荧光衍生法测定邻苯二胺的研究

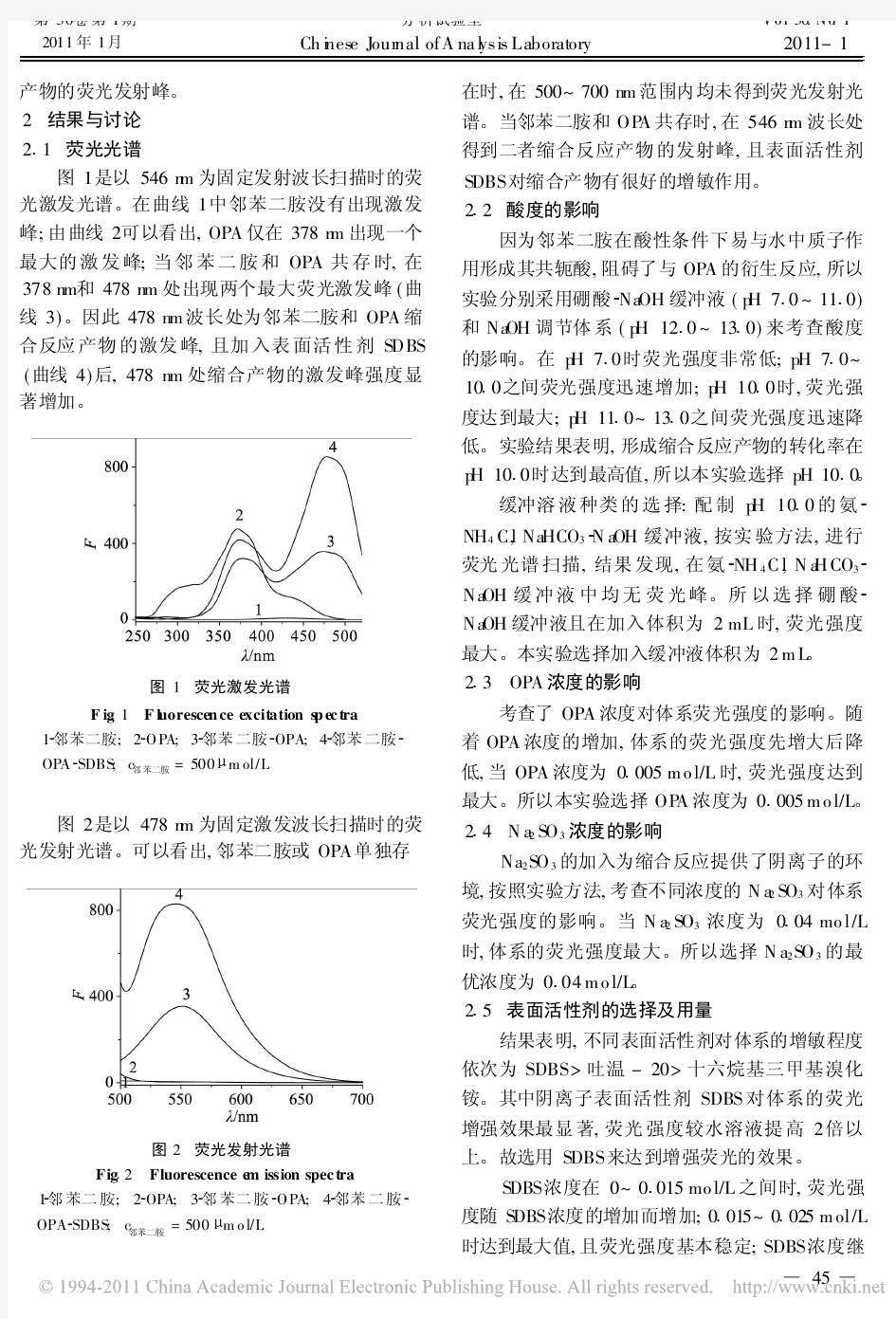
涂膜性能及测量
涂膜性能及测量 1、涂膜的制备 国家标准《GB1727—— 79(88)漆膜一般制备法》中分别列出刷涂法、喷涂法、浸涂法和刮涂法的涂膜制备方法。但在制备时需要依赖操作人员的技术熟练程度,涂膜的均匀性较难保证。采用仪器制备涂膜在当前普遍推行,方法有旋转涂漆法和刮涂器法。 2、涂膜外观及光泽测定 (1)涂膜外观 通常在日光下肉眼观察涂膜的样板有无缺陷,如刷痕、颗粒、起泡、起皱、缩孔等,一般与标准样板对比。 (2)光泽的测定基本上采用两大仪器,即光电光泽计和投影光泽计,前者用得较多。 3、涂膜的鲜映性测定 鲜映性是指涂膜表面反映影象(或投影)的清晰程度,以DOI值表示(distinctness of image)。它能表征与涂膜装饰性相关的一些性能(如光泽、平滑度、丰满度等)的综合效应。它可用来对飞机、汽车、精密仪器、家用电器,特别是高级轿车车身等的涂膜的装饰性进行等级评定。 鲜映性测定仪的关键装置是一系列标准的鲜映性数码板,以数码表示等级,分为0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.2、1.5、2.0共13个等级,称为DOI值。每个DOI值旁印有几个数字,随着DOI值升高,印的数字越来越小,用肉眼越不易辨认。观察被测表面并读取可清晰地看到的DOI值旁的数字,即为相应的鲜映性。 4、涂膜雾影测定 雾影系高光泽漆膜由于光线照射而产生的漫反射现象。雾影光泽仪是一台双光束光泽仪,其中参与光束可以消除温度对光泽以及颜色对雾影值的影响。仪器的主接收器接收漆膜的光泽,而副接收器则接收反射光泽周围的雾影。雾影值最高可达1000,但评价涂料时,雾影
人血免疫球蛋白中甘氨酸含量测定法
人血免疫球蛋白中甘氨酸含量测定法(新增) 本法系依据过量的6-氨基喹啉基-N-羟基琥珀酰亚氨基氨基甲酸酯(AQC)在一定条件下和氨基酸形成稳定的衍生产物(柱前衍生),用高效液相色谱法测定衍生产物,根据衍生产物的含量计算甘氨酸含量。 照高效液相色谱法(附录III B)测定 色谱条件和系统适应性试验用十八烷基硅烷键合硅胶为基质的C18反相色谱柱,颗粒度4μm,内径3.9mm,柱长150mm;柱温为37℃;以140mmol/L乙酸钠、17mmol/L 三乙胺pH 5.65、1μg/mlEDTA二钠盐为流动相A液,以100%乙腈为流动相B液,以纯水为流动相C液,流速为每分钟1.0ml,梯度洗脱32分钟(见梯度表),检测波长为248nm。甘氨酸与相邻色谱峰之间分离度应≥1.5;拖尾因子(T)为0.95~1.40之间(甘氨酸和α-氨基丁酸峰)。重复性RSD≤2.0%(甘氨酸对照品峰面积测量值)。 内标溶液的制备精密称取α-氨基丁酸对照品4.0g, 加超纯水至100ml定容。 对照品溶液的制备(1)精密称取甘氨酸标准2.5g加超纯水至100ml定容。(2)精密量取(1)项氨基酸标准溶液1.0ml,加9.0ml 1.5%磺基水扬酸,混匀静置2小时以上3000rpm离心10分钟,留取上清备用。①分别精密量取上清液50μl、100μl、125μl、150μl、200μl,置10ml容量瓶中,用纯水定容。②分别精密量取①项中各甘氨酸标准溶液0.1 ml,加0.4ml纯水,加内标0.02ml混匀备用。精密量取(2)②项甘氨酸标准溶液10μl放入衍生管中加70μl硼酸缓冲液(pH8~10)涡旋混和并加入20μl AQC衍生剂涡旋混和15秒,备用。 供试品溶液的制备精密量取供试品溶液1.0ml,加9.0ml 1.5%磺基水扬酸,混匀静置2小时以上3000rpm离心10分钟,留取上清备用。(1)精密量取离心上清液1.0ml,置10ml容量瓶中,用纯水定容。(2)精密量取(1)项中供试品溶液0.1 ml,加0.4ml纯水,加内标0.02ml混匀备用。精密量取(2)项供试品溶液10μl放入衍生管中加70μl硼酸缓冲液涡旋混和并加入20μl AQC衍生剂涡旋混和15秒, 备用。 测定法精密量对照品溶液与供试品溶液,分别注入液相色谱仪,记录色谱图32分钟。进样量为10μl。按内标法计算。 梯度表 1
分子荧光法测定水杨酸乙酰水杨酸1
分子荧光法测定水杨酸和乙酰水杨酸 一、实验目的 1、学习荧光分析法的基本原理。 2、掌握应用分子荧光法测定乙酰水杨酸、水杨酸的分析方法。 3、了解F-3501型荧光分光光度计的主要结构及工作原理,掌握其正确的使用方法。 二、实验原理 某些物质经紫外光或波长较短的可见光照射后,会发射出较入射波长更长的荧光。荧光光谱反映了物质的特性,建立在测量荧光光谱基础上的分析方法称为荧光分析法。当进行荧光测定时,总要选择不同波长的光波进行测定,即一个为激发光——物质所吸收的光;另一个为物质吸收后发出的光称为发射光或荧光。 对同一物质而言,在稀溶液(即A = abc < 0.05)中,荧光的强度F与该物质的浓度C 有以下关系: F = 2.3φabcI0 式中φ为荧光过程的量子效率,a为荧光分子的吸光系数,b为试样的吸收光程,I0为入射光的强度。当I0及b 不变时,上式变为: F= KC 其中,K为常数。 荧光分析法具有灵敏度高(一般超过分光光度法2~3个数量级)、取样少、方法快速等特点,现已成为食品、生物医药、天然产品、农业、环境保护、化工等领域中的重要分析方法之一。但由于许多物质本身不会发生荧光,故在使用范围上受到一定的限制。 乙酰水杨酸(ASA,即阿司匹林)水解能生成水杨酸(SA),而在乙酰水杨酸中,或多或少都存在着水杨酸。由于两者都有苯环,也有一定的荧光效率,因而在以三氯甲烷为溶剂的条件下,可用荧光法进行测定。从乙酰水杨酸和水杨酸的激发光谱和荧光光谱中可以发现:乙酰水杨酸和水杨酸的激发波长和发射波长均不同,利用此性质,可在各自的激发波长和发射波长下分别测定。 三、仪器与试剂 1、仪器 F-3501型荧光分光光度计、电子天平、离心机、真空泵、石英比色皿、移液管、棕色容量瓶、比色管、烧杯、砂芯抽滤装置、量筒、洗耳球、洗瓶等。
