z.Scott A D.May G D Expression profiling of UV-and Gamma-irradiated Ambidopsis plantlets through serial analysis of gene expression 2001 7.Inadera H.Hashimot0 S.Dongi H Y WISP-2 as a novel estrogen-responsive gene in human breast cancer cell 2000(01) 8.Xu L L.Shanmugan N.Sesterhenn I A A novel androgen regulated gene,PMEPAI.Iocated on chromosome 20113 exhibit high level expression in protstate 2000(03) 本文链接:https://www.360docs.net/doc/643687011.html,/Periodical_yadxxb-yxkxb200501045.aspx 授权使用:西安交通大学(xajtdx),授权号:fa53fce6-7ae2-4ac8-b779-9e9900a7d328 下载时间:2011年3月1日 基因差异表达技术 真核生物中,从个体的生长、发育、衰老、死亡,到组织的得化、调亡以及细胞对各种生物、理化因子的应答,本质上都涉及基因的选择性表达。高等生物大约有30000个不同的基因,但在生物体内任意8细胞中只有10%的基因的以表达,而这些基因的表达按特定的时间和空间顺序有序地进行着,这种表达的方式即为基因的差异表达。其包括新出现的基因的表达与表达量有差异的基因的表达。生物体表现出的各种特性,主要是由于基因的差异表达引起的。 由于基因的差异表达的变化是调控细胞生命活动过程的核心机制,通过比较同一类细胞在不同生理条件下或在不同生长发育阶段的基因表达差异,可为分析生命活动过程提供重要信息。研究基因差异表达的主要技术有差别杂交(differential hybridization)、扣除(消减)杂交(subtractive hybridization of cDNA,SHD)、mRNA差异显示(mRNA differential display,DD)、抑制消减杂交法(suppression subtractive hybridization,SSH)、代表性差异分析(represential display analysis,RDA)、交互扣除RNA差别显示技术(reciprocal subtraction differential RNA display)、基因表达系列分析(serial analysis of gene expression,SAGE)、电子消减(electronic subtraction)和DNA微列阵分析(DNA microarray)等。 一、差别杂交与扣除杂交 差别杂交(differential hybridization)又叫差别筛选(differential screening),适用于分离经特殊处理而被诱发表达的mRNA的cDNA克隆。为了增加这种方法的有效性,后来又发展出了扣除杂交(subtractive hybridization)或扣除cDNA克隆(subtractive cDNA cloning),它是通过构建扣除文库(subtractive library)得以实现的。 (一)差别杂交 从本质上讲,差别杂交也是属于核酸杂交的范畴。它特别适用于分离在特定组织中表达 随机信号分析 朱华,等北京理工大学出版社2011-07-01 《随机信号分析》是高等学校工科电子类专业基础教材。内容为概率论基础、平稳随机过程、窄带随机过程、随机信号通过线性与非线性系统的理论与分析方法等。在相应的部分增加了离散随机信号的分析。《随即信号分析》的特点侧重在物理概念和分析方法上,对复杂的理论和数学问题着重用与实际的电子工程技术问题相联系的途径及方法去处理。《随即信号分析》配套的习题和解题指南将与《随即信号分析》同期出版。《随即信号分析》适用于电子工程系硕士研究生及高年级本科生,也适用于科技工作者参考。 