医疗器械各国认证要求

医疗器械各国认证要求
医疗器械标准
这些医疗器械标准已被许多国家广泛采用。
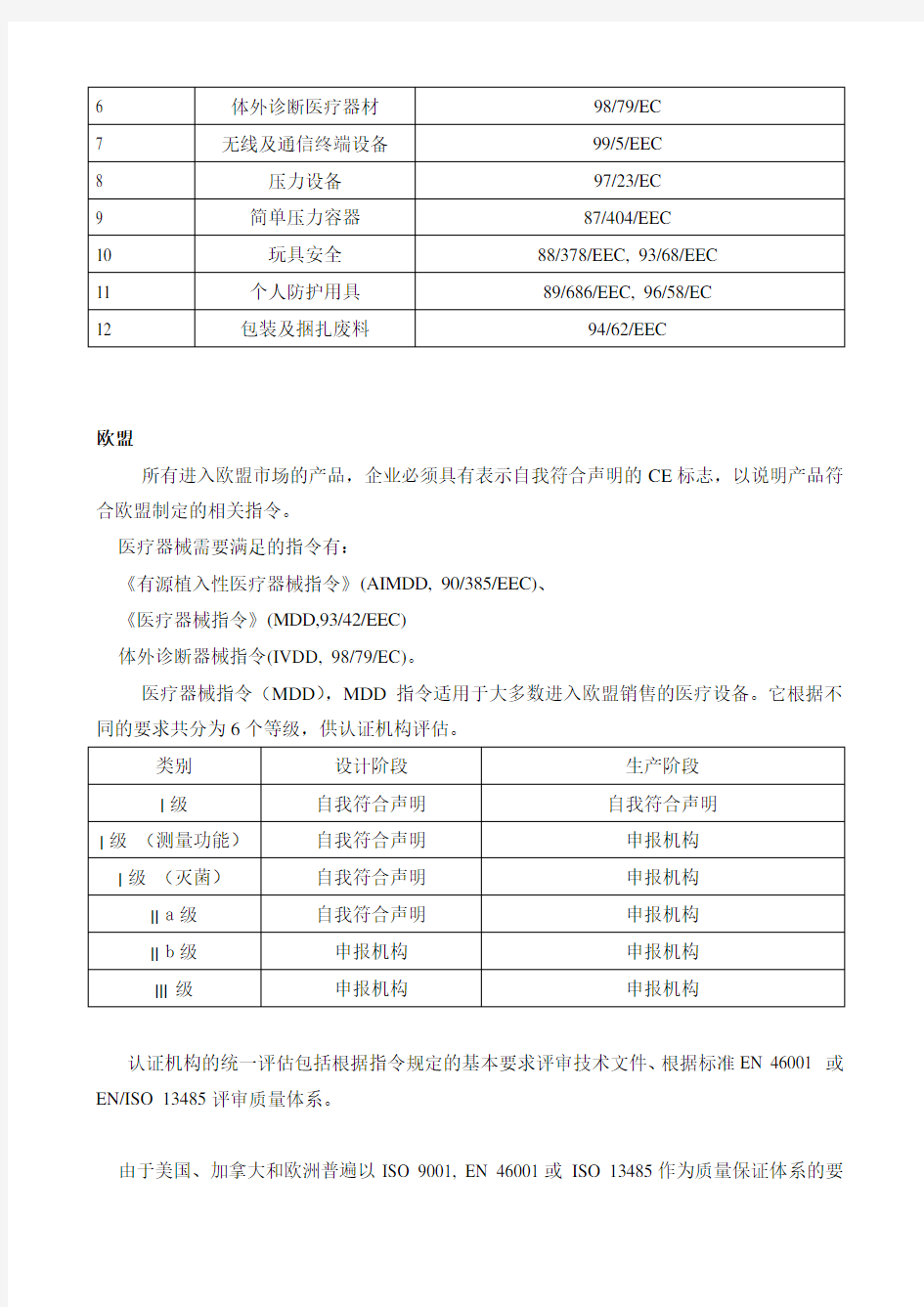
医疗器械指令列表
欧盟指令中涵括许多基本保健及安全规定,以及评估产品符合规定程度的订定程序。每项指令在区域性标准制订机构所订定的调和欧洲标准中均会具体说明详细的基本规定。
因此,相关产品的制造商、进口商及配销商都必须明显标示,产品完全符合每项指令在「基本规定」所列出的保健及安全规定。
形成「新方法」基础的指令,包含了广泛的产品类别(横向指令)或某种特定的产品类别(纵向指令)。
指令标题
欧盟
所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE标志,以说明产品符合欧盟制定的相关指令。
医疗器械需要满足的指令有:
《有源植入性医疗器械指令》(AIMDD, 90/385/EEC)、
《医疗器械指令》(MDD,93/42/EEC)
体外诊断器械指令(IVDD, 98/79/EC)。
医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备。它根据不同的要求共分为6个等级,供认证机构评估。
认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或EN/ISO 13485评审质量体系。
由于美国、加拿大和欧洲普遍以ISO 9001, EN 46001或ISO 13485作为质量保证体系的要
求,故建议质量保证体系的建立均以这些标准为基础。
体外诊断医疗器械指令(IVDD),IVDD的要求与MDD相似,可按以下分类申请:-
北美
在美国,食品和药物管理局(FDA)是监督和管理获准向消费者进行销售的食物,药物,化妆品和医疗器械的法定机构。器械及放射线健康中心(CDRH),作为FDA的一个分支,专管医疗器械。其对医疗器械按不同等级进行不同程度的监管(医疗器械分为I级,II级或III 级,I级作为低风险范畴,而III级属高风险范畴):
在加拿大方面,加拿大医疗器械认证认可机构(CMDCAS)要求医疗器械厂商提前获得经CMDCAS认可的第三方机构,如UL的质量体系审查,证明其质量系统符合CMDCAS的ISO13485/ISO13488标准。对CMDCAS认证的了解对于完成FDA 的质量系统注册(QSR)非常有帮助,因为如上所述的QSR是以ISO 9001和ISO 13485标准为基础的。
大多数属于I级或II级的医疗器械,需要获得510(k)或称上市通告,只有低风险的I级器械,可以豁免510(k)。
FDA 要求准备上市的医疗器械必须具有和已被肯定的器械(指已获准在市场销售的器械)等同的安全性和效力。因此制造商需提供报告说明其产品与市场上同类产品的详细比较情况。
制造商有责任获取和验证已被肯定的器械的相关信息,比如目录,使用说明书和510(k)其它要求的资料信息。
通常,有三种情况需要申请510(k):
①传统审核,适用于推介新器械,申请时需递交适用的性能报告。
②特殊审核,适用于依照设计控制程序作了较小修改的器械。
③简化审核,由制造商提交,制造商必须确保并声明其产品符合现有FDA认可的标准。
FDA 510(k) 审查
从2002年10月1日起,审查需直接向FDA缴纳用户费。经过FDA的初次审查,申请人将收到FDA出示的产品缺陷报告或声明,这个过程通常需时90天。经过改正和/或其它资料的补充后,FDA随后还将再进行为期90天的复审。
要缩短510(k)审查的周期,并减少工作量,第三方510(k)审查是完成审核的另一选择。如您选择诸如UL这样的第三方评审机构,那么整个审查可在四周内完成。
亚洲
亚洲的医疗设备市场是发展潜力最大的市场之一,随着生活质量和保健意识的提高,亚洲的消费者比以前更愿意在保健产品上消费。
日本
医疗设备在日本、中国和韩国拥有最大的消费市场。仅日本,2001年医疗设备的销售额高达230亿美元。
日本的保健体系和美国的完全不同。日本政府制定了严格的产品认证流程,新进入日本市
场的外国医疗设备产品都必须严格遵守。为了进入日本市场,医疗产品厂商必须首先获得两种由日本厚生省(MHLW)颁发的文件——营业执照和上市许可证。
外国厂商必须委托一个在日本已取得营业执照的代理商。国外企业和日本国内的代理商同时负责适用于其产品的进口程序和文件、GMP标准和售后监督的认证工作。
在日本,产品根据不同风险程度(由低到高)分为3类。UL根据日本国内标准如JIS T1001 和JIS T1002为客户提供―类型测试(Type Testing)‖服务。
中国
中国的国家药品监督管理局(CFDA)相当于FDA的角色,负责进口医疗器械的注册和监督工作。除此之外,中国政府的其它代理机构有权调整对某些医疗器械管理的相关规定。国家药监局管理出入关时的检验检疫工作,如为医用X光机、透析器、血液净化装置、心电图机、植入式心脏起搏器和超声仪器签发安全许可证。
随着WTO的加入,中国开始对进口及国内产品实行强制性认证。为符合标准化的要求,相应的医疗器械规章制度也面临着重大的变化。这些变化中包括了对产品的分类、产品安全要求的评估方法、认证标志和认证费用的全面标准化。从2003年8月1日起(原定于2003年5月1日),中国国家认证认可监督委员会将对一些高风险医疗设备实行强制性认证,即CCC认证,正式取代原来的中国电子设备安全认证合格证书,即CCEE认证和中国进口商品安全质量许可证,即CCIB认证。在中国,制造商可直接申请CCC认证或由授权代理机构办理。
韩国
凡在韩国销售的医疗器械,根据其《药品管理法》,必须获得韩国食品药品管理局(KFDA)颁发的境内产品生产和销售许可证。韩国食品药品管理局直接受政府保健福利部(MOHW)管理。目前,国外制造商获得销售许可的唯一途径是通过韩国境内的进口商来申请。许可证具体可分为以下几级:
⑴I 级–上市通告
⑵II 级–上市许可证(包括型号测试);
⑶III 类–上市许可证(包括型号测试和安全性能评估);
II类和III类医疗器械进口商须向KFDA提交相关产品技术资料,相当于向FDA申请上市
通告和/或上市许可证时所需提供的资料。对于一些III类产品,要求进行安全和性能测试,有时还需要临床跟踪。法律规定,生产许可申请必须在从资料提交日起55天内完成复审。近来,韩国接受了第三方审核的概念,现在经KFDA认可的实体可以完成II类产品生产许可的复审工作,但一些放射性仪器除外。
III类产品必须由KFDA认可并具有试验能力的试验室进行―类型测试‖,这一规定与日本申请上市许可证时的―类型测试‖相类似。测试必须完成对产品安全、电磁兼容性(EMC)和性能的测定。所有这些测定工作必须在―类型测试‖阶段完成。并且在产品的技术资料中应当有按照国际标准的测试方法和产品规格的详细说明。国外制造商可用国际电工委员会(IEC)的CB测试报告或符合优良实验实践(GLP)的实验室出具的测试报告。