农药残留分析复习题及答案
农药残留分析 农药残留分析复习题 简答题 1、写出各种硅胶代号及其意义 a.硅胶H—不含粘合剂 b.硅胶G—硅胶和锻石膏混合而成 c.硅胶HF254—不含粘合剂,含荧光 指示剂,254nm呈现强绿色荧光背景 d.硅胶GF254—同时含粘合剂和荧光指示剂,254nm光致发光,呈强的绿色荧光背景 e.硅胶HF254+336—不含粘合剂,含无机荧光剂,在254nm和336nm呈现荧光背景 2、简述农药残留分析常用的气相色谱仪检测器的类型及其特点 (1)火焰离子化检测器(FID)特点:质量型,准通用型;最高操作温度(℃)450最低检测限丙烷<5 pg 碳/s;线性范围107(±10%)主要用途各种有机化合物的分析,对碳氢化合物的灵敏度高 (2)热导检测器(TCD)特点:浓度型,通用型;最高操作温度(℃) 400;最低检测限<400 pg/ml,壬烷:20000 mv?ml/mg;线性范围丙烷:104(±5%);主要用途适用于各种无机气体和有机物的分析,多用于永久气体的分析。 (3)电子俘获检测器(ECD)特点:浓度型,选择型;最高操作温度(℃) 400;低检测限六氯苯<0.04 pg/ml;范围>104;主要用途适合分析含电负性元素或基团的有机化合物,多用于分析含卤素化合物。 (4)氮磷检测器(NPD)特点:质量型,选择型;操作温度(℃) 400;低检测限用用偶氮苯和马拉硫磷的 混合物测定:<0.4 pg氮/s; <0.2 pg磷/s;范围>105;要用途适合于含氮和含磷化合物的分析。 (5)火焰光度检测器(FPD)特点:质量型,选择型;高操作温度(℃) 250;低检测限用十二烷硫醇和 三丁基膦酸酯混合物测定:<20 pg硫/s,<0.9 pg磷/s;性范围硫:>105磷:>106;要用途适合于含硫、含磷和含氮化合物的分析。 3、简述酶抑制法测定农药残留的基本原理 乙酰胆碱酯酶与胆碱能神经的生理功能极为密切。其生理功能是:当神经冲动到达胆碱能神经末梢时,突触小泡内的Ach(乙酰胆碱)外排至突触间隙,作用于突触后膜的胆碱能受体,引起下一级神经元或效应器的激发。在正常生理条件下,Ach完成传递冲动作用后,随即被突触后膜上的AchE在数毫秒内水解,生成乙酸和胆碱,水解产物与加入的试剂反应产生颜色。反之,式样提取剂中含有一定量的有机磷或氨基甲酸酯类农药,酶的活性就被抑制,式样中加入的机制就不能被水解,从而不显色或颜色很浅,用分光光度计测定吸光度,计算出抑制率,就可判断农药残留情况。 4、简述超声波萃取原理 超声波辅助萃取:是利用超声波辐射压强产生的强烈空化效应、机械振动、扰动效应、高的加速度、乳化、扩散、击碎和搅拌作用等多级效应,增大物质分子运动频率和速度,增加溶剂穿透力,从而加速目标成分进入溶剂,促进提取的进行。 名词解释 1、农药残留:指由于农药的应用而残存于生物体、农产品和环境中的农药亲体及其具有毒理学意义的杂 质、代谢转化产物和反应物等所有衍生物的总称。 农药残留毒性:因摄入或长时间重复暴露农药残留而对人、畜以及有益生物产生急性或慢性中毒 2、农药纯品:能够真正反映化合物特性的高纯度物质。 农药标准物质:具有一种或多种足够均匀和很好确定了的特性值,用以校准设备,评价测量方法或给材料赋值的材料或物质。 3、最大残留限制(MRL) :指由食品营养标准委员会推荐的,食品或动物饲料中允许的农药残留物的最大 浓度(毫克/千克)。是根据在毒理学上认为可以接受的食品农药残留量制定的。 每日允许摄入量(ADI) :指在一生中,对消费者健康没有可感知危险的日摄入量。 论述题 1、论述农药标准品常用的制备方法p62 ①重结晶法(适于除去农药原粉中的少量杂质) ②蒸馏法 ③柱层析法(其特点:应用范围广、操作简便、分离效率高) ④区域熔融法 ⑤升华法(此法在农药纯品制备中应用较少) ⑥薄层色谱法(常用来制备毫克级农药纯品) 2、写出样品提取技术的类型及其提取剂的要求 (1)溶剂提取法包括液液提取和固液提取 固液提取常用以下方法:①索式提取法②振荡提取法③组织捣碎法④消化提取法 (2)固相提取法
防腐涂料的性能及检测方法