第一章概率论 1.1 概率空间的概念 1.1.1 古典概率 1.1.2 几何概率 1.1.3 统计概率 1.2 条件概率空间 1.2.1 条件概率的定义 1.2.2 全概率公式 1.2.3 贝叶斯公式 1.2.4 独立事件、统计独立 1.3 随机变量及其概率分布函数 1.3.1 随机变量的概念 1.3.2 离散型随机变量及其分布列 1.3.3 连续型随机变量及其密度函数 1.3.4 分布函数及其基本性质 1.4 多维随机变量及其分布函数 1.4.1 二维分布函数及其基本性质 1.4.2 边沿分布 1.4.3 相互独立的随机变量与条件分布 1.5 随机变量函数的分布 1.5.1 一维随机变量函数的分布 1.5.2 二维随机变量函数的分布 1.5.3 二维正态随机变量函数的变换 1.5.4 多维情况 1.5.5 多维正态概率密度的矩阵表示法 1.6 随机变量的数字特征 1.6.1 统计平均值与随机变量的数学期望值 1.6.2 随机变量函数的期望值 1.6.3 条件数学期望 1.6.4 随机变量的各阶矩 1.7 随机变量的特征函数 1.7.1 特征函数的定义 1.7.2 特征函数的性质 概率论基础 1.概率空间、概率(条件概率、全概率公式、贝叶斯公式) 2.随机变量的定义(一维、二维实随机变量) 3.随机变量的描述: ⑴统计特性 一维、二维概率密度函数、一维二维概率分布函数、边缘分布 概率分布函数、概率密度函数的关系 ⑵数字特征 一维数字特征:期望、方差、均方值(定义、物理含义、期望和方差的性质、三者之间的关系) 二维数字特征:相关值、协方差、相关系数(定义、相互关系) ⑶互不相关、统计独立、正交的定义及其相互关系 4.随机变量函数的分布 △雅柯比变换(随机变量函数的变换一维随机变量函数的单值和双值变换、二维随机变量函数的单值变换) 5、高斯随机变量 一维和二维概率密度函数表达式 高斯随机变量的性质 △随机变量的特征函数及基本性质 、 随机信号的时域分析 1、随机信号的定义 从三个方面来理解①随机过程(),X t ζ是,t ζ两个变量的函数②(),X t ζ是随时间t 变化的随机变量③(),X t ζ可看成无穷多维随机矢量在0,t n ?→→∞的推广 2、什么是随机过程的样本函数?什么是过程的状态?随机过程与随机变量、样本函数之间的关系? 3、随机信号的统计特性分析:概率密度函数和概率分布函数(一维、二维要求掌握) 4、随机信号的数字特征分析(定义、物理含义、相互关系) 一维:期望函数、方差函数、均方值函数。(相互关系) 二维:自相关函数、自协方差函数、互相关函数、互协方差函数(相互关系) 5、严平稳、宽平稳 定义、二者关系、判断宽平稳的条件、平稳的意义、联合平稳定义及判定 6、平稳随机信号自相关函数的性质: 0点值,偶函数,均值,相关值,方差 7、两个随机信号之间的“正交”、“不相关”、“独立”。 (定义、相互关系) 8、高斯随机信号 定义(掌握一维和二维)、高斯随机信号的性质 9、各态历经性 定义、意义、判定条件(时间平均算子、统计平均算子)、平稳性与各态历经性的关系直流分量、直流平均功率、总平均功率、交流平均功率 随机信号的频域分析 1、随机信号是功率信号,不存在傅里叶变换,在频域只研究其功率谱。 功率谱密度的含义,与总平均功率的关系 2、一般随机信号功率谱计算公式与方法 3、平稳随机信号的功率谱密度计算方法 基因表达分析 1、EST(Expressed Sequence Tag)表达序列标签(EST)分析 1、EST基本介绍 1、定义: EST是从已建好的cDNA库中随机取出一个克隆,进行5’端或3’端进行一轮单向自动测序,获得短的cDNA部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20到7000bp不等,平均长度为400bp。 EST来源于一定环境下一个组织总mRNA所构建的cDNA文库,因此,EST也能说明该组织中各基因的表达水平。 2、技术路线: 首先从样品组织中提取mRNA,在逆转录酶的作用下用oligo(dT)作为引物进行RT-PCR 合成cDNA,再选择合适的载体构建cDNA文库,对各菌株加以整理,将每一个菌株的插入片段根据载体多克隆位点设计引物进行两端一次性自动化测序,这就是EST序列的产生过程。 