新的医疗器械使用法已于2003年5月间颁布,取代原先的《药品管理法》,对医疗器械的相关管理作了规定。新法非常接近FDA管理制度。主要包括IDE(器械测试免除,Investigation Device Exemption)和国外制造商直接认可等法律规定。
美国FDA医疗器械认证详解
美国FDA医疗器械认证详解 FDA对医疗器械有明确和严格的定义,其定义如下:“所谓医疗器械是指符合以下条件之仪器、装置、工具、机械、器具、插入管、体外试剂及其它相关物品,包括组件、零件或附件:明确列于National Formulary或the Unite States Pharmacopeia或前述两者的附录中者;预期使用于动物或人类疾病,或其它身体状况之诊断,或用于疾病之治愈、减缓与治疗者;预期影响动物或人体身体功能或结构,但不经由新陈代谢来达到其主要目的者” 只有符合以上定义的产品方被看作医疗器械,在此定义下,不仅医院内各种仪器与工具,即使连消费者可在一般商店购买之眼镜框、眼镜片、牙刷与按摩器等健身器材等都属于FDA 之管理范围。它与国内对医疗器械的认定稍有不同。 根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),Ⅲ类风险等级最高。FDA 将每一种医疗器械都明确规定其产品分类和管理要求,目前FDA医疗器械产品目录中共有1,700多种。任何一种医疗器械想要进入美国市场,必须首先弄清申请上市产品分类和管理要求。 只有符合以上定义的产品方被看作医疗器械,在此定义下,不仅医院内各种仪器与工具,即使连消费者可在一般商店购买之眼镜框、眼镜片、牙刷与按摩器等健身器材等都属于FDA 之管理范围。它与国内对医疗器械的认定稍有不同。 根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),Ⅲ类风险等级最高。FDA 将每一种医疗器械都明确规定其产品分类和管理要求,目前FDA医疗器械产品目录中共有1,700多种。任何一种医疗器械想要进入美国市场,必须首先弄清申请上市产品分类和管理要求。 在明确了以上信息后,企业就可以着手准备有关的申报资料,并按一定程序向FDA申报以获取批准认可。对于任何产品,企业都需进行企业注册(Registration)和产品列名(Listing)。对Ⅰ类产品(占47%左右),实行的是一般控制(General Control),绝大部分产品只需进行注册、列名和实施GMP规范,产品即可进入美国市场(其中极少数产品连GMP也豁免,极少数保留产品则需向FDA递交510(K)申请即PMN (Premarket Notification));对Ⅱ类产品(占46%左右),实行的是特殊控制(Special Control),企业在进行注册和列名后,还需实施GMP和递交510(K)申请(极少产品是510(K)豁免);对Ⅲ类产品(占7%左右),实施的是上市前许可,企业在进行注册和列名后,须实施GMP并向FDA递交PMA (Premarket Application)申请(部分Ⅲ类产品还是PMN)。 对Ⅰ类产品,企业向FDA递交相关资料后,FDA只进行公告,并无相关证件发给企业;对Ⅱ、Ⅲ类器械,企业须递交PMN或PMA,FDA在公告的同时,会给企业以正式的市场准入批准函件(Clearance),即允许企业以自己的名义在美国医疗器械市场上直接销售其产品。至于申请过程中是否到企业进行现场GMP考核,则由FDA根据产品风险等级、管理要求和市场反馈等综合因素决定。 综合以上内容可知,绝大部分产品在进行企业注册、产品列名和实施GMP,或再递交510(K)申请后,即可获得FDA批准上市。
欧洲有源医疗器械相关法规
COMMISSION REGULATION (EU) No 207/2012 of 9 March 2012 on electronic instructions for use of medical devices (Text with EEA relevance) THE EUROPEAN COMMISSION, Having regard to the Treaty on the Functioning of the European Union, Having regard to Council Directive 90/385/EEC of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices ( 1 ), and in particular Article 9(10) thereof, Having regard to Council Directive 93/42/EEC of 14 June 1993 concerning medical devices ( 2 ), and in particular Article 11(14) thereof, Whereas: (1) For some medical devices the provision of instructions for use in electronic form instead of in paper form can be beneficial for professional users. It can reduce the environmental burden and improve the competitiveness of the medical devices industry by reducing costs, while maintaining or improving the level of safety. (2) Such possibility of providing instructions for use in elec - tronic form instead of in paper form should be limited to certain medical devices and accessories intended to be used in specific conditions. In any case, for reasons of safety and efficiency users should always have the possi -bility to obtain those instructions for use in paper form on request. (3) In order to reduce potential risks as far as possible, the appropriateness of the provision of instructions for use in electronic form should be subject to a specific risk assessment by the manufacturer. (4) In order to ensure that users have