盐酸氨溴索杂质分析及杂质色谱图
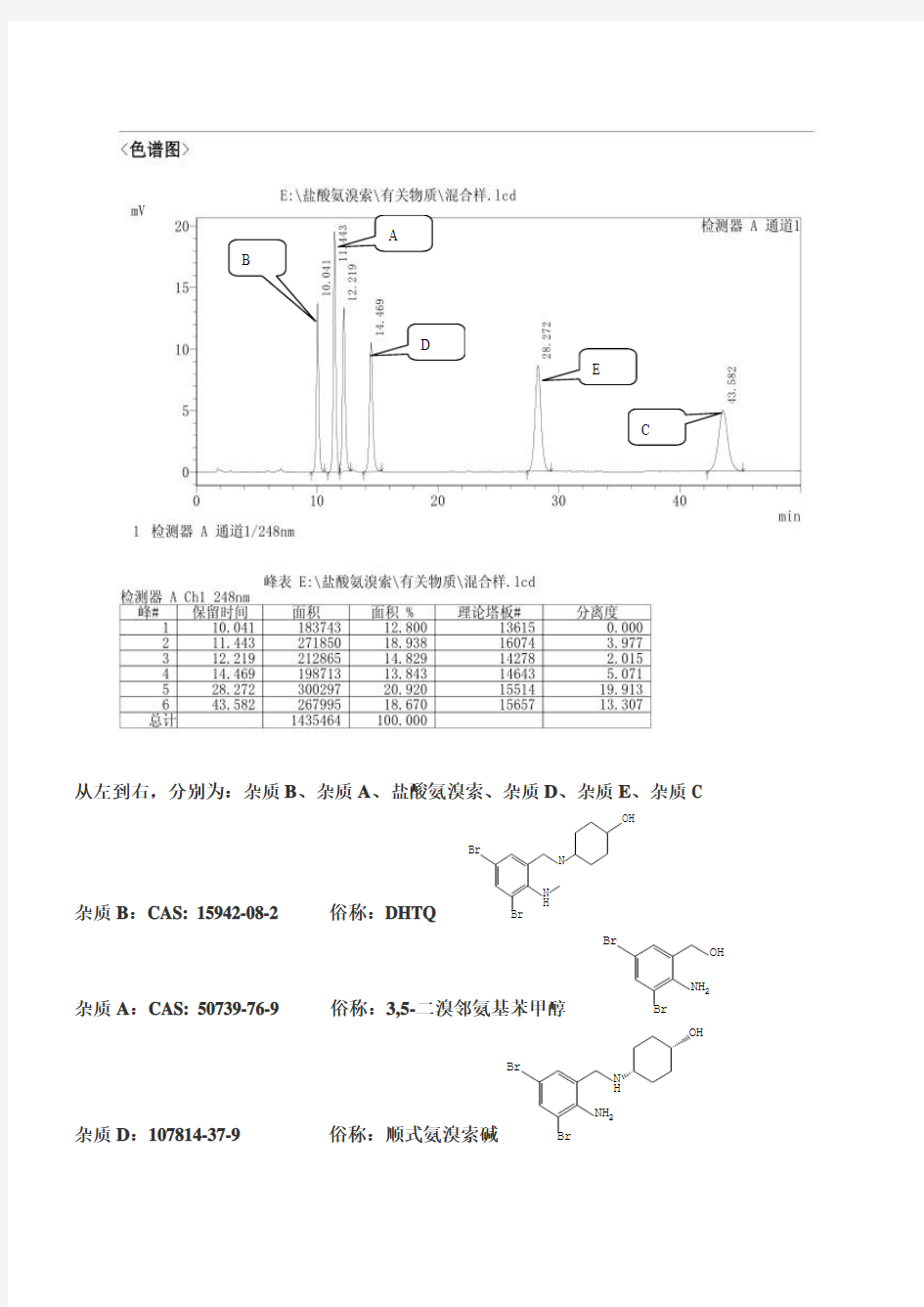

从左到右,分别为:杂质B、杂质A、盐酸氨溴索、杂质D、杂质E、杂质C
杂质B:CAS: 15942-08-2 俗称:DHTQ
N
N
H
Br
Br
OH
杂质A:CAS: 50739-76-9 俗称:3,5-二溴邻氨基苯甲醇
NH2 Br
Br
OH
杂质D:107814-37-9 俗称:顺式氨溴索碱
N
H
NH2
Br
Br
OH
B
A
D
E
C
杂质E:CAS: 50910-55-9 俗称:3,5-二溴邻氨基苯甲醛
NH2 Br
Br
杂质C:CAS: 50910-53-7 俗称:氨溴索席夫碱
N
NH2
Br
Br
OH
气相色谱仪原理(图文详解)
气相色谱仪原理(图文详解) 什么是气相色谱 本章介绍气相色谱的功能和用途,以及色谱仪的基本结构。 气相色谱(GC)是一种把混合物分离成单个组分的实验技术。它被用来对样品组分进行鉴定和定量测定: 基子时间的差别进行分离 和物理分离(比如蒸馏和类似的技术)不同,气相色谱(GC)是基于时间差别的分离技术。 将气化的混合物或气体通过含有某种物质的管,基于管中物质对不同化合物的保留性能不同而得到分离。这样,就是基于时间的差别对化合物进行分离。样品经过检测器以后,被记录的就是色谱图(图1),每一个峰代表最初混合样品中不同的组分。 峰出现的时间称为保留时间,可以用来对每个组分进行定性,而峰的大小(峰高或峰面积)则是组分含量大小的度量。 图1典型色谱图
系统 一个气相色谱系统包括 可控而纯净的载气源.它能将样品带入GC系统进样口,它同时还作为液体样品的气化室色谱柱,实现随时间的分离 检测器,当组分通过时,检测器电信号的输出值改变,从而对组分做出响应 某种数据处理装置图2是对此作出的一个总结。 样品 载气源一^ 进样口一^ 色谱柱一^ 检测器一_ 数据处理」 图2色谱系统 气源 载气必须是纯净的。污染物可能与样品或色谱柱反应,产生假峰进入检测器使基线噪音增大等。推荐使用配备有水分、烃类化合物和氧气捕集阱的高纯载气。见图
钢瓶阀 若使用气体发生器而不是气体钢瓶时,应对每一台GC都装配净化器,并且使气源尽可能靠近仪器的背面。 进样口 进样口就是将挥发后的样品引入载气流。最常用的进样装置是注射进样口和进样阀。注射进样口 用于气体和液体样品进样。常用来加热使液体样品蒸发。用气体或液体注射器穿透隔垫将样品注入载气流。其原理(非实际设计尺寸)如图4所示。
怎样分析气相色谱图
在实际工作中,当我们拿到一个样品,我们该怎样定性和定量,建立一套完整的分析方法是关键,下面介绍一些常规的步骤: 1、样品的来源和预处理方法 GC能直接分析的样品通常是气体或液体,固体样品在分析前应当溶解在适当的溶剂中,而且还要保证样品中不含GC不能分析的组分(如无机盐),可能会损坏色谱柱的组分。这样,我们在接到一个未知样品时,就必须了解的来源,从而估计样品可能含有的组分,以及样品的沸点范围。如果样品体系简单,试样组分可汽化则可直接分析。如果样品中有不能用GC直接分析的组分,或样品浓度太低,就必须进行必要的预处理,如采用吸附、解析、萃取、浓缩、稀释、提纯、衍生化等方法处理样品。 2、确定仪器配置 所谓仪器配置就是用于分析样品的方法采用什么进样装置、什么载气、什么色谱柱以及什么检测器。 一般应首先确定检测器类型。碳氢化合物常选择FID检测器,含电负性基团(F、Cl等)较多且碳氢含量较少的物质易选择ECD检测器;对检测灵敏度要求不高,或含有非碳氢化合物组分时,可选择TCD检测器;对于含硫、磷的样品可选择FPD检测器。 对于液体样品可选择隔膜垫进样方式,气体样品可采用六通阀或吸附热解析进样方法,一般色谱仅配置隔膜垫进样方式,所以气体样品可采用吸附-溶剂解析-隔膜垫进样的方式进行分析。 根据待测组分性质选择适合的色谱柱,一般遵循相似相容规律。分离非极性物质时选择非极性色谱柱,分离极性物质时选择极性色谱柱。色谱柱确定后,根据样本中待测组分的分配系数的差值情况,确定色谱柱工作温度,简单体系采用等温方式,分配系数相差较大的复杂体系采用程序升温方式进行分析。 常用的载气有氢气、氮气、氦气等。氢气、氦气的分子量较小常作为填充柱色谱的载气;氮气的分子量较大,常作为毛细管气相色谱的载气;气相色谱质谱用氦气作为载气。 3、确定初始操作条件 当样品准备好,且仪器配置确定之后,就可开始进行尝试性分离。这时要确定初始分离条件,主要包括进样量、进样口温度、检测器温度、色谱柱温度和载气流速。进样量要根据样品浓度、色谱柱容量和检测器灵敏度来确定。样品浓度不超过10mg/mL时填充柱的进样量通常为1-5uL,而对于毛细管柱,若分流比为50:1时,进样量一般不超过2uL。进样口温度主要由样品的沸点范围决定,还要考虑色谱柱的使用温度。原则上讲,进样口温度高一些有利,一般要接近样品中沸点最高的组分的沸点,但要低于易分解温度。
热分析考试考试)20121210)