3、EST数据的优点和缺点: (1)相对于大规模基因组测序而言,EST测序更加快速和廉价。 (2)EST数据单向测序,质量比较低,经常出现相位的偏差。 (3)EST只是基因的一部分,而且序列里有载体序列。 (4)EST数据具有冗余性。 (5)EST数据具有组织和不同时期特异性。 4、EST数据的应用 EST作为表达基因所在区域的分子标签因编码DNA序列高度保守而具有自身的特殊性质,与来自非表达序列的标记(如AFLP、RAPD、SSR等)相比,更可能穿越家系与种的限制。因此,EST标记在亲缘关系较远的物种间比较基因组连锁图和比较质量性状信息是特别有用的。同样,对于一个DNA序列缺乏的目标物种,来源于其他物种的EST也能用于该物种有益基因的遗传作图,加速物种间相关信息的迅速转化。具体说,EST的作用表现在: SAGE 技术 MRNA 结合到微珠子上(Microscopic Bead and mRNA) mRNA 转录成DNA(mRNA binds to bait and is copied into DNA) 用酶切开DNA的一小段(An enzyme cuts the DNA) 另一个酶定在DNA末端以便切下一小段(An enzyme locks onto the DNA and cuts off a short tag),这一小段就被视为这个基因的标签 两个标签连在一起(Two tags are linked together) 在末端的定位分子被切掉(Enzymes cut off the "Docking Molecules") 都连成一条线(Di-Tags are combined into large concatemers) DNA上所携带的遗传信息,需要通过RNA为中介体,合成出组织和正常生理功能所需要的蛋白质,这个过程被称为基因的表达。在生物体中不同的组织和器官所表达的基因群是不一样的,我们把基因群的表达状况称为基因表达谱。目前,高通量地研究基因表达谱的方法主要有两种,即生物芯片和基因表达串联分析(serial analysis of gene expression, SAGE)。基因芯片所能检测的基因必须是已知的基因,放在芯片上几种基因的探针就只能检测这几种基因的表达谱;相比之下,SAGE能以远高于DNA芯片的精确度和重复性来检测在病理条件下基因表达谱的改变,而不必考虑所检测的基因是已知的还是未知的。因此在检测疾病相关的新基因,特别是无法用基因芯片进行检测的低表达量致病基因时,SAGE是目前的最佳手段,无可取代。 SAGE技术为Genzyme公司所拥有的专利技术。其技术简介如下: SAGE技术得以建立的理论基础 首先,一段来自于任一转录本特定区域的"标签"(Tag),即长度仅9-14bp的短核苷酸序列,就已包含足够的信息以特异性地确定该转录本。例如:一个9碱基的序列能有49=262144种不同的排列组合,而人类基因组据估计仅编码80000种转录本,因此在理论上每一个9碱基标签就能够代表一种转录本的特征序列。 第二,如果将短片段标签相互连接、集中形成长的DNA分子,则对该克隆进行 基因表达及其分析技术 生命现象的奥秘隐藏在基因组中,对基因组的解码一直是现代生命科学的主流。基因组学研究可以说是当今生命科学领域炙手可热的方向。从DNA 测序到SNP、拷贝数变异(copy number variation , CNV)等DNA多态性分析,到DNA 甲基化修饰等表观遗传学研究,生命过程的遗传基础不断被解读。 基因组研究的重要性自然不言而喻。应该说,DNA 测序技术在基因组研究 中功不可没,从San ger测序技术到目前盛行的新一代测序技术(Next Gen eration Seque ncing NGS)到即将走到前台的单分子测序技术,测序技术是基因组解读最重要的主流技术。而基因组测序、基因组多态性分析、DNA 甲基化修饰等表观遗传分析等在基因组研究中是最前沿的课题。但是基因组研究终究类似“基因算命”,再清晰的序列信息也无法真正说明一个基因的功能,基因功能的最后鉴定还得依赖转录组学和蛋白组学,而转录作为基因发挥功能的第一步,对基因功能解读就变得至关重要。声称特定基因、特定SNP、特定CNV、特定DNA修饰等与某种表型有关,最终需要转基因、基因敲除、突变、 RNAi 、中和抗体等技术验证,并必不可少要结合基因转录、翻译和蛋白修饰等数据。 