access to the instructions for use, appropriate information about access to those instructions for use in electronic form and about the right to request the instructions for use in paper form, should be provided. (5) To ensure unconditional access to the instructions for use in electronic form and to facilitate the communication of updates and of product alerts, the instructions for use in electronic form should also be available through a website. (6) Regardless of the language obligations imposed on manufacturers by the law of the Member States, manu -facturers who provide instructions for use in electronic form should indicate on their website in which Union languages those instructions are available. (7) Except for medical devices of Class I, as defined in Annex IX to Directive 93/42/EEC, the fulfilment of the obligations laid down in this Regulation should be reviewed by a notified body during the procedure applicable for conformity assessment based on a specific sampling method. (8) As the protection of the right to privacy of natural persons with respect to the processing of personal data should be ensured by manufacturers and notified bodies as well, it is appropriate to provide that websites containing instructions for use of a medical device fulfil the requirements of Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995 on the protection of individuals with regard to the processing of personal data and on the free movement of such data ( 3 ). (9) In order to ensure safety and consistency, instructions for use in electronic form which are provided in addition to complete instructions for use in paper form should be covered by this Regulation as regards limited requirements in relation to their contents and websites. (10) It is appropriate to provide for a deferred application of this Regulation so as to facilitate the smooth transition to the new system and to allow all operators and Member States time to adapt to it. (11) The measures provided for in this Regulation are in accordance with the opinion of the Committee set up by Article 6(2) of Directive 90/385/EEC, HAS ADOPTED THIS REGULATION: Article 1 This Regulation establishes the conditions under which the instructions for use of medical devices referred to in point 15 of Annex 1 to Directive 90/385/EEC and in point 13 of Annex I to Directive 93/42/EEC may be provided in electronic form instead of in paper form. It also establishes certain requirements concerning instructions for use in electronic form which are provided in addition to complete instructions for use in paper form relating to their contents and websites. Article 2 For the purposes of this Regulation, the following definitions shall apply: (a) ‘instructions for use’ means information provided by the manufacturer to inform the user of the device of its safe and proper use, of its expected performances and of any ( 1 ) OJ L 189, 20.7.1990, p. 17. ( 2 ) OJ L 169, 12.7.1993, p. 1. ( 3 ) OJ L 281, 23.11.1995, p. 31.