热分析习题 一、填空(10分,共10题,每题1分)。 1、差热分析是在程序控温条件下,测量样品坩埚与坩埚间的温度差与温 度的关系的方法。(参比) 2、同步热分析技术可以通过一次测试分别同时提供-TG或 -TG两组信号。(DTA-TG ,DSD-TG) 3、差示扫描量热分析是在程序控温条件下,测量输入到物质与参比物的功率差与温度的关 系的方法,其纵坐标单位为。(mw或mw/mg) 4、硅酸盐类样品在进行热分析时,不能选用材质的样品坩埚。(刚玉) 5、差示扫描量热分析根据所用测量方法的不同,可以分类为热流型DSC 与 型DSC。(功率补偿) 6、与差热分析(DTA)的不同,差示扫描量热分析(DSC)既可以用于定性分析,又可以 用于分析。(定量) 7、差热分析(DTA)需要校正,但不需要灵敏度校正。(温度) 8、TG热失重曲线的标注常常需要参照DTG曲线,DTG曲线上一个谷代表一个失重阶段, 而拐点温度显示的是最快的温度。(失重) 9、物质的膨胀系数可以分为线膨胀系数与膨胀系数。(体) 10、热膨胀系数是材料的主要物理性质之一,它是衡量材料的好坏的一个重要指 标。(热稳定性) 二、名词解释 1.热重分析答案:在程序控温条件下,测量物质的质量与温度的关系的方法。 2.差热分析答案:在程序控温条件下,测量物质与参比物的温度差与温度的关系的方法。 3.差示扫描量热分析答案:在程序控温条件下,测量输入到物质与参比物的功率差与温度的关系的方法。 4.热膨胀分析答案:在程序控温条件下,测定试样尺寸变化与温度或时间的关系的方法。 三、简答题 1.DSC与DTA测定原理的不同 答案:DSC是在控制温度变化情况下,以温度(或时间)为横坐标,以样品与参比物间温差为零所需供给的热量为纵坐标所得的扫描曲线。DTA是测量T-T 的关系,而DSC是保持T = 0,测定H-T 的关系。两者最大的差别是DTA只能定性或半定量,而DSC的结果可用于定量分析。DTA在试样发生热效应时,试样的实际温度已不是程序升温时所控制的温度(如
仪器分析气相色谱分析习题+答案.doc
气相色谱习题 一 . 选择题 ( ) 1.色谱图上一个色谱峰的正确描述是( ) A. 仅代表一种组分 ; B. 代表所有未分离组分 ; C. 可能代表一种或一种以上组分; D. 仅代表检测信号变化( )2.下列保留参数中完全体现色谱柱固定相对组分滞留作用的是( ) A. 死时间 ; B. 保留时间 ; C.调整保留时间; D.相对保留时间 ( )3.气-液色谱系统中,待分离组分的k值越大,则其保留值: A. 越大; B. 越小; C.不受影响; D.与载气流量成反比 ( )4.关于范第姆特方程式,正确的说法是: A. 最佳线速这一点,塔板高度最大; B. 最佳线速这一点,塔板高度最小; C. 塔板高度最小时,线速最小; D.塔板高度最小时,线速最大 ( )5.根据范第姆特方程式H=A+B/u+Cu,下列说法正确的是: A.H 越大,则柱效越高,色谱峰越窄,对分离有利; B. 固定相颗粒填充越均匀,则柱效越高; C. 载气线速越高,柱效越高; D. 载气线速越低,柱效越高 ( )6.在范第姆特方程式中,涡流扩散项主要受下列哪个因素影响 A. 载体填充的均匀程度 ; B.载气的流速大小; C.载气的摩尔质量; D.固定液的液膜厚度
( )7.用气相色谱法定量分析试样组分时,要求分离达98%,分离度至少为: ( )8.在气相色谱中,当两组分未能完全分离时,我们说: A. 柱效太低; B. 柱的选择性差; C.柱的分离度低; D. 柱的容量因子大 ( )9.分离非极性组分和极性组分混合物时,一般选用极性固定液,这是利用极性固定液的: A. 氢键作用; B. 诱导效应; C.色散作用; D.共轭效应 ( )10.苯和环已烷的沸点分别是80.10 °C 和 80.81 ° C,都是非极性分子。气相色谱分析中,若采用极性固定 液,则保留时间关系是: A. 苯比环已烷长; B. 环已烷比苯长; C. 二者相同; D. 无法确定 ( )11. 已知苯的沸点为80.10 ° C,环已烷的沸点为80.81 °C。当用邻苯二甲酸二壬酯作固定液分析这二种组 分时,环已烷比苯先出峰,其原因是固定液与被测组分间的: A. 静电力; B. 诱导力; C.色散力; D.氢键力 ( )12.使用热导池检测器时,一般选用H 2或He作载气,这是因为它们: A. 扩散系数大; B. 热导系数大; C.电阻小; D. 流量大 ( )13.氢火焰离子化检测器优于热导检测器的主要原因是: A. 装置简单; B. 更灵敏; C.可以检出许多有机化合物; D.较短的柱能够完成同样的分离
色谱分析中各种图谱现象的判断
色谱分析中各种图谱现象的判断 色谱分析中各种图谱现象的判断 可能产生的原因及处理办法 一.基线噪音 1 流动池脏,用极性试剂清洗。当有填料进入,拆开流通池。 2 检测器灯有问题,如能量偏低,更换氘灯。 3 周期性的波动,则起源于泵的脉冲,检修泵或更换垫片等。 4 温度对检测器的影响,控制温度。 5 气泡经过检测器,用大流量冲洗。 6 可能难出峰的样品连续不断出来,用强极性流动相冲柱。 7 流动相本底高,如水的纯度不够,换超纯水。或试剂纯度不够,换色谱纯的试剂。 二.基线漂移(上漂和下漂) 1 柱中的流动相没有平衡,延长平衡时间,尤其在流动相中添加了有紫外吸收的添加剂。 2 在梯度洗脱中,基线上漂是正常的,在空白梯度中有可能是柱子中有杂质洗出。其次是流动相中有干扰物,换流动相。 3 温度不稳定(示差检测器),控温。 4 在等度分析中,样品缓慢洗出,改变淋洗液强度或用梯度分析。 5 样品进入检测器,吸附在池中,可能每进样一次,本底一次比一次高,很少见。 三.倒峰的产生和消除 1.柱切换的脉冲效应,一般不是很明显,必要时考虑换阀。
2.在低波长分析时,流动相本底比较高时,而样品用本底低的流动相溶解,肯定出现倒峰,其程度同进样量和本底差有关。解决办法,用流动相溶解样品,减少进样量,消除倒峰的影响。高波长时,影响比较小。 3.如果倒峰不影响峰的分离,对外标法定量不影响。但影响面积归一化法。 4.样品中有比流动相本底低的物质存在,如无机盐等,将出倒峰。这种情况下,倒峰的位置不一定在死体积位置出现(大多数在死体积位置出现)。 四.鬼峰的产生和消除 1.样品分析时峰没出完,在下一针或下下一针出现,判断办法,延长分析时间,计算可能出现的保留时间。然后调整流动相。 2.连续进样,在某个位置出现忽高忽低的峰,最可能是进样针污染,清洗进样针,注意污垢的干扰,有些样品易残留在针管里。可重新取样分析。 3.定量管污染,处理方法同上。 4.在死体积位置出现的小峰,可能是柱切换造成的。 5.流动相与样品溶剂不一致,也会出现鬼峰,尤其在低波长时,出现位置在死体积的地方。 6.气泡,如果有小气泡通过流通池,也出现随机的假峰,大气泡存在,其出现的峰往往直上直下,脱气解决。 7.样品发生变化反应,重新取样快速分析。