防腐涂料的性能及检测方法 防腐涂料是油漆涂料中必不可少的一种涂料,对物体起到防腐蚀的作用,保护物体的使用寿命,在农业、工业等各个领域发挥着越来越重要的作用。下面介绍防腐涂料的性能以及检测方法: 防腐涂料性能 1、耐水性 耐水性是指防腐涂料涂膜抵抗水的破坏能力的量度。其测试是在规定的条件下,将涂膜试板浸泡在水中,观察其有无发白、失光、起泡、脱落等现象。以及恢复原状态的难易程度。这将直接影响涂膜的使用寿命。其检测方法可按GB/T1733《漆膜耐水性测定法》中规定进行。 2、耐盐水性 耐盐水性是指防腐涂料涂膜对盐水侵蚀的抵抗能力。可以用耐盐水试验判断涂膜产品的防护性能。其检测方法可按GB/T1763-89《漆膜耐盐水试剂性测定法》或GB16834-89《船舶漆耐盐水性的测定》中规定进行。 3、耐石油制品性 耐石油制品性是指防腐涂料涂膜抵抗石油制品(即汽油、润滑油、和溶剂等)的破坏能力的量度。其检测方法可按GB/T1734-93《漆膜耐汽油性测定法》或HG/T3343《漆膜耐油性测定法》中规定进行。 4、耐湿热性 耐湿热性是指防腐涂料涂膜抵抗湿热环境破坏的能力。在涂膜耐腐蚀性的检测中,耐湿热性的检测往往与耐盐雾性试验同时进行。其检测方法可按GB/T1740-89《漆膜耐湿热性测定法》或GB/T19893-92《色漆和清漆耐湿热性的测定连续冷凝浸水法》中规定进行。5、耐盐雾性 耐盐雾性是指防腐涂料涂膜抵抗盐雾侵蚀的能力。是涂膜耐腐蚀性关健指标,也是模拟大气中的盐雾腐蚀加速试验方法。其检测方法可按GB/T1771《色漆和清漆耐中性盐雾性能的测定》中规定进行。 6、耐化学试剂性 耐化学试剂性是指防腐涂料涂膜抵抗酸、碱和盐及其它化学药品破坏的能力。其检测方
甘氨酸检验标准操作规程
1. 目的 建立甘氨酸检验标准操作规程,规范操作。 2. 范围 适用于甘氨酸的检验。 3. 依据: 中国药典2010版二部。 4. 职责 4.1 起草:QC 审核:质量保证部负责人批准人:质量管理负责人。 4.2QC实施本规程。 4.3QA监督本规程的实施。 5. 内容 5.1 性状 本品为白色至类白色结晶性粉末;无臭,味甜。 本品在水中易溶,在乙醇或乙醚中几乎不溶。 5.2 鉴别 5.2.1 鉴别(1) 5.2.1.1 试液及仪器 一般实验仪器。 5.2.1.2 分析步骤 取本品与甘氨酸对照品各适量,分别加水溶解并稀释制成每1ml中约含10mg的溶液,作为供试品溶液与对照品溶液。吸取上述两种溶液各2μl分别点于同一硅胶G薄层板上,以正丙醇-氨水(7:3)为展开剂,展开约10cm,晾干,在80℃干燥30分钟,喷以茚三酮的正丙醇溶液(1→100),在105℃加热至斑点出现,立即检视。供试品溶液所显主斑点的位置和颜色应与对照品溶液的主斑点的位置和颜色相同。 5.2.2 鉴别(2)
5.2.2.1 试液及仪器 一般实验仪器和红外可见分光光度仪。 5.2.2.2 分析步骤 用红外分光光度仪检测,采用压片法,取残渣约1~1.5mg,置玛瑙研钵中,加入干燥的 溴化钾或氯化钾细粉约200~300mg(与供试品的比约为200:1)作为分散剂,充分研磨混匀,置于直径为13mm的药片模具中,使铺展均匀,抽真空约2min,加压至(0.8×106)kPa(约8~10T/㎝2),保持压力2min,撤去压力并放弃后取出制成的供试片,目视检测,片子应呈透明状,其中样品分布应均匀,并无明显的颗粒状样品。亦可采用其他直径的压膜制片,样品与分散剂的用量需相应调整以制得浓度适合的片子。测定。本品的红外光吸收图谱应与对照的图谱(光谱集929图)一致。 5.3 检查 5.3.1 酸度 5.3.1.1 试液及仪器 一般实验仪器。 5.3.1.2 分析步骤 取本品1.0g,加水20ml溶解后,依法测定(附录Ⅵ H),pH值应为5.6~6.6。 5.3.2 溶液的透光率 5.3.2.1 试液及仪器 一般实验仪器和紫外-可见分光光度仪。 5.3.2.2 分析步骤 取本品1.0g,加水20ml溶解后,照紫外-可见分光光度法(附录ⅣA),测定时,以配制供试品溶液的同批溶剂为空白校零,采用1 c m的石英吸收池,在430nm波长处测定透光率,不得低于98.0%。 5.3.3 氯化物 5.3.3.1 试液及仪器 一般实验仪器。 稀硝酸:取確酸105ml,加水稀释至1000ml,即得。本液含HN03 应为9.5%~10.5%。 硝酸银试液:即硝酸银滴定液。 5.3.3.2 分析步骤 取本品1.0g,加水溶解使成25ml,再加稀硝酸10ml;溶液如不澄清,应滤过;置50ml 纳氏比色管中,加水使成约40ml,摇匀,即得供试品溶液。另取7.0ml标准氯化钠溶液〔称取
2341农药残留量测定法
2341 农药残留量测定法 第五法药材及饮片(植物类)中禁用农药多残留测定法 1. 气相色谱-串联质谱法 色谱条件用(50%苯基)-甲基聚硅氧烷为固定液的弹性石英毛细管柱(柱长为30m,柱内径为0.25mm,膜厚度为0.25μm)。进样口温度250℃,不分流进样。载气为高纯氦气(He)。进样口为恒压模式,柱前压力为146kPa。程序升温:初始温度60℃,保持1分钟,以每分钟10℃的速率升温至160℃,再以每分钟2℃ ) , % 监测离子对、碰撞电压(CE)见附表2。为提高检测灵敏度,可根据保留时间分段监测各农药。 3. 对照溶液的制备 3.1 混合对照品溶液的制备精密量取禁用农药混合对照品溶液(已标示各相关农药品种的浓度)1ml,置20ml量瓶中,用乙腈稀释至刻度,摇匀,即得。