(完整版)医疗器械全部法规汇总2018.1.8
目录 1. 医疗器械监督管理条例 (3) 2. 医疗器械注册管理办法 (28) 3. 体外诊断试剂注册管理办法 (47) 4. 体外诊断试剂注册管理办法修正案 (70) 5. 医疗器械说明书和标签管理规定 (71) 6. 医疗器械分类规则 (78) 7. 医疗器械通用名称命名规则 (87) 8. 医疗器械临床试验质量管理规范 (90) 9. 关于印发医疗器械应急审批程序的通知 (116) 10. 关于印发创新医疗器械特别审批程序(试行)的通知 (119) 11. 关于第一类医疗器械备案有关事项的公告 (133) 12. 医疗器械召回管理办法 (154) 13. 关于发布第一类医疗器械产品目录的通告 (165) 14. 关于发布医疗器械产品技术要求编写指导原则的通告 (231) 15. 关于实施《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》有关事项的通知235 16. 关于发布免于进行临床试验的第二类医疗器械目录的通告 (242) 17. 关于发布免于进行临床试验的第三类医疗器械目录的通告 (356) 18. 关于发布需进行临床试验审批的第三类医疗器械目录的通告 (378) 19. 关于印发医疗器械检验机构开展医疗器械产品技术要求预评价工作规定的通知 (382) 20. 关于公布医疗器械注册申报资料要求和批准证明文件格式的公告 (385) 21. 关于公布体外诊断试剂注册申报资料要求和批准证明文件格式的公告 (414) 22. 关于发布体外诊断试剂临床试验技术指导原则的通告 (435) 23. 关于发布体外诊断试剂说明书编写指导原则的通告 (450) 24. 关于实施第一类医疗器械备案有关事项的通知 (461) 25. 关于印发境内第三类和进口医疗器械注册审批操作规范的通知 (465) 26. 关于印发境内第二类医疗器械注册审批操作规范的通知 (476) 27. 关于发布医疗器械优先审批程序的公告 (492) 28. 关于发布创新医疗器械特别审批申报资料编写指南的通告 (499) 29. 关于发布医疗器械审评沟通交流管理办法(试行)的通告 (506) 30. 医疗器械生产监督管理办法 (517) 31. 医疗器械生产质量管理规范 (534) 32. 关于发布医疗器械生产质量管理规范附录无菌医疗器械的公告 (549) 33. 关于发布医疗器械生产质量管理规范附录植入性医疗器械的公告 (560) 34. 关于发布医疗器械生产质量管理规范附录体外诊断试剂的公告 (574) 35. 关于医疗器械生产日常监督现场检查工作指南的通知 (587) 36. 关于医疗器械生产经营备案有关事宜的公告 (596) 37. 关于实施《医疗器械生产监督管理办法》和《医疗器械经营监督管理办法》有关事项的通知 (607)
医疗器械FDA认证详细操作流程和操作术语
医疗器械FDA认证详细操作流程和操作术语 一、概述 据了解,大多数国家都对医疗器械制定了相应的法规,以保护公民的安全。不同国家对进入本国市场的医疗器械,要求各不相同。FDA510(K)就是由美国食品和药物管理局(简称FDA)制定的美国市场标准。随着我国医疗器械行业的发展和加入世界贸易组织,美国器械市场的大门已向我国厂商敞开。据海关统计,2002年我国对美国出口了超过5亿美元的医疗器械,成为其继欧盟、墨西哥、日本和瑞士之后的第五大进口来源国。尽管如此,据官方统计,从1976年至今,FDA共受理、批准超过10万份510(k)申请,而其中由中国申请人提交的只有40份,这与我国作为美国重要医疗器械供应国的地位极不相称。究其原因,主要是由于国内厂商向美国出口医疗器械时大多让美国的经销商或代理来申请FDA510(K),最终并没有直接以自己名义获得FDA授权。 510(K)的含义是市场预投放登记,对应药品和化妆品(FD&C)行动委员会和21CFR807的510(k)章节,故称510(K)文件,它所覆盖的范围包括食品、药品、化妆品和医疗器械。 为了在美国上市医疗器械,制造商必须经过两个评估过程其中之一(如果器械没有被510(k)豁免): 二、上市前通告[510(k)(如果器械没有被510(k)豁免),或者上市前批准(PMA) 大多数在美国进行商业分销的医疗器械都是通过上市前通告[510(k)]的形式得到批准的。在某些情况下,在1976年5月28日之前合法上市
的器械,既不要求递交510(k)也不要求递交PMA。 510(k)文件是向FDA递交的上市前申请文件,目的是证明申请上市的器械与不受上市前批准(PMA)影响的合法上市器械同样安全有效,即为等价器械(substantially equivalent)。申请者必须把申请上市的器械与现在美国市场上一种或多种相似器械对比,得出并且支持等价器械的结论。合法上市器械是在1976年5月28日之前合法上市的器械(preamendment device),或者从III类器械中分入II或I类的器械,或者通过510(k)程序发现与这样的器械等价的器械,或者通过自动的III类器械定义的评价建立的器械。与之等价的器械被称为“predicate device(s)”。申请者必须提交描述性的数据,必要的时候,要提交性能数据来说明器械是predicate device的等价器械。再次说明,510(k)的数据是显示相似性的数据,即,新器械与predicate device的等价程度 三、等价器械 510(k)不像PMA那样要求合理的安全性和有效性的证明,而是要求等价器械的证明。等价器械就是新的器械与predicate device一样安全有效。与predicate device相比,如果符合下列条件,就认为器械是等价器械:--与predicate device有相同的使用目的,具有相同的技术性能,或者 --与predicate device有相同的使用目的,具有不同的技术性能,但是并没有增加安全性和有效性的问题,并且证明人证明器械与合法上市器械一样安全有效。 所谓等价器械并不是说新的器械与predicate devices必须完全相同。等价器械是关于使用目的、设计、使用的或传送的能源、材料、性能、安全