热分析常用方法及谱图
常用的热分析方法 l热重法(Thermogravimetry TG) l 差示扫描量热仪(Differential Scanning Calorimetry DSC)l 差热分析(Differential Thermal Analysis DTA) l 热机械分析(Thermomechanical Analysis TMA) l 动态热机械法(Dynamic Mechanical Analysis DMA) 谱图分析的一般方法 《热分析导论》刘振海主编 《分析化学手册》热分析分册 TGA DSC 分析图谱的一般方法——TGA 1. 典型图谱 分析图谱的一般方法——TGA的实测图谱
I、PVC 35.26% II、Nylon 6 25.47% III、碳黑14.69% IV、玻纤24.58% 已知样品的图谱分析 与已知样品各方面特性结合起来分析 如:无机物(黏土、矿物、配合物)、生物大分子、高分子材料、金属材料等热分析谱图都有各自的特征峰。 与测试的仪器、条件和样品结合起来分析 仪器条件样品 应用与举例 TGA DSC/DTA TMA 影响测试图谱结果的因素——测试条件 TGA 升温速率 样品气氛
扫描速率 样品气氛 升温速率对TGA 曲线的影响 气氛对TGA 曲线的影响 PE TGA-7 测试条件: 扫描速率:10C/min 气氛:a. 真空 b. 空气 流量:20ml/min 样品:CaCO3(AR) 过200目筛,3-5mg 扫描速率对DSC/DTA曲线的影响气氛对DSC/DTA曲线的影响 气氛的性质
两个氧化分解峰 曲线b: 一个氧化分解峰, 和一个热裂解峰 影响测试图谱结果的因素——样品方面 TGA/DSC/DTA 样品的用量 样品的粒度与形状 样品的性质 样品用量对TGA/DSC/DTA曲线的影响 样品的粒度与形状对曲线的影响——TGA/DSC/DTA 样品的性质对曲线的影响——TGA/DSC/DTA TGA/ DSC/DTA 热分析曲线的形状随样品的比热、导热性和反应性的不同而不同。即使是同种物质,由于加工条件的不同,其热谱图也可能不同。如PET树脂,经过拉伸过的PET树脂升温结晶峰就会消失。 PET 树脂的DSC 曲线 TGA应用 成分分析 无机物、有机物、药物和高聚物的鉴别与多组分混合物的定量分析。游离水、结合水、结晶水的测定,残余溶剂或单体的测定、添加剂的测定等。 热稳定性的测定 物质的热稳定性、抗氧化性的测定,热分解反应的动力学研究等 居里点的测定 磁性材料居里点的测定 可用TGA测量的变化过程
气相色谱分析方法的建立
气相色谱分析方法的建立
内标法与外标法 一、内标法 什么叫内标法?怎样选择内标物? 内标法是一种间接或相对的校准方法。在分析测定样品中某组分含量时,加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。 内标法在气相色谱定量分析中是一种重要的技术。使用内标法时,在样品中加入一定量的标准物质,它可被色谱拄所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。采用内标法定量时,内标物的选择是一项十分重要的工作。理想地说,内标物应当是一个能得到纯样的己知化合物,这样它能以准确、已知的量加到样品中去,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质(如化学结构、极性、挥发度及在溶剂中的溶解度等)、色谱行为和响应特征,最好是被分析物质的一个同系物。当然,在色谱分析条什下,内标物必须能与样品中各组分充分分离。需要指出的是,在少数情况下,分析人员可能比较关心化台物在一个复杂过程中所得到的回收率,此时,他可以使用一种在这种过程中很容易被完全回收的化台物作内标,来测定感兴趣化合物的百分回收率,而不必遵循以上所说的选择原则。 在使用内标法定量时,有哪些因素会影响内标和被测组分的峰高或峰面积的比值? 影响内标和被测组分峰高或峰面积比值的因素主要有化学方面的、色谱方面的和仪器方面的三类。 由化学方面的原因产生的面积比的变化常常在分析重复样品时出现。 化学方面的因素包括: 1、内标物在样品里混合不好; 2、内标物和样品组分之间发生反应, 3、内标物纯度可变等。 对于一个比较成熟的方法来说,色谱方面的问题发生的可能性更大一些,色谱上常见的一些问题(如渗漏)对绝对面积的影响比较大,对面积比的影响则要小一些,但如果绝对面积的变化已大到足以使面积比发生显著变化的程度,那么一定有某个重要的色谱问题存在,比如进样量改变太大,样品组分浓度和内标浓度之间有很大的差别,检测器非线性等。进样量应足够小并保持不变,这样
色谱简单流程方框图