基因实现功能的第一步就是转录为mRNA或非编码RNA,转录组学主要研究基因转录为RNA 的过程。在转录研究中,下面几点是必须考虑的: 1,基因是否转录(基因是否表达)及基因表达水平高低(基因是低丰度表达还是中、高丰度表达)。特定基因有时候在一个细胞中只有一个拷贝的表达,而表达量会随细胞类型不同或发育、生长阶段不同或生理、病理状态不同而改变。因此任何基 利用实时定量PCR和2-△△CT法分析基因相对表达量 METHODS 25, 402–408 (2001) Analysis of Relative Gene Expression Data Using Real-Time Quantitati ve PCR and the 2-△△CT Method Kenneth J. Livak* and Thomas D. Schmittgen?,1 *Applied Biosystems, Foster City, California 94404; and ? Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Pullman, Washington 99164-6534 摘要: 现在最常用的两种分析实时定量PCR 实验数据的方法是绝对定量和相对定量。绝对定量通过标准曲线计算起始模板的拷贝数;相对定量方法则是比较经过处理的样品和未经处理的样品目标转录本之间的表达差异。2-△△CT方法是实时定量P CR 实验中分析基因表达相对变化的一种简便方法,即相对定量的一种简便方法。本文介绍了该方法的推导,假设及其应用。另外,在本文中我们还介绍了两种2-△△CT衍生方法的推导和应用,它们在实时定量 PCR 数据分析中可能会被用到。 关键词:反转录PCR 定量PCR 相对定量实时PCR Taqman 反转录 PCR (RT-PCR )是基因表达定量非常有用的一种方法(1 - 3 )。实时PCR 技术和RT-PCR 的结合产生了反转录定量 PCR 技术(4 ,5 )。实时定量 P CR 的数据分析方法有两种:绝对定量和相对定量。绝对定量一般通过定量标准曲线来确定我们所感兴趣的转录本的拷贝数;相对定量方法则是用来确定经过不同处理的样品目标转录本之间的表达差异或是目标转录本在不同时相的表达差异。 绝对定量通常在需要确定转录本绝对拷贝数的条件下使用。通过实时 PCR 进行绝对定量已有多篇报道(6 - 9 ),包括已发表的两篇研究论文(10,11 )。在有些情况下,并不需要对转录本进行绝对定量,只需要给出相对基因表达差异即可。显然,我们说 X 基因在经过某种处理後表达量增加 2.5 倍比说该基因的表达从1000 拷贝/ 细胞增加到2500 拷贝/ 细胞更加直观。 第8章基因表达数据分析 基因芯片或DNA微阵列等高通量检测技术的发展,可以从全基因组水平定量或定性检测基因转录产物mRNA,获取基因表达的信息。由于生物体中的细胞种类繁多,同时基因表达具有时空特异性,因此,基因表达数据要比基因组数据更为复杂、数据量更大、数据的增长速度更快。基因表达数据中蕴含着基因调控的规律,可以反映细胞当前的生理状态,例如(??)是否恶化、(??)是否对药物有效等。对基因表达数据的分析是生物信息学的重大挑战之一,也是DNA微阵列能够推广应用的关键环节之一。 基因表达数据分析的对象是在不同条件下,全部或部分基因的表达数据所构成的数据矩阵。通过对数据矩阵的分析,回答一些生物学问题,例如,基因的功能是什么?在不同条件或不同细胞类型中,哪些基因的表达存在差异?在特定的条件下,哪些基因的表达发生了显著改变,这些基因受到哪些基因的调节,或者调控哪些其它的基因?哪些基因的表达是条件特异性的,根据它们的行为可以判断细胞的状态(正常或癌变)????等等。对这些问题的回答,结合其他生物学知识和数据有助于阐明基因的调控路径和基因之间的调控网络。揭示基因调控路径和网络是生物学和生物信息学共同关注的目标,是系统生物学(Systems Biology,在附录中增加解释条目!)研究的核心内容。目前,对基因表达数据的分析主要是在三个逐渐复杂的层次上进行:1、分析单个基因的表达水平,根据在不同实验条件下,该基因表达水平的变化,来判断它的功能,例如可以确定肿瘤类型特异基因。