2018一类医疗器械注册流程
综普咨询,一类医疗器械注册团队,您值得信赖 2018一类医疗器械注册流程 医疗器械,是指单独或者组合使用于人体的仪器、设备、器具、材料或者其他物品,包括所需要的软件;其用于人体体表及体内的作用不是用药理学、免疫学或者代谢的手段获得,但是可能有这些手段参与并起一定的辅助作用。 国产医疗器械注册过程需提交的资料 (1) 境内医疗器械注册申请表 (2)医疗器械生产企业资格证明 (3)产品技术报告 (4)安全风险分析报告 (5)适用的产品标准及说明 (6)产品性能自测报告 (7)医疗器械检测机构出具的产品注册检测报告 (8)医疗器械临床试验资料 (9)医疗器械说明书 (10)产品生产质量体系考核(认证)的有效证明文件 (11)所提交材料真实性的自我保证声明 步骤/方法 01确定申办产品型号 02进行产品分类 03受理资料所需要的证件
综普咨询,一类医疗器械注册团队,您值得信赖04熟悉企业的产品文件 05技术标准制定 06省局进行申报,取得受理 07医疗器械审评中心技术审评 08行政审批 09取得证书 特别提示
综普咨询,一类医疗器械注册团队,您值得信赖医疗器械注册周期主要分为四个段:标准撰写及备案、资料准备时间、产品检测时间、SFDA资料审查和批准时间。其中资料准备的时间取决于产品生产者;产品检测的时间国家规定为45个工作日(电气类产品为60工作日,涉及生物学检测的产品时间取决于试验项目和相应试验周期);资料审查及注册证打印的时间国家规定为105个工作日(一次性通过的情况下)。如产品资料在审查过程中需要补充资料,审查时间就会停止,在接到补充资料后重新开始计时。根据经验,I类产品注册周期,从资料基本到齐算起,注册周期6个月左右。II、III类产品注册周期,从资料基本到齐算起,注册周期10-14个月左右、三类植入产品注册周期,从资料基本到齐算起,注册周期12-15个月左右。 综普咨询,一家备案技术团队,为了保证服务质量,公司已建有高素质的资深技术团队,和前端申报顾问团队。并且已在国产特殊化妆品和进口化妆品备案注册,国产非特殊化妆品备案,国内化妆品出口检测认证,消毒产品批件注册、保健食品备案申报、一类医疗器械批文注册等业务方面,积累了丰富的经验和具有明显优势!欢迎咨询综普为您提供合理的备案服务!
医疗器械各国认证要求
医疗器械各国认证要求 医疗器械标准 这些医疗器械标准已被许多国家广泛采用。 医疗器械指令列表 欧盟指令中涵括许多基本保健及安全规定,以及评估产品符合规定程度的订定程序。每项指令在区域性标准制订机构所订定的调与欧洲标准中均会具体说明详细的基本规定。 因此,相关产品的制造商、进口商及配销商都必须明显标示,产品完全符合每项指令在「基本规定」所列出的保健及安全规定。 形成「新方法」基础的指令,包含了广泛的产品类别(横向指令)或某种特定的产品类别( 纵向指令)。 指令标题
欧盟 所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE标志,以说明产品符合欧盟制定的相关指令。 医疗器械需要满足的指令有: 《有源植入性医疗器械指令》(AIMDD, 90/385/EEC)、 《医疗器械指令》(MDD,93/42/EEC) 体外诊断器械指令(IVDD, 98/79/EC)。 医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备。它根据不同的要求共分为6个等级,供认证机构评估。 认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或EN/ISO 13485评审质量体系。 由于美国、加拿大与欧洲普遍以ISO 9001, EN 46001或ISO 13485作为质量保证体系的要求,故建议质量保证体系的建立均以这些标准为基础。 体外诊断医疗器械指令(IVDD),IVDD的要求与MDD相似,可按以下分类申请:-
北美 在美国,食品与药物管理局(FDA)就是监督与管理获准向消费者进行销售的食物,药物,化妆品与医疗器械的法定机构。器械及放射线健康中心(CDRH),作为FDA的一个分支,专管医疗器械。其对医疗器械按不同等级进行不同程度的监管(医疗器械分为I级,II级或III级,I级作为低风险范畴,而III级属高风险范畴): 在加拿大方面,加拿大医疗器械认证认可机构(CMDCAS)要求医疗器械厂商提前获得经CMDCAS认可的第三方机构,如UL的质量体系审查,证明其质量系统符合CMDCAS的ISO13485/ISO13488标准。对CMDCAS认证的了解对于完成FDA 的质量系统注册(QSR)非常有帮助,因为如上所述的QSR就是以ISO 9001与ISO 13485标准为基础的。 大多数属于I级或II级的医疗器械,需要获得510(k)或称上市通告,只有低风险的I级器械,可以豁免510(k)。 FDA 要求准备上市的医疗器械必须具有与已被肯定的器械(指已获准在市场销售的器械)等同的安全性与效力。因此制造商需提供报告说明其产品与市场上同类产品的详细比较情况。 制造商有责任获取与验证已被肯定的器械的相关信息,比如目录,使用说明书与510(k)其它要求的资料信息。 通常,有三种情况需要申请510(k): ①传统审核,适用于推介新器械,申请时需递交适用的性能报告。 ②特殊审核,适用于依照设计控制程序作了较小修改的器械。 ③简化审核,由制造商提交,制造商必须确保并声明其产品符合现有FDA认可的标准。 FDA 510(k) 审查
欧盟医疗器械CE认证监管模式简介