色谱简单流程方框图:1..典型流程中的各部件 离开来。 3.色谱操作条件选择 最佳流速的选择: 从速率理论方程式知道,载气流速对柱效有明显的影响。如果从小到大改变载气线速,那么它和理论板高H的关系如图(1)所示: H H μμ 图(1)板高H与载气线速μ关系图 曲线的最低点,即H最小则柱效最高,此点对应的流速即是最佳线速度。对N 来说, 2 则为600~720cm/min。在实际工作中,往往采用稍高于最佳线速为420~600cm/min;而H 2 最佳线速的流速,以缩短分析时间。对于一个内径为4mm的填充柱,载气流速多选用50~80ml/min。 4.固定相的使用温度范围任何一种固定相,都有其使用温度范围。如柱温超过其上限,则固定相会流失或分解,使柱寿命缩短甚至失效,而且污染检测器;如果低于其下限, 加大,而使传质阻力增高,柱效降低。则固定液粘度变大,使组分在液相中的扩散系数D L 往往还会出现异常现象,表现为峰形不正常。如果低于固定液的凝固点时,则其已不是液相了,失去了分配能力。一般说来,提高柱温,各组分的挥发度都增加,分配系统变小而组分靠拢,溶剂效率降低,不利于分开。但操作速度快,分析周期短;降低柱温,有利于分离。但柱温太低,组分蒸气在两相中的扩散传质速率大为减小,分配不能迅速达到平衡,致使峰形变宽、柱效下降,并延长分析时间。甚至组分蒸气会冷凝下来,使分析不能正常进行。 5.汽化温度
对气化温度的要求:应有足够的温度和热容量使被测试样瞬时汽化。一般高于柱温50℃以上,或比样品中组分的最高沸点高出20~40℃;试样在该温度下,不被分解。 汽化温度不足的危害:峰形变宽、峰不对称,降低柱效及分离度;峰形异常,不能重复。 汽化温度过高的危害:样品分解,出现极为复杂的峰图,同样给以假象;汽化室橡皮垫变粘,易漏气; 6.检测温度 应保证样品组分蒸气不被冷凝,一般不低于柱温;要考虑检测器对温度的要求。如火焰离子化检测器,温度不能低于100℃,防止水蒸汽冷凝,否则会破坏离子室的绝缘性,出现异常现象;要考虑温度对检测器灵敏度的影响,如热导检测器的温度高,则灵敏度降低。 注意事项: a.色谱先通载气再开电源,关机时先关电源,在各温度降至室温后再关载气。 b. 使用氢火焰检测器,不点火,为了安全严禁打开氢气气路,换气时先断电源,打开柱箱门,让柱箱内温度下降后再换气瓶, c.热导检测器,以氢气为载气,系统应试漏,尾气必须排到室外,先通载气,后开桥 流电源,换气时应关闭桥流电源。 d.用气瓶应先开总阀,再开减压器阀,关闭时先关减压器阀,后关总阀。 e.使用电脑时应先按显示器电源,后按主机电源,严禁在开机的状态下插拔电缆,不 能随意使用电脑的光驱、软驱,以防电脑感染病毒。 f.用六通阀时要轻开轻闭,不要用力过度,造成六通阀损坏。 g.色谱开机状态下,应经常注意色谱操作条件的变化,出现问题时应尽己所能及时处 理,如不能处理的应向有关技术人员或部门反映,使问题及时得到解决。 SP-2305气相色谱仪操作规程 1、适用范围:SP-2305(1#、6#)适用于酯柱色谱分析。 2、仪器设备:配有热导检测器的色谱、电脑、色谱数据处理工作站。 3、试剂和材料:氢气(纯度≥%),乙醚:AR,6201载体(φ~),5A分子筛。 4、色谱柱:柱长3m,内径4mm不锈钢管。 固定相(酯柱)的配制:按固定液邻苯二甲酸二乙酯/6201载体(φ~)=1:5的比例称取配柱所需的固定液和载体。将称好的固定液用乙醚充分溶解,倒入称好的载体,使载
色谱分析谱图
A5000气相色谱工作站分析报告 样品信息: 样品名称: 乙酸乙酯、甲苯盲样样品编号: 样品来源: 省职防院邮寄采样人: 稀释倍数: 0.0 样品量: 0.0 含量单位: 取样时间: 仪器条件: 仪器名称: 气相色谱仪柱子型号: FFAP 检测器: FID 积分参数: 最小值: 10.00 漂移: 0.02 mV/min 噪声: 0.05 mV 最小峰宽: 2.00 S 相对窗宽: 5% 计算方式: 峰面积 色谱条件: 柱箱温度: 50 (℃)程序升温载气流速: 30 (ml/min) 检测器温度: 130 (℃)空气流速: 300 (ml/min) 气化室温度: 200 (℃)氢气流速: 30 (ml/min) 谱图: 分析结果: 定量方法:外标法 序号组分名保留时间峰面积峰高含量峰型 1 二硫化碳 3.91 9726895 366254 9726895 BB 2 乙酸乙酯0.00 0 0 0.000000 BB
3 甲苯0.00 0 0 0.000000 BB 谱图: 分析结果: 定量方法:归一法 序号组分名保留时间峰面积峰高含量峰型 1 二硫化碳 3.87 9287219 363551 9287219 BB 2 乙酸乙酯 5.40 67436 4449 25.265 BB 3 甲苯8.2 4 63476 13403 8.777 B B 谱图:
分析结果: 定量方法:外标法 序号组分名保留时间峰面积峰高含量峰型 1 二硫化碳 3.88 9515607 362744 9515607 BB 2 乙酸乙酯 5.42 68086 4510 25.508 B B 3 甲苯8.25 58293 13600 8.061 BB 谱图: 分析结果: 定量方法:外标法 序号组分名保留时间峰面积峰高含量峰型 1 二硫化碳 3.88 9231735 354067 9231735 BB 2 乙酸乙酯 5.41 67415 4556 25.256 B B 3 甲苯8.25 59548 13601 8.235 BB 谱图:
色谱图