采用的分析方法可以是统计学中的假设检验等。2、考虑基因组合,将基因分组,研究基因的共同功能、相互作用以及协同调控等。多采用聚类分析等方法。3、尝试推断潜在的基因调控网络,从机理上解释观察到的基因表达谱。多采用反工程的方法。 本章首先介绍基因表达数据的来源和预处理方法;然后介绍基因表达数据分析的主要方法,即表达差异分析和聚类分析;最后简单介绍从基因表达数据出发研究基因调控网络的一些经典模型。 8.1 基因表达数据的获取 基因表达数据反映的是直接或间接测量得到的基因转录产物mRNA在细胞中的拷贝数或者水平(转录??),这些数据可以用于分析哪些基因的表达发生了改变,它们有何相关性,在不同条件下基因是如何受影响的。它们在医学临床诊断、药物疗效判断、揭示疾病发生机制等方面有重要的应用。目前检测mRNA水平的方法有DNA微阵列、基因芯片、基因表达串行化分析(Serial analysis of gene expression,SAGE)、RT-PCR、EST测序等。目前,最主要的表达数据来自于基因芯片或cDNA微阵列,它们的原理是相同的,利用4种核苷酸之间两两配对互补的特性,使两条在序列上互补的单链形成双链,这个过程被称为杂交。基本技术是:在一个约1cm2大小的玻璃片上,将称为探针的核苷酸片段固定在上面,这个过程称为芯片制备;从细胞或组织中提取mRNA,通过RT-PCR合成荧光标记的cDNA,与芯片杂交;用激光显微镜或荧光显微镜检测杂交后的芯片,获取荧光强度,分析细胞中的mRNA的相对水平。 高等真核生物的基因组一般具有80 000~100 000个基因,而每一个细胞大约只表达其中的15%[1]。基因在不同细胞间及不同生长阶段的选择性表达决定了生命活动的多样性,如发育与分化、衰老与死亡、内环境稳定、细胞周期调控等。比较细胞间基因表达的差异为我们揭示生命活动的规律提供了依据。 由于真核细胞mRNA 3′端一般含有Poly(A)尾,因此现有的方法基本上都是利用共同引物将不同的mRNA反转录成cDNA,以cDNA为对象研究基因表达的差异。1992年Liang等[2]建立了一种差异显示反转录PCR法(differential display reverse transcription PCR,DDRT-PCR),为检测成批基因表达的差异开辟了新天地。迄今为止已出现了大量应用该技术的研究报道[3,4]。然而,尽管应用DDRT-PCR方法已经取得了不少成果,而且该方法还在不断改进之中,但它仍然存在几个难以解决的问题:(1) 重复率低,至少有20%的差异条带不能被准确重复[5];(2) 假阳性率可以高达90%[6];(3) 获得的差异表达序列极少包含编码信息。近年来,针对DDRT-PCR方法的不足,又有几种新的检测差异表达基因的方法出现,现仅就这方面的进展做一简要介绍。 1.基因表达指纹(gene expression fingerprinting,GEF):GEF技术使用生物素标记的引物Bio-T13合成cDNA第一链,用dGTP对其进行末端加尾,再以富含C的引物引发合成cDNA第二链。用限制性内切酶消化双链cDNA,以交联有抗生物素蛋白的微球捕获cDNA3′端,以T4DNA连接酶连接同前述内切酶相对应的适配子,并以Bio-T13及适配子中的序列作为新的引物进行特异的PCR 扩增,得到大量的特异cDNA片段。适配子末端被32P-dATP标记后,固定于微球上的cDNA片段经过一系列酶切,产生的酶切片段从微球表面释放出来,其中那些含有标记末端的片段经凝胶电泳后构成mRNA指纹图谱。通过分析不同细胞间的指纹图谱就能得到差异表达的序列[7]。GEF技术所需的工作量较DDRT-PCR明显减少,由于用酶切反应替代了条件不严格的PCR反应,其重复性也较好,假阳性率低,并且所获得的片段中包含有一定的编码信息。GEF技术最大的缺点在于电泳技术的局限。由于它的指纹图谱要显示在同一块电泳胶上,经过几轮酶切之后常会得到1 000~2 000条电泳带,而现有的PAGE电泳很少能分辨超过400条带,故只有15%~30%的mRNA能够被辨认出来,因此得 荧光定量PCR 在基因表达分析中的应用 所谓基因表达就是指在特定的时刻某种我们感兴趣的基因在组织或细胞中的mRNA 的表达数量。