欧盟医疗器械CE认证监管模式简介 一、欧盟简介 欧洲联盟(European Union),简称欧盟(EU),是由欧洲共同体(European Communities) 发展而来,是一个集政治实体和经济实体于一身、在世界上具有重要影响的区域一体化组织。1991年12月,欧洲共同体马斯特里赫特首脑会议通过《欧洲联盟条约》,通称《马斯特里赫特条约》(简称《马约》)。1993年11月1日,《马约》正式生效,欧盟正式诞生。欧盟的宗旨是―通过建立无内部边界的空间,加强经济、社会的协调发展和建立最终实行统一货币的经济货币联盟,促进成员国经济和社会的均衡发展‖,―通过实行共同外交和安全政策,在国际舞台上弘扬联盟的个性‖。目前欧盟有27个成员国(Member State),4个自由贸易区域(European Free Trade Area)。 欧盟的主要组织机构有: 欧洲理事会(European Council) ,即首脑会议,由欧盟成员国国家元首或政府首脑及欧盟委员会主席组成。是欧盟的最高权力机构。理事会下设有总秘书处。 欧盟委员会(Commission of European Union) ,是欧盟的常设执行机构。负责实施欧盟条约和欧盟理事会做出的决定,处理日常事务,代表欧盟对外联系和进行贸易等方面的谈判。 欧洲议会(European Parliament) ,是欧洲联盟的执行监督、咨询机构,在某些领域有立法职能,并有部分预算决定权。欧洲议会可以2/3多数弹劾欧盟委员会,迫其集体辞职。 二、医疗器械的相关法规文件 目前,欧盟已颁布实施的医疗器械指令有三个,包括: 1、有源植入医疗器械指令(EC-Directive 90/385/EEC)。该指令适用于心脏起搏器、可植入的胰岛素泵等有源植入医疗器械,于1993年1月1日生效,1995年1月1日强制实施。 2、医疗器械指令(EC-Directive 93/42/EEC)。该指令适用于除90/385 EEC指令和98/79 EEC指令规定以外的一般医疗器械,于1995年1月1日生效,并于1998年6月14日强制实施。 3、体外诊断医疗器械指令(EC-Directive 98/79/EEC)。该指令适用于血细胞计数器、妊娠检测装置等体外诊断用医疗器械,于1998年12月7日生效,2003年12月7日强制实施。 上述指令是欧盟范围内统一执行的医疗器械管理法规,其法律地位相当于中国的《医疗器械监督管理条例》和日本的药事法(The Pharmaceutical Affairs Law)。 三个医疗器械指令虽然颁布的时间不同,但相互关联。医疗器械指令(EC-Directive 93/42/EEC)是在有源植入医疗器械指令(EC-Directive 90/385/EEC)的基础上制订的,二者又同为体外诊断医疗器械指令(EC-Directive 98/79/EEC)的编写基础。三个指令的格式、内容、基本要求大致相同,并针对医疗器械的不同特点而规定了特殊条款。当新颁布的指令对已有指令的基本要求进行修改时,已有指令同时进行相应修订。 医疗器械指令(EC-Directive 93/42/EEC)由23项条款和12个附录组成,其主要内容为: 1、定义和范围(Definitions,scope) 2、上市与投入使用(Placing on the market and putting into service) 该条款中规定制造商需采取所有必要的措施,确保医疗器械在依照设计的目的安装、维护和使用时不会危及患者、使用者或相关人员的安全及健康。 3、基本要求(Essential requirements) 该条款中规定医疗器械必须符合指令附录Ⅰ中的基本要求。 4、医疗器械的自由流通和特殊用途的医疗器械(Free movement,devices intended for special purposes) 该条款中规定各成员国不能对符合指令规定的临床研究用器械、定制器械和带有CE标记的医疗器械产品设置流通障碍。同时规定定制器械、参展器械和临床研究用器械在使用时可无需带有CE标记。 5、可参考的标准(Reference to standards) 6、标准与技术法规委员会(Committee on Standards and Technical Regulations) 该条款规定依据83/189/EEC号指令第五条所设立的委员会应协助欧盟委员会工作。
申请医疗器械注册证流程细解
申请医疗器械注册证流程细解
申请医疗器械注册证流程细解 医疗器械注册证(II类,非体外诊断试剂类) 一、非体外诊断医疗器械申请材料目录: 资料编号1、医疗器械注册申请表; 资料编号2、医疗器械生产企业资格证明; 资料编号3、产品技术报告; 资料编号4、安全风险分析报告; 资料编号5、适用的产品标准及说明;(应有检测机构签章) 资料编号6、产品性能自测报告; 资料编号7、有承检资质的医疗器械检测机构出具的产品注册检测报告;(原件) 资料编号8、医疗器械临床试验资料;(原件,具体提交方式见《注册管理办法》附件12) 资料编号9、医疗器械说明书;
资料编号10、产品生产质量体系考核(认证)的有效证明文件;(原件) 资料编号11、所提交材料真实性的自我保证声明。 二、非体外诊断医疗器械申请材料要求: (一)申报资料的一般要求: 1、格式要求:(1)申请材料的同一项目的填写应一致;(2)申请材料应使用A4规格纸张打印;(3)申请材料应清晰、整洁,每份申请材料均应装订并加盖企业公章,并按照申请材料目录的顺序装订成册;(4)在每项文件的第一页作一标签,或用带标签的隔页纸分隔,并标明项目编号;(5)用档案袋将报送的材料装好,档案袋需使用封面(格式见“档案袋封面格式.doc”),在袋面标明生产企业名称、地址、产品名称、联系人及电话,并加注申请材料审核的医疗器械注册申请事务人员姓名(需亲笔签名),联系方式,如是医疗器械注册专员请提供姓名(需亲笔签名)、联系方式及备案凭证号。