3.2气相色谱-串联质谱法分析用内标溶液的制备取磷酸三苯酯对照品适量,精密称定,加乙腈溶解并制成每1ml含1.0mg的溶液,即得。精密量取适量,加乙腈制成每1ml含0.1μg的溶液。 3.3 空白基质溶液的制备取空白基质样品,同供试品溶液的制备方法处理制成空白基质溶液。 3.4 基质混合对照溶液的制备分别精密量取空白基质溶液1.0ml(6份),置氮吹仪上,40℃水浴浓缩至约0.6ml,分别加入混合对照品溶液10μl、20μl、50μl、100μl、150μl、200μl,加乙腈稀释至l ml,涡旋混匀,即得。 4. 供试品溶液的制备 4.1 直接提取法 取供试品粉末(过三号筛)5g,精密称定,加氯化钠1g,立即摇散,再加入乙腈50ml,匀浆处理2分钟(转速不低于每分钟12000转),离心(每分钟4000转),分取上清液,沉淀再加乙腈50ml,匀浆处理1分钟,离心,合并两次提取的上清液,减压浓缩至约3~5ml,放冷,用乙腈稀释至10.0ml,摇匀,即得。 4.2 快速样品处理法(QuEChERS)法 取供试品粉末(过三号筛)3g,精密称定,置50ml聚苯乙烯具塞离心管中,加入1%冰醋酸溶液15ml,涡旋使药粉充分浸润,放置30分钟,精密加入乙腈15ml,涡旋使混匀,置振荡器上剧烈振荡(每分钟500次)5分钟,加入无水硫酸镁与无水乙酸钠的混合粉末(4:1)7.5g,立即摇散,再置振荡器上剧烈振荡(每分钟500次)3分钟,于冰浴中冷却10分钟,离心(每分钟4000转)5分钟,取上清液9ml,置预先装有净化材料的分散固相萃取净化管[无水硫酸镁900mg,N-丙基乙二胺300mg,十八烷基硅烷键合硅胶300mg,硅胶300mg,石墨化碳黑90mg]中,涡旋使充分混匀,置振荡器上剧烈振荡(每分钟500次)5分钟使净化完全,离心(每分钟4000转)5分钟,精密吸取上清液5ml,置氮吹仪上于40℃水浴浓缩至约0.4ml,加乙腈稀释至1.0ml,涡旋混匀,滤过,取续滤液,即得。 4.3固相萃取法 固相萃取净化方式包括以下三种: 方式一:量取直接提取法制备的供试品溶液3~5ml,置于装有分散型净化材料的净化管[无水硫酸镁1200mg,N-丙基乙二胺300mg,十八烷基硅烷键合硅胶
3 溶液法测定极性分子的偶极矩
实验3 溶液法测定极性分子的偶极矩 1 目的要求 (1) 用溶液法测定乙酸乙酯的偶极矩。 (2) 了解偶极矩与分子电性质的关系。 (3) 掌握溶液法测定偶极矩的主要实验技术。 2 基本原理 (1) 偶极矩与极化度:分子结构可以近似地看成是由电子云和分子骨架(原子核及内层电子)所构成。由于其空间构型的不同,其正负电荷中心可以是重合的,也可以不 重合。前者称为非极性分子,后者称为极性分子。 图18-1电偶极矩示意图 图18-2极性分子在电场作用下的定向 1912年德拜提出“偶极矩” μ 的概念来度量分子极性的大小,如图18-1所示, 其定义是 d q ?=μ (1-1) 式中,q 是正负电荷中心所带的电量; d 为正负电荷中心之间的距离;μ 是一个 向量,其方向规定为从正到负。因分子中原子间的距离的数量级为10-10m ,电荷的数量级为10-20C ,所以偶极矩的数量级是10-30C ·m 。 通过偶极矩的测定,可以了解分子结构中有关电子云的分布和分子的对称性,可以用来鉴别几何异构体和分子的立体结构等。 极性分子具有永久偶极矩,但由于分子的热运动,偶极矩指向某个方向的机会均等。所以偶极矩的统计值等于零。若将极性分子置于均匀的电场E 中,则偶极矩在电场的作用下,如图Ⅱ-29-2所示趋向电场方向排列。这时我们称这些分子被极化了。极化的程度可用摩尔转向极化度P 转向来衡量。 转向 P 与永久偶极矩2μ的值成正比,与绝对温度T 成反比。 kT N P 3432μπ ?=转向 kT N μ π ?=9 4 (1-2) 式中:k 为玻兹曼常数,N 为阿伏加德罗常数。
在外电场作用下,不论极性分子或非极性分子,都会发生电子云对分子骨架的相对移动,分子骨架也会发生形变。这称为诱导极化或变形极化。用摩尔诱导极化度P 诱 导 来衡量。显然P 诱导可分为二项,即电子极化度P 电子和原子极化度P 原子,因此P 诱导=P 电子 +P 原子。P 诱导与外电场强度成正比,与温度无关。 如果外电场是交变场,极性分子的极化情况则与交变场的频率有关。当处于频率 小于1010s -1的低频电场或静电场中,极性分子所产生的摩尔极化度P 是转向极化、电子极化和原子极化的总和。 原子电子转向P P P P ++= (1-3) 当频率增加到1012~1014的中频(红外频率)时,电子的交变周期小于分子偶极矩的松弛时间,极性分子的转向运动跟不上电场的变化,即极性分子来不及沿电场方向定向,故转向P =0,此时极性分子的摩尔极化度等于摩尔诱导极化度诱导P 。当交变电场的频率进一步增加到>1015秒-1的高频(可见光和紫外频率)时,极向分子的转向运动和分子骨架变形都跟不上电场的变化。此时极性分子的摩尔极化度等于电子极化度电子 P 。 因此,原则上只要在低频电场下测得极性分子的摩尔极化度P ,在红外频率下测得极性分子的摩尔诱导极化度诱导P ,两者相减得到极性分子摩尔转向极化度转向P ,然后代入(18-2)式就可算出极性分子的永久偶极矩μ来。 (2) 极化度的测定:克劳修斯、莫索和德拜从电磁场理论得到了摩尔极化度P 与介电常数ε之间的关系式: ρ εεM P ?+-= 21 (1-4) 式中,M 为被测物质的分子量;ρ为该物质在TK 下的密度;ε可以通过实验测定。 但(1-4)式是假定分子与分子间无相互作用而推导得到的。所以它只适用于温度不太低的气相体系,对某些物质甚至根本无法获得气相状态。因此后来提出了用一种溶液来解决这一困难。溶液法的基本想法是,在无限稀释的非极性溶剂的溶液中,溶质分子所处的状态和气相时相近,于是无限稀释溶液中溶质的摩尔极化度∞2P ,就可以看作为(1-4)式中的P 。 海台斯纳特首先利用稀释溶液的近似公式。
食品中农药残留的检测方法