chromatogram 样品流经色谱柱和检测器,所得到的信号-时间曲线,又称色谱流出曲线(elution profile)。 色谱图是指被分离组分的检测信号随时间分布的图象。色谱图形状随色谱方法和检测记录的方式不同而不同,迎头色谱和顶替色谱的色谱图为一系列台阶;在洗脱法色谱中,若采用微分型检测器时,分离组分的检测信号随时间变化的图形为近似于高斯分布的一组色谱峰群,色谱图的纵坐标为检测器的响应信号,横坐标为时间、体积或距离。 [编辑本段] 相关术语 ⊕色谱图(chromatogram)--样品流经色谱柱和检测器,所得到的信号-时间曲线,又称色谱流出曲线(elution profile). ⊕基线(base line)--流动相冲洗,柱与流动相达到平衡后,检测器测出一段时间的流出曲线。一般应平行于时间轴。 ⊕噪音(noise)――基线信号的波动。通常因电源接触不良或瞬时过载、检测器不稳定、流动相含有气泡或色谱柱被污染所致。 ⊕漂移(drift)基线随时间的缓缓变化。主要由于操作条件如电压、温度、流动相及流量的不稳定所引起,柱内的污染物或固定相不断被洗脱下来也会产生漂移。 ⊕色谱峰(peak)--组分流经检测器时相应的连续信号产生的曲线。流出曲线上的突起部分。正常色谱峰近似于对称性正态分布曲线(高斯Gauss曲线)。不对称色谱峰有两种:前延峰(leading peak)和脱尾峰(tailing peak ).前者少见。 ⊕拖尾因子(tailing factor,T)--T=B/A,用以衡量色谱峰的对称性。也称为对称因子(symmetry factor)或不对称因子(asymmetry factor)《中国药典》规定T 应为0.95~1.05。T<0.95为前延峰,T>1.05为拖尾峰。 ⊕峰底――基线上峰的起点至终点的距离。 ⊕峰高(Peak height,h)――峰的最高点至峰底的距离。 ⊕峰宽(peak width,W)--峰两侧拐点处所作两条切线与基线的两个交点间的距离。W=4σ。 ⊕半峰宽(peak width at half-height,Wh/2)--峰高一半处的峰宽。W h/2=2. 355σ。 ⊕标准偏差(standard deviation, σ)--正态分布曲线x=±1时(拐点)的峰宽之半。正常峰宽的拐点在峰高的0.607倍处。标准偏差的大小说明组分在流出色谱柱过程中的分散程度。σ小,分散程度小、极点浓度高、峰形瘦、柱效高;反之,σ大,峰形胖、柱效低。
实验 醇系物的气相色谱分析
实验10 醇系物的气相色谱法定性、定量分析 【实验目的】 (1)理解用已知纯物质对照定性的方法。 (2)理解用气相色谱归一化法进行定量分析的方法和特点。 (3)了解CP-3800气相色谱仪的使用及软件的操作。 (4)掌握微量进样器进样技术。 (5)了解程序升温气相色谱法的原理及基本特点。 【实验原理】 气相色谱法是以气体作为流动相(简称载气)的色谱法。 气相色谱法具有如下的特点: 1.高效能、高选择性可分离性质相似的多组分混合物,如同系物、同分异构体等;分离制备高纯物质,纯度可达99.99%。 2.灵敏度高可检出10-13-10-11g的物质; 3.分析速度快通常一个样品的分析可在几分钟到几十分钟内完成; 4.应用范围广气体样品、低沸点、易挥发或可转化为易挥发的液体或固体样品,不仅可分析有机物,也可以分析部分无机物。 气相色谱仪一般由气路系统、进样系统、分离系统、检测记录系统和温度控制系统(图中未显示)五部分组成(见图1-8-1)。 1.气路系统包括气源(高压气瓶)、气体净化、 气体流量控制等 图1-8-1 气相色谱过程示意图 1.高压钢瓶 2.减压阀 3.净化管 4.流量控制 5.进样口 6.色谱柱 7.检测器
部分组成,其作用是为仪器提供纯洁、稳定的载气。常用的载气有氮气和氢气,也可用氦气、氩气或空气。 2.进样系统包括进样装置和气化室。其作用是将样品在进入色谱柱前迅速气化,并定量转入到色谱柱中。要想获得良好的分离结果,进样速度应极快,且样品应在气化室瞬间气化。液体样品一般都采用微量进样器,可根据进样量的不同选用不同体积的进样器。对气化室的要求是热容量要大,温度要足够高且无催化效应。 3.分离系统该部分由色谱柱组成,是色谱仪的心脏,其作用是分离样品。色谱柱分为填充柱和毛细管柱两种: (1)填充柱:由不锈钢或玻璃作为柱管,内填固定相制成,一般内径为2~4mm,长1~3m。形状有U型和螺旋型两种。 (2)毛细管柱又叫空心柱。毛细管材料可以是不锈钢、玻璃或石英。内径有0.53mm、0.32mm、0.25mm等几种规格,长度一般为10~30m。它的固定相可以直接涂布或通过化学交联键合在预先经过处理的管壁上。按照所用的色谱柱不同又可分为:填充柱色谱和毛细管柱色谱。 4.温度控制系统在气相色谱法中,温度直接影响到色谱柱的分离选择、检测器的灵敏度和稳定性。因此在仪器中主要是对色谱柱箱、气化室、检测器三处的温度进行控制。 5.检测和放大记录系统当样品经色谱柱分离后,各组分按保留时间不同随载气进入检测器,检测器将有关各组分含量的信息转化为易于测量的信号(一般为电信号),经过必要的放大传递给记录仪,最后得到该样品的色谱流出曲线。
气相色谱分析中色谱峰的特征
气相色谱分析中色谱峰的特征 质检部油品分析小班的色谱岗的样品分析,都是使用气相色谱仪和液相色谱仪等等。在色谱分析中,色谱图中的色谱峰不仅是我们得出最终数据的主要依据,而且一定程度上还可以精准的反映出仪器的性能和平稳性。因此,作为分析人员必须深入和充分的理解色谱峰的特征,以进一步提高分析数据的准确性。 为理解色谱峰的特征,首先必须能清楚气相色谱分析、色谱图和色谱峰之间的关系。 气相色谱(GC) 是一种把混合物分离成单个组分的实验技术它被用来对样品组分进行鉴定和定量测定。和物理分离(比如蒸馏和类似的技术)不同,气相色谱(GC) 是基于时间差别的分离技术。气相色谱仪将分析的样品分离、鉴定后,由记录仪绘出样品中各个组分的流出曲线图,即色谱图。色谱图是以组分的流出时间(t)为横坐标,以检测器对各组分的电讯号响应值(mv)为纵坐标。色谱图上可得到一组色谱峰,每个峰代表样品中的一个组分(见下图)。 图1 色谱图 对于一个色谱峰,我们可以获得以下四个基本的测量数据特征: (1)进样后到色谱峰被检测到的时间——定性分析 色谱图中色谱峰的出峰时间反映了被分析的组分因与色谱柱中固定相发生相互作用,而在色谱柱中滞留的时间,从本质上更准确的表达了被分析组分的保留特性。色谱峰的峰位与气相色谱分离过程的热力学性质密切相关,是进行气相色谱定性分析的主要依据。 (2)色谱峰的大小——定量分析