2、医疗器械注册申请表、产品标准一式两份,其他资料各一份。(附件1~附件5另附,无需与整套申请材料一起装订) 3、各项(上市批件、标准、检测报告、说明书)申报资料中的产品名称应与申请表中填写的产品名称实质性内容相对应。若有商品名,应标注商品名。申报资料应当使用中文,根据外文资料翻译的申报资料,应当同时提供原文。 4、申报资料受理后,企业不得自行补充申请,但属于《医疗器械注册管理办法》第三十八条规定情形的,可以补充申请。 5、生产企业在提交注册申报资料时,应同时提交医疗器械注册申请表、产品注册标准及备案说明书、标签和包装标识的电子文本(Word格式,具体要求见《关于调整医疗器械说明书备案内容的通知》(食药监办[2008]125号),其内容须与纸质文件的内容相一致。电子文本可通过移动存储设备(U盘或光盘)形式提交。 6、办理医疗器械注册申请事务的人员应当受生产企业委托,应提交生产企业负责人身份证
CE医疗器械认证流程介绍
CE医疗器械认证流程介绍 步骤一、确定并分析出口器械,确定它是否在欧盟的3个医疗器械指令的范围内。 因为CE认证过程比较复杂,因此寻找合适的医疗器械咨询公司配合如奥咨达公司,将会缩短产品进入欧洲市场的时间和减少认证成本。 步骤二、确认适用的基本要求 指令规定,任何医疗器械必须满足相关指令中所规定的预期用途,所以对制造商来说,首先要做的而且是最重要的事情就是确认所有的适用于其产品的基本条件。 步骤三、确认任何有关的欧洲协调标准 协调标准是由欧洲标淮委员会(CEN)和欧洲电气技术委员会 (CENELEC)制定的公布在欧盟官方杂志上的标准,对于某种医疗器械来说,可能有多种协调标准适用于它,因此在确认哪些协调标准适用于某种产品对应十分仔细。 步骤四、产品分类 根据指令附录IX的分类规则,医疗器械分成4类,即I、IIa、IIb和III 类,不同类型的产品,其获得CE标志的途径(符合性评价程序)不同,因此对制造商来说,如何准确地确定其产品的类型,是十分关键的。 步骤五、确定相应的符合性评价程序 对于IIa、IIb和III类医疗器械的制造商来说,存在着如何选择符合性评价程序途径的问题。主要的区别是选择型式试验的方式,还是选择质量体系的方式,这两种途径各有其特点,制造商应根据自己的实际情况选择最为适合的途径。 步骤六、确保产品满足基本要求或协调标准的要求并且使证据文件化 制造商应能提出充分的证据(如,由认证机构或其他检测机构依据协调标准进行的检测等)来证明产品符合基本要求。制造商应建立质量体系、进行产品测试、准备技术文件。 步骤七、选择认证机构审核 对于IIa、IIb和III类医疗器械,以及无菌的或具有测量功能的I类医疗器械,应选择一个认证机构并进行符合性评价程序。在欧盟官方杂志上公布的认证机构名单上,对每个认证机构可以从事的医疗器械认证范围以及可进行的符合
ISO13485医疗器械质量管理体系认证的流程
ISO13485中文叫“医疗器械质量管理体系用于法规的要求”由于医疗器械是救死扶伤、防病治病的特殊产品,仅按ISO9000标准的通用要求来规范是不够的,为此ISO组织颁布了ISO13485:1996版标准(YY/T0287 和YY/T0288),对医疗器械生产企业的质量管理体系提出了专用要求,为医疗器械的质量达到安全有效起到了很好的促进作用。2017年11月为止的执行版本是ISO13485:2016《医疗器械质量管理体系用于法规的要求》。名称和内容相较以前版本有所改变。 ISO13485认证分为初次认证、年度监督检查和复评认证等,具体如下: 一、初次认证 1、企业将填写好的《ISO13485认证分申请表》,认证中心收到申请认证材料后,会对文件进行初审,符合要求后发放《受理通知书》。 2、现场检查一周前将检查组组成和检查计划正式报企业确认。 3、现场检查按环境标志产品保障措施指南的要求和相对应的环境标志产品认证技术要求进行。 4、检查组根据企业申请材料、现场检查情况、产品环境行为检验报告撰写环境标志产品综合评价报告,提交技术委员会审查。 5、认证中心收到技术委员会审查意见后,汇总审查意见。 6、认证中心向认证合格企业颁发环境标志认证证书,组织公告和宣传。 7、获证企业如需标识,可向认证中心订购;如有特殊印制要求,应向认证中心提出申请并备案。 8、年度监督审核每年一次。 二、年度监督检查 1、认证中心根据企业认证证书发放时间,制订年检计划,提前向企业下发年检通知。企业按合同要求缴纳年度监督管理费,认证中心组成检查组,到企业进行现场检查工作。 2、现场检查时,对需要进行检验的产品,由检查组负责对申请认证的产品进行抽样并封样,送指定的检验机构检验。 3、检查组根据企业材料、检查报告、产品检验报告撰写综合评价报告,报认证中心总经理批准。 4、年度监督检查每年一次。三、复评认证3年到期的企业,应重新填写《ISO13485认证分申请表》,连同有关材料报认证中心。其余认证程序同初次认证。 认证材料 1.申请方授权代表签署的产品质量认证申请书、质量体系认证申请书; 2.申请单位质量手册,必要时提供企业的程序文件; 3.申请认证的产品或质量体系覆盖的产品标准;
二类医疗器械注册所需资料及详细步骤
二类医疗器械注册所需资料及详细步骤 所需资料: 1、医疗器械注册申请表 2、医疗器械生产企业资格证明 3、产品技术报告 4、安全风险分析报告 5、适用的产品标准及说明 6、产品性能自测报告 7、有承检资质的医疗器械检测机构出具的产品注册检测 报告 8、医疗器械临床试验资料 9、医疗器械说明书 10、产品生产质量体系考核(认证)的有效证明文件 11、所提交材料真实性的自我保证声明 注册步骤: a)产品技术报告(产品名称、产品型号/规格及其划分说明; 性能指标:能客观判定的成品的功能性、安全性指标及质量控制相关指标,符合国家标准/行业标准,但不包括产品设计开发的评价性内容;检验方法:具有可重现性和可操作性。) b)注册检测(要求检测公司具有相应的检测资质,我方提供