食品中农药残留的检测方法 1 波谱法 该方法是根据有机磷农药中某些官能团或水解、还原产物与特殊的显色剂在特定条件下发生氧化、磺酸化、酯化、络合等化学反应,产生特定波长的颜色反应来进行定性或定量(限量) 测定。 2.色谱法 2.1 薄层色谱法(TLC) 薄层色谱法是一种成熟的、应用也较广的微量快速检测方法。它在农药残留测定技术上有它独特的用处,它既是重要的分离手段,又是定性、定量的分析方法。 检测过程一般先用适宜的溶剂提取有机磷农药,经纯化浓缩后,在薄层硅胶板上分离展开,显色后与标准的有机磷农药比较Rf 值进行定性测定或用仪器进行定量测定。 2.2 气相色谱法(GC) 该方法是利用经提取、纯化、浓缩后的有机磷农药注入气相色谱柱,程序化升温汽化后,不同的有机磷农药在固相中分离,经不同的检测器检测扫描绘出气相色谱图,通过保留时间来定性,通过峰或峰面积与标准曲线对照来定量。一次可同时测定多组份,简便快捷,灵敏度高,准确性也好。而色谱条件的最佳设定是气相色谱技术的关键。 2.3 高效液相色谱法(HPLC) 高效液相色谱法是在液相色谱柱层析的基础上,引入气相色谱理论并加以改进而发展起来的色谱分析方法。高效液相色谱法在农药残留分析的应用越来越广泛,是因为高效液相色谱法能适合分析沸点高而不太容易汽化、热不稳定和强极性农药及其代谢产物;且可以与柱前提取、纯化及柱后荧光衍生化反应和质谱等联用,易实现分析自动化;同时一些新型检测器的问世在一定程度上提高了高效液相色谱法的检测灵敏度。与气相色谱法相比,不仅分离效能好,灵敏度高,检测速度快,而且应用面广。 3 酶抑制法 有机磷农药对哺乳动物中毒作用的基础,通常与它们抑制中枢和周围神经系
甘氨酸含量测定和甲硫氨酸含量测定
甘氨酸含量测定 甲硫氨酸含量测定 一、方法原理 根据《中国药典》甘氨酸含量测定:取本品约70mg,精密称定,加无水甲酸1.5ml使溶解,加冰醋酸50ml,照电位滴定法,用高氯酸滴定液(0.1mol/L)滴定。每1ml高氯酸滴定液(0.1mol/L)相当于7.507mg的甘氨酸。 甘氨酸含量:药典2010版二部86页 二、实验仪器及推荐配置 雷磁自动电位滴定仪 231-01 pH玻璃电极217-01甘汞参比电极 三、实验试剂 1、滴定液:0.1mol/L高氯酸溶液配制 取无水冰醋酸(按含水量计算,每1g水加醋酐 5.22ml)750ml,加入高氯酸(70%-72%)8.5ml,摇匀,在室温下缓缓滴加醋酐23ml,边加边摇,加完后摇匀,放冷,加无水冰醋酸适量使成1000ml,摇匀,放置24小时。 2、基准纯邻苯二甲酸氢钾:在105℃干燥至恒重 3、样品 四、样品分析及结果计算 1、标定 取在105℃干燥至恒重的基准邻苯二甲酸氢钾约0.16g,精密称定,加无水冰醋酸20ml使溶解。在自动电位滴定仪上滴定至终点,每1ml高氯酸滴定液(0.1mol/L)相当于20.42mg的邻苯二甲酸氢钾。 按下列公式计算 C HClO4=0.1(m邻苯二甲酸氢钾/0.02042)/V HClO4 0.1 –高氯酸标准溶液理论浓度(mol/L) C HClO4–高氯酸标准溶液浓度(mol/L) V HClO4 - 滴定至终点时消耗高氯酸的体积(ml) m邻苯二甲酸氢钾- 称取的邻苯二甲酸氢钾质量(g) 0.02042 - 与1.00ml高氯酸标准溶液[c(HClO4)0.1mol/L]相当的邻苯二甲酸氢钾的质量(g)2、样品分析 取样品约70mg,精密称定,加无水甲酸1.5ml使溶解,加冰醋酸50ml,在雷磁
中国药品检验标准操作规范2010年版中药补充部分24有机磷类农药残留量测定法
有机磷类农药残留量测定法 1 简述 很多有机磷类农药具有毒性,其残留严重危及人体健康。《中国药典》2010年版一部收载了有机磷类农药(对硫磷、甲基对硫磷、乐果、氧化乐果、甲胺磷、久效磷、二嗪农、乙硫磷、马拉硫磷、杀扑磷、敌敌畏、乙酰甲胺磷)的测定方法。 本法通过提取、净化和富集等步骤制备供试品溶液,采用气相色谱法,氮磷检测器测定。 2 仪器与用具 2.1 气相色谱仪,带有氮磷检测器(NPD),载气为高纯氮(纯度>99.9999%)。 2.2 超声仪。 2.3 旋转蒸发仪。 2.4 多功能真空样品处理器(如SUPELCO,isiprep TM DL)。 2.5 活性炭小柱(120~400目,石墨碳填料0.25g,内径0.9cm,3ml)。 2.6 氮吹仪(如Organomation Associates,Inc.,N-EV AP TM 112 nitrogen evaporator)。 2.7 色谱柱:DB-17MS或HP-5弹性石英毛细管柱(30m×0.25mm×0.25μm)或类似极性的毛细管柱。 2.8 具塞锥形瓶、250ml平底烧瓶、棕色量瓶、移液管等。 3 试药与试液 3.1 无水硫酸钠为分析纯。 3.2 乙酸乙酯、正己烷(农残级或分析纯试剂经过全玻璃蒸馏装置重蒸馏,经气相色谱法确认,符合农残检测的要求)。 3.3 农药对照品:对硫磷、甲基对硫磷、乐果、氧化乐果、甲胺磷、久效磷、二嗪农、乙硫磷、马拉硫磷、杀扑磷、敌敌畏、乙酰甲胺磷,由国家标准物质研究中心及农业部环境保护科研检测所提供,其纯度大于99%;也可以使用国际认可的、纯度要求等符合规定的进口标准物质。 4 色谱条件与系统适用性试验 进样口温度:220℃;检测器温度:300℃。不分流进样。程序升温:初始120℃,每fenzh 10℃升至200℃。每分钟5℃升至240℃,保持2min,每分钟20℃升至270℃,保持0.5min。