众所周知,很多的疾病(如肿瘤)的发生发展、很多药物的作用机理、很多生物的代谢调控作用等都和基因表达的变化有关,因此对基因表达进行精确定量是十分重要的。过去为了对mRNA 进行定量有了各种各样的方法,如Southern 杂交、Northern 杂交、原位杂交、传统PCR 等,但是我们也都知道这些技术灵敏性较差,重复性不好,操作比较烦琐,已经无法满足现在科研和检测的需要,于是荧光定量PCR 技术也就应运而生了。荧光定量PCR 技术能对核酸进行精确定量,因此大大提高了在基因表达的准确性和灵敏度,深受用户的青睐,广泛的应用于肿瘤研究、药物筛选、功能基因组研究等各个领域,目前已经成了很多科研文章发表的重要实验内容。 基因表达分析中常见到的重要问题 1、要检测的基因 基因表达分析的目的就是检测某种我们感兴趣的基因在不同组织或细胞中的表达差异。荧光定量PCR 技术可以对核酸物质的含量进行精确的定量,也就成了研究基因表达差异的一把利器。 在基因表达分析实验中要检测两个基因,一个是目的基因和另一个是看家基因。之所以要引入看家基因是由于不能确定要比较的样品所用的组织起始量相同。就是说比如有的老师提取正常样品的基因时用了100个细胞,而提取病变样品时只用了10个细胞,这时候的基因表达差异可能是由于提取时候的样品细胞数不同引起的,为了纠正这种误差,我们选用认为在两个样本中表达量不变的基因作为内参照,来去除这带来的干扰。例如,要研究某个基因在肿瘤样品和正常样品中的基因表达差异。我们在实验中发现我们选择研究的正常样品中的看家基因的表达量是肿瘤样品中的10倍,就认为正常样品的细胞数就是肿瘤样品细胞数的10倍,那么在肿瘤样品中目的基因的基因表达量应该乘以10倍,才能和正常样品进行比较。 2、计算基因表达差异 基因表达差异的计算是通过所得到的Ct 值来计算的,要计算两个样品(待测样品和对照样品)的目的基因的表达差异必须检测得到4个Ct 值:待测样品和对照样品中目的基因和看家基因的Ct 值。 那么基因表达差异应该计算为 基因表达差异=2(△Ct1-△Ct2) 目的基因 看家基因 待测样品 对照样品 △Ct1 △Ct2 基因表达谱分析技术 1、微阵列技术(microarray) 这是近年来发展起来的可用于大规模快速检测基因差别表达、基因组表达谱、DNA序列多态性、致病基因或疾病相关基因的一项新的基因功能研究技术。其原理基本是利用光导化学合成、照相平板印刷以及固相表面化学合成等技术,在固相表面合成成千上万个寡核苷酸“探针”(cDNA、ESTs或基因特异的寡核苷酸),并与放射性同位素或荧光物标记的来自不同细胞、组织或整个器官的DNA或mRNA反转录生成的第一链cDNA进行杂交,然后用特殊的检测系统对每个杂交点进行定量分析。其优点是可以同时对大量基因,甚至整个基因组的基因表达进行对比分析。包括cDNA芯片(cDNA microarray)和DNA 芯片(DNA chips)。 cDNA芯片使用的载体可以是尼龙膜,也可以是玻片。当使用尼龙膜时,目前的技术水平可以将20000份材料点在一张12cm×18cm的膜上。尼龙膜上所点的一般是编好顺序的变性了的双链cDNA片段。要得到基因表达情况的数据,只需要将未知的样品与其杂交即可。杂交的结果表示这一样品中基因的表达模式,而比较两份不同样品的杂交结果就可以得到在不同样品中表达模式存在差异的基因。杂交使用的探针一般为mRNA的反转录产物,标记探针使用32PdATP。如果使用玻片为载体,点阵的密度要高于尼龙膜。杂交时使用两种不同颜色的荧光标记不同的两份样品,然后将两份样品混合起来与一张芯片杂 交。洗去未杂交的探针以后,能够结合标记cDNA的点受到激发后会发出荧光。通过扫描装置可以检测各个点发出荧光的强度。对每一个点而言,所发出的两种不同荧光的强度的比值,就代表它在不同样品中的丰度。一般来讲,显示出来的图像中,黄色的点表示在不同的样品中丰度的差异不大,红色和绿色的点代表在不同样品中其丰度各不相同。使用尼龙膜为载体制作cDNA芯片进行研究的费用要比玻片低,因为尼龙膜可以重复杂交。检测两种不同的组织或相同组织在不同条件下基因表达的差异,只需要使用少量的尼龙膜。