色谱峰的大小指峰高或峰面积的大小,其和每个组分在样品中的含量相关。即若色谱峰的峰面积大,则该峰代表的组分在样品中的含量高;反之,则该峰代表的组分在样品中的含量低。色谱峰的峰高或峰面积是气相色谱进行定量分析的重要依据。 (3)色谱峰的宽窄——色谱柱柱效的高低 色谱峰的宽窄可用来说明色谱分离过程的动力学性质——色谱柱柱效率的高低,色谱峰形愈窄说明柱效愈高,色谱峰形愈宽表明柱效愈低,但是色谱峰的宽窄只能定性的表达柱效。 (4)色谱峰间的距离——色谱柱的选择性 在色谱图上,两个色谱峰之间的距离大,表明色谱柱对各组分的选择性好;两个色谱峰之间的距离小,表明色谱柱对各组分的选择性差。 深入和充分的理解色谱峰特征,是为了进一步获得色谱分析的重要信息,保证分析数据准确无误与及时。
实验-醇系物的气相色谱分析
实验10醇系物的气相色谱法定性、定量分析 【实验目的】 (1)理解用已知纯物质对照定性的方法。 (2)理解用气相色谱归一化法进行定量分析的方法和特点。 (3)了解CP-3800气相色谱仪的使用及软件的操作。 (4)掌握微量进样器进样技术。 (5)了解程序升温气相色谱法的原理及基本特点。 【实验原理】 气相色谱法是以气体作为流动相(简称载气)的色谱法。 气相色谱法具有如下的特点: 1.高效能、高选择性可分离性质相似的多组分混合物,如同系物、同分异构体等;分离制备高纯物质,纯度可达99.99%。 2.灵敏度高可检出10-13-10-11g的物质; 3.分析速度快通常一个样品的分析可在几分钟到几十分钟内完成; 4.应用范围广气体样品、低沸点、易挥发或可转化为易挥发的液体或固体样品,不仅可分析有机物,也可以分析部分无机物。 气相色谱仪一般由气路系统、进 样系统、分离系统、检测记录系 统和温度控制系统(图中未显示) 五部分组成(见图1-8-1)。 1.气路系统包括气源(高压气 图1-8-1 气相色谱过程示意图瓶)、气体净化、气体流量控制等 1.高压钢瓶 2.减压阀 3.净化管
部分组成,其作用是为仪器提供纯洁、稳定的载气。常用的载气有氮气和氢气,也可用氦气、氩气或空气。 2.进样系统包括进样装置和气化室。其作用是将样品在进入色谱柱前迅速气化,并定量转入到色谱柱中。要想获得良好的分离结果,进样速度应极快,且样品应在气化室瞬间气化。液体样品一般都采用微量进样器,可根据进样量的不同选用不同体积的进样器。对气化室的要求是热容量要大,温度要足够高且无催化效应。 3.分离系统该部分由色谱柱组成,是色谱仪的心脏,其作用是分离样品。色谱柱分为填充柱和毛细管柱两种: (1)填充柱:由不锈钢或玻璃作为柱管,内填固定相制成,一般内径为2~4mm,长1~3m。形状有U型和螺旋型两种。 (2)毛细管柱又叫空心柱。毛细管材料可以是不锈钢、玻璃或石英。内径有0.53mm、0.32mm、0.25mm等几种规格,长度一般为10~30m。它的固定相可以直接涂布或通过化学交联键合在预先经过处理的管壁上。按照所用的色谱柱不同又可分为:填充柱色谱和毛细管柱色谱。 4.温度控制系统在气相色谱法中,温度直接影响到色谱柱的分离选择、检测器的灵敏度和稳定性。因此在仪器中主要是对色谱柱箱、气化室、检测器三处的温度进行控制。 5.检测和放大记录系统当样品经色谱柱分离后,各组分按保留时间不同随载气进入检测器,检测器将有关各组分含量的信息转化为易于测量的信号(一般为电信号),经过必要的放大传递给记录仪,最后得到该样品的色谱流出曲线。
液相色谱系统图谱相关知识
液相色谱系统图谱相关知识 点击次数:429 发布时间:2009-7-17 谱图的各种问题 液相色谱系统的许多问题都能在谱图上反映出来。其中有一些问题可以通过改变设备参数得到解决;而其他的问题必须通过修改操作程序来解决。对于色谱柱和流动相的正确选择是得到好的色谱图的关键。 A、峰拖尾 原因 解决方法 1、筛板阻塞 1、a、反冲色谱柱 b、更换进口筛板 c、更换色谱柱 2、色谱柱塌陷 2、填充色谱柱 3、干扰峰 3、a、使用更长的色谱柱 b、改变流动相或更换色谱柱 4、流动相PH选择错误 4、调整PH值。对于碱性化合物,低PH值更有利于得到对称峰 5、样品与填料表面的溶化点发生反应 图 5、a、加入离子对试剂或碱性挥发性修饰剂 b、更改色谱柱 B、峰前延 原因 解决方法
1、柱温低 1、升高柱温 2、样品溶剂选择不恰当 2、使用流动相作为样品溶剂 3、样品过载 3、降低样品含量 4、色谱柱损坏 4、见A1、A2 C、峰分叉 原因 解决方法 1、保护柱或分析柱污染 图 1、取下保护柱再进行分析。如果必要更换保护柱。如果分析柱阻塞,拆下来清洗。如果问题仍然存在,可能是柱子被强保留物质污染,运用适当的再生措施。如果问题仍然存在,入口可能被阻塞,更换筛板或更换色谱柱。 2、样品溶剂不溶于流动相 2、改变样品溶剂。如果可能采取流动相作为样品溶剂。 D、峰变形 原因 解决方法 1、样品过载 1、减少样品载量 E、早出的峰变形 原因 解决方法 1、样品溶剂选择不恰当 1、a、减少进样体积 b、运用低极性样品溶剂
F、早出的峰拖尾程度大于晚出的峰 原因 解决方法 1、柱外效应 1、a、调整系统连接(使用更短、内径更小的管路) b、使用小体积的流通池 G、K’增加时,脱尾更严重 原因 解决方法 1、二级保留效应,反相模式 1、a、加入三乙胺(或碱性样品) b、加入乙酸(或酸性样品) c、加入盐或缓冲剂(或离子化样品) d、更换一支柱子 2、二级保留效应,正相模式 2、a、加入三乙胺(或碱性样品) b、加入乙酸(或酸性样品) c、加入水(或多官能团化合物) d、试用另一种方法 3、二级保留效应,离子对 3、加入三乙胺(或碱性样品) H、酸性或碱性化合物的峰拖尾 原因 解决方法 1、缓冲不合适 1、a、使用浓度50-100mM的缓冲液