理论板数按敌敌畏峰计算应不低于6000,两个相邻色谱峰的分离度应大于1.5。 5 操作方法 5.1 对照品储备液的制备精密称取对硫磷、甲基对硫磷、乐果、氧化乐果、甲胺磷、久效磷、二嗪农、乙硫磷、马拉硫磷、杀扑磷、敌敌畏、乙酰甲胺磷农药对照品适量,用醋酸乙酯分别制成每1ml约含100μg的溶液,即得。 5.2 混合对照品储备液的制备精密量取上述各对照品储备液1ml,置20ml棕色量瓶中,加乙酸乙酯稀释至刻度,摇匀,即得。 5.3 混合对照品溶液的制备精密量取上述混合对照品储备液,用乙酸乙酯制成每1ml 分别含0.1μg、0.5μg、1μg、2μg、5μg的溶液,即得。 5.4 供试品溶液的制备药材取供试品粉末(过二号筛)约5g,精密称定,加无水硫酸钠5g,加入乙酸乙酯50~100ml,冰浴超声处理3min,放置,取上层液滤过,药渣加乙酸乙酯30~50ml,冰浴超声处理2min,放置,滤过,合并两次滤液,用少量乙酸乙酯洗涤滤纸及残渣,与上述滤液合并。取滤液于40℃下减压浓缩至近干,用乙酸乙酯转移至5ml量瓶中,并稀释至刻度,精密量取1ml,置活性炭小柱[120~400目,0.25g,内径0.9cm(如Supelclean ENVI-Carb SPE Tubes,3ml活性炭小柱),用乙酸乙酯5ml预洗]上,置多功能真空样品处理器上,用正己烷-乙酸乙酯(1:1)的混合溶液5ml洗脱,收集洗脱液,置氮吹仪
稀溶液法测定偶极矩
华南师范大学实验报告 学生姓名学号 2 专业化学(师范)年级、班级2009级化6 课程名称结构化学实验项目稀溶液法测定偶极矩 实验类型验证综合实验时间2011 年12 月 2 日 实验指导老师实验评分 一、实验目的 1.掌握溶液法测定偶极矩的主要实验技术 2.了解偶极矩与分子电性质的关系 3.测定正丁醇的偶极矩 二、实验原理 1.偶极矩与极化度 分子结构可以近似地看成是由电子云和分子骨架(原子核及内层电子)所构成。由于空间构型的不同,其正负电荷中心可能重合,也可能不重合。前者称为非极性分子,后者称为极性分子。
1912年,德拜提出“偶极矩”的概念来度量分子极性的大小,其定义是 qd =→ μ ① 式中,q 是正负电荷中心所带的电量;d 为正负电荷中心之间的距离;→ μ是一个矢量,其方向规定为从正到负。因分子中原子间的距离的数量级为10-10 m ,电荷的数量级为10-20 C ,所以偶极矩的数量级是10-30 C ·m 。 通过偶极矩的测定,可以了解分子结构中有关电子云的分布和分子的对称性,可以用来鉴别几何异构体和分子的立体结构等。 极性分子具有永久偶极矩,但由于分子的热运动,偶极矩指向某个方向的机会均等。所以偶极矩的统计值等于零。若将极性分子置于均匀的电场E 中,则偶极矩在电场的作用下,趋向电场方向排列。这时称这些分子被极化了。极化的程度可以用摩尔转向极化度P μ来衡量。P μ与永久偶极矩μ的平方成正比,与绝对温度T 成反比。 kT 9μ πN 4P A μ= ② 式中,k 为波兹曼常数;NA 为阿弗加德罗常数;T 为热力学温度;μ为分子的永久偶极矩。 在外电场作用下,不论极性分子或非极性分子,都会发生电子云对分子骨架的相对移动,分子骨架也会发生形变。这称为诱导极化或变形极化。用摩尔诱导极化度P 诱导来衡量。显然,P 诱导可分为两项,即电子极化度P e 和原子极化度P a ,因此 P 诱导 = P e + P a ③
1L--色氨酸含量的测定
1L--色氨酸含量的测定 1.1实验仪器与试剂 色氨酸样品,甲醇,STI501液相色谱仪梯度系统,UV501紫外检测器,7725I手动进样阀,N2000色谱工作站,0.03%KH2PO4溶液 1.2HPLC色谱分析条件 流动相为0.03%KH2PO4溶液(A)-甲醇(B),线性梯度淋洗,流速1.0mL/min,柱温35℃,检测波长276nm。 流动相时间(t/min) 5101520 A(%)80908070 B(%)20102030 1.3标准溶液的配制 精密称取色氨酸标准标准品50mg,置100ml容量瓶中,振摇,用流动相溶解到刻度,作为供试品溶液,另取色氨酸样品适量,同法操作。 1.4标准直线的制作 精密称取色氨酸标准样品适量,分别稀释制成每1ml中含色氨酸50.0、100.0、200.0、400.0、600.0、800.0、1000.0μg的溶液,注入液相色谱仪,以色氨酸峰面积A为纵坐标,浓度C为横坐标,制作回归标准直线。 1.5发酵液中样品中色氨酸含量的测定 取发酵液,稀释配制成约500μg/ml的溶液,摇匀,过滤,取25μl进样,在高效液相色谱仪下测量,记录峰值面积,作图。 2L-精氨酸含量的测定 2.1实验仪器与试剂 L-精氨酸标准样品,乙腈,STI501液相色谱仪梯度系统,UV501紫外检测器,7725I 手动进样阀,N2000色谱工作站,磷酸二氢铵。 PH计,磷酸,