技术相关资料、注册检验的样品及产品技术;检测公测出检测报告和预评价报告) c)临床试验(临床试验;临床试验合同或协议、临床试验方 案、临床试验报告) d)产品生产质量体系考核(认证)的有效证明文件 e)安全风险分析报告、产品性能自测报告…….. 备注: 医疗器械注册申请表(包括电子版和纸质版,已有表格)、医疗器械生产企业资格证明(生产许可证和营业执照)、 安全风险分析报告(包括能量危害、生物学危害、环境危害、有关使用的危害和由功能失效、维护不周及老化引起的危害等方面的风险分析、风险控制与防范措施等,有相应的表格)、 适用的产品标准及说明(产品的标准可为国家标准、行业标准或注册产品标准文本;采用国家和行业标准的,要提交所采纳的国家标准或行业标准的有效文本及采标说明,采用注册产品标准作为产品标准的,提交注册产品标准正式文本及其编制说明) 产品性能自测报告(产品标准中要求的出厂检测,包括产品名称、规格型号、产品编号或批号、生产日期、样品数量、抽样基数;检测依据、检测项目、标准要求、检测结果、结
医疗器械相关法规汇总
一、主要注册法规 《医疗器械标准管理办法》(试行)(局令第31号) 2002年1月4日发布,2002年5月1日实施 《境内第三类和进口医疗器械注册文件受理标准》(国药监械[2002]18号) 2002年1月22日发布并实施 关于执行《医疗器械标准管理办法》有关事项的通知(国药监械[2002]223号) 2002年7月2日发布并实施 《关于发布医疗器械注册产品标准编写规范的通知》(国药监械[2002]407号) 2002年11月7日发布并实施 《医疗器械临床试验规定》(国家食品药品监督管理局第5号令) 2004年1月17日发布,2004年4月1日实施 《关于医疗器械产品注册后说明书更改备案有关事项的公告》(国食药监械[2004]55号)2004年3月11日发布并实施 国家食品药品监督管理局第10号令《医疗器械说明书、标签和包装标识管理规定》 2004年7月8日发布并实施 《医疗器械注册管理办法》(国家食品药品监督管理局第16号令) 2004年8月9日发布并实施 关于向生产企业提供境内第三类、境外医疗器械注册产品标准副本有关操作程序的通知(食药监械函[2005]42号) 2005年7月5日发布并实施 关于《医疗器械注册管理办法》重新注册有关问题的解释意见(国食药监械[2006]284号)2006年6月26日发布并实施 《医疗器械产品注册技术咨询管理规范》
2007年3月26日发布并实施 《医疗器械注册证书纠错-技术审评环节管理规范》 2007年3月26日发布并实施 《医疗器械说明书备案审查规范》 2007年4月30日发布并实施 《关于印发医疗器械生物学评价和审查指南的通知》(国食药监械[2007]345号) 2007年6月15日发布并实施 《关于医疗器械注册证书变更申请有关事项的通知》(国食药监械[2007]778号) 2007年12月25日发布并实施 关于执行GB 《医用电气设备第一部分:安全通用要求》有关事项的通知(国食药监械[2008]314号) 2008年6月26日发布,2008年7月1日实施 关于印发进一步加强和规范医疗器械注册管理暂行规定的通知(国食药监械[2008]409号)2008年7月23日发布并实施 《关于清理医疗器械注册管理文件有关问题的通知》(国食药监械[2008]518号) 2008年9月16日发布并实施 《关于境外医疗器械标签和包装标识有关问题的通知》(国食药监械[2008]634号) 2008年11月3日发布并实施 《执行《关于进一步加强和规范医疗器械注册管理的暂行规定》中涉及重新注册产品注册申报资料的说明》 2009年2月23日发布并实施 《医疗器械技术审评中心关于无源植入性医疗器械产品注册申报若干技术问题的说明》 2009年7月30日发布并实施
医疗器械各国认证要求(终审稿)
医疗器械各国认证要求公司内部档案编码:[OPPTR-OPPT28-OPPTL98-OPPNN08]
医疗器械各国认证要求 医疗器械标准 这些医疗器械标准已被许多国家广泛采用。 医疗器械指令列表? 欧盟指令中涵括许多基本保健及安全规定,以及评估产品符合规定程度的订定程序。每项指令在区域性标准制订机构所订定的调和欧洲标准中均会具体说明详细的基本规定。 因此,相关产品的制造商、进口商及配销商都必须明显标示,产品完全符合每项指令在「基本规定」所列出的保健及安全规定。 形成「新方法」基础的指令,包含了广泛的产品类别(横向指令)或某种特定的产品类别(纵向指令)。 指令标题
欧盟 所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE标志,以说明产品符合欧盟制定的相关指令。 医疗器械需要满足的指令有: 《有源植入性医疗器械指令》(AIMDD, 90/385/EEC)、 《医疗器械指令》(MDD,93/42/EEC) 体外诊断器械指令(IVDD, 98/79/EC)。 医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备。它根据不同的要求共分为6个等级,供认证机构评估。
认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或 EN/ISO 13485评审质量体系。 由于美国、加拿大和欧洲普遍以ISO 9001, EN 46001或 ISO 13485作为质量保证体系的要求,故建议质量保证体系的建立均以这些标准为基础。 体外诊断医疗器械指令(IVDD),IVDD的要求与MDD相似,可按以下分类申请:- 北美 在美国,食品和药物管理局(FDA)是监督和管理获准向消费者进行销售的食物,药物,化妆品和医疗器械的法定机构。器械及放射线健康中心(CDRH),作为FDA的一