N2000色谱工作站标准操作规程及谱图处理
N2000色谱工作站标准操作规程 目的:制订N2000色谱工作站的标准操作规程。 适用范围: N2000色谱工作站。 责任: N2000色谱工作站操作人员按本规程操作,检验室主任监督本规程的执行。程序: 1. 配置。 1.1 硬件 CPU奔腾II 内存64MB并配备一个光驱的计算机,并加一个色谱工作站数据采集卡及打印机。 1.2 软件在中文Windows98环境下运行,由N2000软件组成。 2. 开机及系统设置。 2.1 打开显示器及计算机主机的开关,计算机自检完毕后,进入桌面屏幕。 2.2 双点击N2000在线色谱工作站,待工作站运行后,出现“打开通道1”或“打开通道2”画面,在1或2或两者旁边点击,打上一个“√”,再单击“OK”即可以进入N2000型在线色谱工作站。 2.3 出现“实验信息”方法,数据采集的按钮。 2.3.1 单击实验信息,可以用中文或英文输入实验标题、实验人姓名、实验单位、实验简介。而实验时间与实验方法,工作站会自动帮你填好。 2.3.2 单击“方法”,进入编辑实验方法,屏幕下方出现采集控制、积分、组分表谱图显示,报告编辑,仪器条件6个按钮。 2.3.2.1单击“采集控制”,在屏幕上可以输入采样结束时间,文件保存方式,如是自动方式,并在文件前缀的空白处输入一个有特征性的文字,便于以后色谱文件的管理,还可选择标样保存路径,样品保存路径,采样结束是否要自动积分及采样结束后是否自动打印报告,完毕后,单击“采用”按钮。
2.3.2.2单击“积分”,在屏幕上可以选择积分参量(面积或高度),积分方法(归一法内标法,外标法等。)在还可插入、删除修改分析参数,包括峰宽斜率、样品重量、漂移、最小面积、时间变参、锁定时间、停止时间,当修改这些参数后,再单击“OK”退出,最后单击“采用”,整个“积分”编辑完毕。 2.3.2.3单击“组分”,在屏幕上单击“谱图”按钮,在此对话框中选择一个谱图文件,单击打开按钮。打开目标谱图文件后,按下shift键,再单击所要计算的色谱锋,然后单击插入按钮,弹出一个对话窗口,在上面可以输入参数,包括组分名,范围及是否有内标峰,再点电击“确定”同理单击删除和修改按钮,可以删除或修改已输入的组分信息。最后单击“OK”按钮,另外,还可以点击全选按钮,工作站即自动将所有的谱峰信息列在组分表内,可输入组分名,待输完后,点击采用按钮。接着曲线校正,单击校正按钮,出现另一对话框,在此对话框中,单击组分含量,又在此对话框中,输入含量(样品与标准),单击OK退出,再点击加入标样按钮,在此对话框中,选择一个标样图谱文件,点击打开按钮即可。同时还可以输入另一组组分含量,并加入标样,其操作与第一组组分含量输入相同,单击校正完毕按钮即完成。 2.3.2.4单击“谱图显示”在屏幕上输入时间显示范围与电压显示范围的最大及最小值,显示颜色,注释内容(一般是保留时间)。最后单击采用。 2.3.2.5单击“报告编辑”在此屏幕可以根据不同需要设计编辑报告菜单打印出来。 2.3.2.5.1单击谱图尺寸,可输入谱图需要的大小。 2.3.2.5.2单击实验信息,可选择实验人姓名、实验单位、实验日期、实验简介。 2.3.2.5.3单击页首/页脚,可在页首或页脚输入需要增加内容。 2.3.2.5.4单击报告内容,可选择积分方法、积分表、时间程序表、组分表、积分结果。 2.3.2.5.5单击谱图显示,可选择网格显示,基线显示,注释内容(包括洋名,峰高保留时间,含量,峰面积和无注释)另外,窗口左上角还有三个按钮可扶速选择预
综合热分析
寒假—综合热分析 物质加热后发生化学的或物理的变化时,会表现出吸热、放热等能量的转变,或重量、体积等的变化,不同的物质有不同的组成和结构,加热后有特定的热效应,当物质发生相变化时,就会在特定热效应中反应出来。因此,可以用对物质加热的方法进行相分析。 热重法 材料在加热过程中脱水、氧化、蒸发、升华或燃烧等都会发生重量的变化。 调节和控制加热速度,记录材料重量变化与时间或温度的关系、重量变化的大小,称为热重分析。 热差分析 用二种物质在一定的温度范围内加热,其中一种物质加热后不发生相变化,如果另一种物质加热过程也无相变化,则二种物质之问无热量差;如果其中有一种物质在加热过程中产生相变化,由于吸热或放热,会产生与另一种物质的热量差,即差热。量测产生差热时的温度和差热大小,可以定性或定量分析该物质。加热时无相变化的物质称为参比样 一、脱水 以各种不同状态存在于材料中的水.在加热后失水时要吸收热量,因此不同状态的水的脱除为吸热反应。材料结构不同,水的存在形态不同,则脱水吸热的温度也不同。脱水后,材料失重二脱水温度取决于水在物质中的结合力。 二、分解 加热后,物质由一种化合物变成二种以上的化合物称为分解,破坏了原来的结构,吸收热量成为破坏动能。分解温度和吸收的热量取决于晶格结合的牢固程度。 三、结晶 物质由无定形转变为晶态,即无序→有序,内能减少,放出热量。如果结晶破坏转变为非晶态,则为吸热反应。