2.2HPLC色谱分析条件 流动相:以磷酸二氢铵溶液(称取磷酸二氢铵1.15g,加水800m溶解后,用磷酸调节pH值至 2.0±0.1,加水稀释至1000ml)-乙腈为流动相,线性梯度淋洗;柱温为30℃检测波长为206nm;进样量:25μl。 2.3标准直线的制作 精密称取精氨酸标准标准品50mg,置100ml容量瓶中,振摇,用流动相溶解到刻度,作为供试品溶液,另取色氨酸样品适量,同法操作。精密称取精氨酸标准样品适量,分别稀释制成每1ml中含精氨酸50.0、100.0、200.0、400.0、600.0、800.0、1000.0μg的溶液,注入液相色谱仪,以精氨酸峰面积A为纵坐标,浓度C为横坐标,制作回归标准直线。 2.4发酵液中样品中精氨酸含量的测定 取发酵液,稀释配制成约500μg/ml的溶液,摇匀,过滤,取25μl进样,在高效液相色谱仪下测量,记录峰值面积,作图。 3L-甘氨酸含量的测定 3.1实验仪器和试剂 乙酸铵溶液;乙腈;L-甘氨酸标准样品;STI501液相色谱仪梯度系统,UV501紫外检测器,7725I手动进样阀,N2000色谱工作站。 3.2HPLC色谱分析条件 流动相:A为0.02%mol/L乙酸铵溶液(PH=6.5):B为乙腈,线性梯度淋洗;流速:110mL/min;柱温:30℃;进样量:20μL。 梯度洗脱表3 流动相时间(t/min) 102030405060 A(%)806040406080 B(%)204060604020 3.3标准直线的制作 方法同2.2节
蔬菜中农药残留检测方法研究
蔬菜中农药残留检测方法研究 【摘要】随着栽培技术的不断进步,农药残留的问题越来越严重,对消费者的身体健康构成了严重威胁。开展蔬菜中农药残留检测方法的研究是控制农药残留保证食品安全的基础,具有重大的意义。本文介绍了蔬菜中农药残留检测的各种方法并对前景进行了展望。 【关键词】蔬菜、农药残留、检测、研究进展 随着栽培技术的不断进步,蔬菜的生长期已越来越短,而随着环境污染的加剧,蔬菜的病虫害也越来越重,绝大部分蔬菜需要连续多次放药后才能成熟上市。农药污染较重的有叶类蔬菜,其中韭菜、油菜受到的污染比例最大。茄果类蔬菜如青椒、番茄等,嫩荚类蔬菜如豆角等,鳞茎类蔬菜如葱、蒜、洋葱等,农药的污染相对较小。农药残留监测体系的建立,对农药残留的监测手段和检测水平提出了更高要求,并促进了农药残留快速检测方法的研究和应用进展,使农药残留检测技术朝着更加快速方便、灵敏可靠的方向发展,逐渐以农药残留专业检测机构的少量检测为中心,向现场检测及实验室的大量检测辐射翻。 1 仪器分析法 由于农药的活性成分大多是小分子有机化合物,故多使用气相色(GC,)~41、高效液相色谱(HPLC,)~、气相色谱一质谱联用(GC-MS)嘲和高效液相色谱一质谱联用(HPLC—Ms)同等技术。其中研究最多的是色质联用技术。因为色质联用特别适合于多种标样残留分析,所以国外把它也划为农药残留快速检测技术之列。大部分农药(如有机氯、有机磷、拟除虫菊酯等)残留可使用GC—MS检测昀,检出限一般为1~10 b~g/kg,但对分子量较大、极性或热不稳定性太强的农药及其化合物,GC-MS不适用,需采用高效液相色谱一质谱联用(HPLC-MS)和其他的方法来检测。 1.1 固相萃取技术 固相萃取法是1种基于液相色谱分离机制的样品制备方法,已广泛应用于农药残留检测工作。它根据液相分离、解析、浓缩等原理,使样品溶液混合物通过柱子后,样品中某一组分保留在柱中,选择合适的溶剂把保留在柱中的组分洗脱下来,从而达到分离、净化的目的。SPE克服了液一液萃取技术及一般柱层析的缺点,具有高效、简便、快速、安全、重复性好、便于前处理自动化等特点。根据柱中填料大体可分为吸附型(如硅胶、大孔吸附树脂等)、分配型(c。,c 、苯基柱等)和离子交换型。1L.R_odriguez等人采用固相萃取法通过改变移动相中缓冲液的浓度、pH值、表面活性剂的浓度和类型对蔬菜中的木精、笨基苯酚、锑比灵和有机磷残留量进行分析,结果表明:pH9.2,缓冲液中含有4mmoUL硼酸和75mmol/L胆酸钠能够得到最好的结果。 1.2 固相微萃取 加拿大Waterloo大学Pawliszyn 1990年首创的一种无需溶剂的萃取技术,它是在固相萃取的基础上发展起来的一种新型的预处理技术。SPME技术由固相萃取技术(SPE)发展而来,对目标化合物有较好的选择性,并且有较高的灵敏度,
阿司匹林的检查
阿司匹林片剂的检测 一.阿司匹林体外含量测定 1.两步滴定法测定阿司匹林片的含量: 取阿司匹林片样品10片,精密称定,研磨细,精密称取片粉适量(约相当于阿司匹林0.3g),放在锥形瓶中,加入中性乙醇20mL,振摇,使阿司匹林充分溶解,加 入3滴酚酞指示剂,用氢氧化钠滴定液(0.1mol/L)滴定至溶液呈粉红色。 在上述供试品溶液中,精密加入氢氧化钠滴定液(0.1mol/L)40mL,在水浴上边振摇边加热15min后,迅速放冷至室温,用硫酸迪斯尼工业(0.05mol/L)滴定,并将滴定结果用空白试验进行校正。每1mL的氢氧化钠滴定液(0.1mol/L)相当于18.02mg的C9H8O4(阿司匹林)。 标示量%=(V0-V)*F*T*W平/(W*标示量)*100% V0为空白实验消耗的硫酸滴定液的体积(mL);V为样品测定时消耗的硫酸滴定液的体积(mL);F为硫酸滴定液的浓度校正因子;T为氢氧化钠滴定液的滴定度(mg/mL);W为供试品片粉的取样量(g);W平为供试品的平均片重(g);标示量为片剂的规格(mg/片)。 2.仪器分析法 2.1 Hplc法测定阿司匹林片的含量 色谱条件:ODS Cl8 Hypersil(150 mm*4.6mm,5 um)色谱柱,以水一乙腈一磷酸(76:24:0.5)为流动相;检测波长为276 nm;流速1.3 mL/min;进样量20μL;理论板数不低于10 000。 溶液的制备:供试品制备精密量取样品适量置50 mL容量瓶中,加流动相适量,超声使其溶解,用流动相稀释至刻度(得到200μg/mL的溶液),备用。 对照品的制备:精密称取阿司匹林对照品100mg于50 mL容量瓶中,加流动相适量,超声使其溶解,用流动相稀释至刻度(得2 000μg/mL的溶液)+-,备用。空白溶液制备除阿司匹林外,精取处方量的辅料,配置空白样品溶液。按供试品制备方法制备对照品溶液。通过外标法计算样品浓度。 C=A*Co*D/Ao C为样品浓度(mg/mL);A和Ao分别为样品和对照品溶液的峰面积;D为稀释倍数 (mL)。 2.2 紫外分光光度计法测量阿司匹林片的含量 2.2.1标准溶液系列和样品溶液的配制 分别取0.5g/ml乙酰水杨酸标准溶液0.00mL(空白溶液),0.50mL,1.00mL,1.50mL,2.00mL和阿司匹林样品溶液 1.00mL置于25mL的容量瓶中,分别加入7%