但是利用玻片制作的cDNA芯片灵敏度更高,而且可以使用2种探针同时与芯片杂交,从而降低了因为杂交操作带来的差异;缺点是无法重复使用还必须使用更为复杂的仪器。 Guo等(2004)将包含104个重组子的cDNA文库点在芯片上,用于检测拟南芥叶片衰老时的基因表达模式,得到大约6200差异表达的ESTs,对应2491个非重复基因。其中有134个基因编码转录因子,182个基因预测参与信号传导,如MAPK级联传导路径。Li等(2006)设计高密度的寡核苷酸tiling microarray方法,检测籼稻全基因组转录表达情况。芯片上包含13,078,888个36-mer寡核苷酸探针,基于籼稻全基因组shot-gun测序的序列合成,大约81.9%(35,970)的基因发生转录事件。Hu等(2006)用含有60,000寡核苷酸探针(代表水稻全部预测表达基因)的芯片检测抗旱转基因植株(过量表达SNAC1水稻)中基因的表达情况,揭示大量的逆境相关基因都是上升表达的。 2、基因表达系列分析(Serial analysis of gene expression, SAGE) 随机信号分析习题一 概率:P( 1),P(1 2)。 2. 设(X,Y) 的联合密度函数为 f XY(x,y)e (x y), x 0, y 0, 0 , other 求P 0 X 1,0 Y 1 。 3. 设二维随机变量(X,Y) 的联合密度函数为 1 1 2 2 f XY(x, y) 1exp 12(x22xy 5y2) 求:(1)边沿密度f X(x) ,f Y(y) (2)条件概率密度f Y|X (y|x),f X|Y(x|y) 4. 设离散型随机变量X 的可能取值为1,0,1,2 ,取每个值的概率都为1/4 ,又设随机变 量Y g(X) X3X 。 (1)求Y 的可能取值 ( 2)确定Y 的分布。 (3)求E[Y] 。 5. 设两个离散随机变量X ,Y 的联合概率密度为: 111 f XY(x,y) 3(x 2) (y 1) 3(x 3) (y 1) 3(x A) (y A) 试求:(1) X 与Y 不相关时的所有A 值。 (2) X 与Y 统计独立时所有A值。 6. 二维随机变量( X ,Y )满足: X cos Y sin 为在[0,2 ]上均匀分布的随机变量,讨论X ,Y 的独立性与相关性。 7. 已知随机变量X 的概率密度为f (x),求Y bX 2的概率密度f (y)。 1. 设函数F(x),试证明F(x) 是某个随机变量的分布函数。并求下列 8. 两个随机变量X1,X 2 ,已知其联合概率密度为f(x1,x2),求X1 X 2的概率密度? 9. 设X 是零均值,单位方差的高斯随机变量,y g(x) 如图,求y g(x) 的概率密度 f Y(y) W X 2 Y 2 Z X 2 设X ,Y是相互独立的高斯变量。求随机变量W和Z的联合概率密度函数。 11. 设随机变量W 和Z 是另两个随机变量X 和Y 的函数 WXY Z 2(X Y) 已知f XY(x,y) ,求联合概率密度函数f WZ( ,z) 。 ,axb ba 0,其它 1)求X 的特征函数, X( ) 。 2)由X( ),求E[X]。 13. 用特征函数方法求两个数学期望为0,方差为1,互相独立的高斯随机变量X1和X2之和的概率密度。 14. 证明若X n依均方收敛,即l.i.m X n X,则X n必依概率收敛于X。 n 12. 设随机变量X 为均匀分布,其概率密度f X (x) 15. 设{ X n}和{Y n} (n 1,2,L ) 为两个二阶矩实随机变量序列,X 和Y 为两个二阶矩实随 机变量。若l.i.m X n n n X ,l.i.m Y n Y,求证lim E{X m X n} E{XY} 。nm 10. 设随机变量W 和Z x基因表达谱芯片的数据分析
基因表达的检测的几种方法
matlab随机信号分析常用函数
基因表达系列分析技术及其应用
基因差异表达技术
随机信号分析
《随机信号分析基础》总复习提
基因表达分析
基因表达系列分析(Serial Analysis of Gene Expression,SAGE)技术
基因表达及分析技术
利用实时定量PCR和2-△△CT法分析基因相对表达量
基因表达数据分析
基因表达差异分析方法进展
基因表达分析
基因表达谱分析技术
随机信号分析习题