硫酸盐对混凝土的侵蚀: 分为化学侵蚀与物理侵蚀。化学侵蚀主要是硫酸盐与水泥水化产物发生化学反应导致混凝土膨胀破坏。物理侵蚀是指硫酸盐结晶对混凝土产生的破坏,这种破坏来自于盐结晶后体积膨胀,其本身未与水泥的水化产物发生化学反应。硅酸盐水泥主要水化产物有水化硅酸钙、水化硅酸钙凝胶、氢氧化钙和水化铝酸钙。 硫酸盐侵蚀是一个复杂的物理化学过程,它是典型的膨胀性腐蚀。以硫酸钠为例,当硫酸根离子的浓度较低时,主要膨胀性产物为钙矾石当硫酸根的浓度很高时,还会生成另一种膨胀产物石膏。其反应如下: 3CaO·Al 2O 3 ·CaSO 4 ·18H 2 O+2CaSO 4 +14H 2 O → 3CaO·Al 2 O 3 ·3CaSO 4 ·32H 2 O(钙矾石) Na 2S0 4 ·10H 2 O+Ca(OH) 2 → CaSO 4 ·2H 2 O+2NaOH+8H 2 O(石膏) Biczok等认为,对硫酸钠侵蚀而言,当硫酸盐浓度比较小时(< 1000mg/L SO 4 2-)侵蚀产物以钙矾石为主,而在高浓度下(> 8000mg/L SO42-)以石膏为主,在1000 —8000mg/L SO 4 2-范围内,石膏和钙矾石都被观察到。 钙矾石(3CaO·Al 2O 3 ·3CaSO 4 ·32H 2 O)在87℃时失去6个结晶水,135℃时失去21~ 22 个结晶水,225℃时失去全部结晶水。石膏(CaSO 4·2H 2 O)在165.6℃转变为CaS04。 (1/2)H 2O,在233 . 7 ℃时转变为无水CaSO 4 ,也有文献报道是123℃和130℃。 因试验原材料、试验条件和仪器型号及参数设置等的不同,不同文献得出的结论也有所差异。 综合目前文献可知 钙钒石主要脱水温度区间是80—130℃ 石膏的主要脱水温度区间是130—150℃ 420-500℃区间的峰对应Ca(OH) 2 的分解 700-850℃区间的峰对应CaCO 3 分解 综合热分析曲线:
液相色谱中的谱图
[ m V ] [ m V ] 组分名保留时间(min) 峰高(mV) 峰面积(mV.sec) 浓度样品含量(%) 1 Unknown 4.60833 257.65 4409.59 0.0000 0.0000
[m V ] 组分名 保留时间(min) 峰高(mV) 峰面积(mV.sec) 浓 度 样品含量(%) 1 Unknown 6.67167 140.58 5693.75 0.0000 0.0000 [M in u te s ] [m V ] 组分名 保留时间(min) 峰高(mV) 峰面积(mV.sec) 浓 度 样品含量(%) 1 Unknown 3.19000 362.36 4302.9 2 0.0000 0.0000 2 Unknown 4.22000 5.40 47.35 0.0000 0.0000
[ m V ] [ m V ] 组分名保留时间(min) 峰高(mV) 峰面积(mV.sec) 浓度样品含量(%) 1 Unknown 6.92917 166.01 4469.95 0.0000 0.0000
[ m V ] 1 Unknown 3.93500 208.03 4337.48 0.0000 0.0000 2 Unknown 4.67000 6.80 109.69 0.0000 0.0000 [ m V ] 组分名保留时间(min) 峰高(mV) 峰面积(mV.sec) 浓度样品含量(%) 1 Unknown 4.50333 244.05 4441.79 0.0000 0.0000 2 Unknown 5.97750 3.34 51.14 0.0000 0.0000
高效液相色谱中异常峰分析完整版
高效液相色谱中异常峰 分析 HUA system office room 【HUA16H-TTMS2A-HUAS8Q8-HUAH1688】
异常峰分析 异常的色谱峰指的是色谱图中的无峰或出现负峰、宽峰、双峰、肩峰、峰形不对称等情况。异常峰是色谱实验工作中最棘手的问题。这些峰严重影响色谱分析的结果。色谱图中不可能有纯正的高斯对称峰 , 轻微的拖尾是正常的 , 这是由分离系统所决定的。在此仅对几种异常峰进行分析。 1.峰前或峰后有小峰的分析 产生原因大致分为以下几种情形 (1) 样品不纯。可改变不同的流动相和色谱柱 ,对样品进行分离比较 , 选择合适的分离条件。 (2) 分析柱或保护柱柱头塌陷。此情况较常见 ,可更换分析柱或保护柱后对峰形进行比较。柱头塌陷时往往所有的峰都会出现峰分裂。对色谱柱再生和清洗可以改善分离效果。 (3) 色谱柱柱容量下降。当长时间使用后 , 有一些强保留组分吸附在柱子中 , 不大的进样量往往就会出现峰分裂。用强洗脱能力的溶剂清洗色谱柱 , 或更换色谱柱可使问题得到改善。 (4) 样品溶剂与流动相不匹配或进样体积过大。当样品溶剂比流动相极性大时 , 有时即使进样体积很小 , 也容易出现峰变形和裂分现象。建议用流动相溶解样品。
(5) 流动相不恰当。此情况较罕见 , 有些样品在特定的色谱条件下可能存在结构的动态平衡 , 而出双峰 , 这种双峰是无法分离完全的 , 改变色谱条件尤其是p H 值会使峰形正常。 (6) 样品分解。不稳定的样品在色谱分离过程中变成其他物质而出现双峰。这时需改变样品处理方法或色谱分离条件。 2.负峰分析 在色谱分析中有时会出现负峰或倒峰 , 如图 3 中的大峰的左下就有一负峰。出现这种现象可能是由以下几种原因引起的 , 可针对不同情况进行排除 , 进而使问题得到解决。 (1) 流动相吸收本底值过高。此时可适当改变检测波长。 (2) 进样过程中进入空气。进行排气处理 , 直到基线平稳再进样。 (3) 样品组分的吸收低于流动相。可改变流动相或改变检测波长。 (4) 配制样品的溶液与流动相不一样。重新配制样品 , 用与流动相一样的溶剂配制或稀释样品。 3.前沿、拖尾峰分析 拖尾:1 干扰峰,优化条件分离;2 色谱柱塌陷,更换色谱柱; 3 流动相pH不合适,调节pH值;4 管路没有接好,存在较大的死体积,可以重新接一下。 前沿:1 溶剂选择不合适,选择合适的溶剂;2 样品过载,降低进样量; 3 柱温太低,升高柱温;4 色谱柱损坏,更换色谱柱;