有机磷农药残留的危害及检测方法
有机磷农药残留的危害及检测方法 姓名:梅增健学号:201008011053 班级:10生工一班 摘要:近年来由于农药的大量使用致使食品的农药残留问题倍受关注,提高农药残留分析检测技术是解决农药残留问题的重要手段。在农药残留检测中样品前处理技术又是检测过程中耗时最长,最容易出现误差的步骤。因而,发展样品前处理技术能提高农药残留检测的效率和准确率。本文综述了近年来食品中农药有机磷残留分析中超临界流体萃取法、固相萃取、固相微萃取和凝胶渗透色谱几种样品前处理技术。 关键词:农药残留;有机磷农药残留;检测;危害 正文: 一、有机磷类农药的危害及特性: 有机磷类农药自问世到现在已有70 年的历史。因为高效、快速、广谱等特点, 有机磷类农药一直在农药中占有很重要的位置, 对世界农业的发展起了很重要的作用。我国已生产和使用的有机磷类农药达数10种之多,其中最常用的有敌百虫、敌敌畏、乐果、马拉硫磷等。但随着这些有机磷类农药的广泛被使用, 暴露出了很多问题,如高残留、毒性强等,尤其在环保意识日益增强的今天,其暴露的问题也引起了人们的高度重视。部分非持久有机磷类农药在某些环境条件下也会有较长的残留期, 并在动物体内产生蓄积。如马拉硫磷是一种高选择性有机磷类农药,在环境中的残留不容忽视,水体中已有检出。马拉硫磷对水生生物属高毒农药, 对人免疫功能也具有一定的毒性作用, 已成为水环境中重要的监测项目。大多数有机磷类农药都属于磷酸酯类或硫代磷酸酯类化合物,其中有机磷酸脂类化合物纯品多为油状,少数为结晶固体。常用剂型有乳剂、油剂、粉剂及颗粒剂等。有机磷类农药的中毒特征是血液中胆碱酯酶活性下降, 胆碱酯酶的活性受到抑制,导致神经系统机能失调,从而使一些受神经系统支配的脏器,如心脏、支气管、肠、胃等发生功能异常。 二、有机磷农药的检测前处理 1、超临界萃取技术 所谓超临界是物质的一种特殊流体状态:当气液平衡的物质升温升压时,热膨胀会引起液体密度减少,压力升高又会使气相密度变大,当温度和压力达到某一点时,气液两相界面消失成一均相体系,此点即为临界点;当物质的温度和压力高于临界点时,就处于超临界状态,此时该物质为超临界流体。超临界萃取技术(supercritical fluid extraction,SFE)是用超临界液体作为萃取剂,根据样品中组分在不同压力和温度条件下溶解能力变化的性质,通过改变萃取剂流体的压力,从而将不同组分萃取出来。 等流体代替有机溶剂,对人体无害、绿色环保,且SFE最大优点是使用CO 2 有效地防止了热敏性物质的氧化和逸散,提高了样品回收率,因而较适合于萃取热不稳定物质和有毒化合物[1]。D.H.Kim等[2]已将SFE用于萃取面粉里的有机磷残留,避免了样品浓缩和分析过程的干扰,提高了萃取速度,60min有7g的样品萃取量,且联用GC-NPD分析后,检测限低至10ng/s。
耐水性试验
漆膜耐水性测定法 1 范围 本标准适用于油漆涂层(底漆、中涂漆、色漆、清漆及相关产品的单一涂层或多图层体系即复合涂层)耐水性的测定,测定的是涂膜在达到规定的试验时间后表面的变化情况,以漆膜表面变化(失光、变色、起泡、溶胀、变脆、软化、起皱、脱落、生锈等)现象和恢复时间表示其耐水性能。 2 术语 2.1漆膜失光:漆膜的颜色因气候环境的影响而降低的现象。 2.2漆膜变色:漆膜的颜色因受气候环境的影响而逐渐变浅或发生变化的现象。 2.3漆膜起泡:涂层因局部失去附着力而离开基体(底材或其他涂层)鼓起,使漆膜呈现圆形的凸出变形。泡内可焊液体、蒸汽、其他气体或结晶物。 2.4漆膜溶胀:漆膜经受液体浸泡后,由于液体渗入整个漆膜,而使其发生增厚、变软的现象。 2.5漆膜软化:漆膜经受液体浸泡后,由于溶胀而硬度明显变低的现象。 2.6漆膜脱落:一道或多道涂层脱离其下涂层,或者涂层完全脱离底材的现象。 2.7漆膜生锈:漆膜下面的钢铁表面局部或整体产生红色或黄色的氧化铁层的现象,它常伴随有漆膜的气泡、开裂、片落等变态。 2.8漆膜起皱:漆膜呈现多少有规律的或无规律的小波幅波纹形式的皱纹,它可深及部分或全部膜厚。 2.9漆膜变脆:漆膜进行加速破坏性试验,其漆膜柔韧性变坏的现象。 2.10破坏现象:漆膜在试验过程中其漆膜表面呈现的各种变坏的现象。例如:漆膜气泡、失光、变色、溶胀、起皱、生锈等现象。 3 试验设备和仪器 3.1试验设备:与试验用水接触的所有部分均应由惰性材料或不生锈材料制成。 注:通常情况下,漆膜耐水性能试验常用设备室恒温水浴锅。 3.1.1水槽:应有适宜的大小(合适的尺寸为700mm*400mm*400mm),配有盖子和恒温加热系统。同时利用水的充分搅拌时槽中各点水的流速和水文基本一致,并能保持一定的页面高度为宜。