能带结构,态密度

能带结构和态密度图的绘制及初步分析
前几天在QQ的群中和大家聊天的时候,发现大家对能带结构和态密度比较感兴趣,我做计算已经有一年半了,有一些经验,这里写出来供大家参考参考,希望能够对初学者有所帮助,另外写的这些内容也不可能全都正确,只希望通过表达出来和大家进行交流,共同提高。
MS这个软件的功能确实是比较强,但是也有一些地方不尽如人意的地方。(也可能是我对一些结果不会分析所致,有些暂时不能解决的问题在最后一部分提出,希望大家来研究
研究,看看有没有实现的可能性)。
能带结构、态密度和布居分析是很重要的内容,在
分析能带结构和态密度的时候,往往是先作图,然后分
析。
软件本身提供的作图功能并不是很强,比如说能带结构
(只能带只能做point图和line图),不美观不说,对于
每一个能带的走势也不好观察,感觉无从下手。所以我
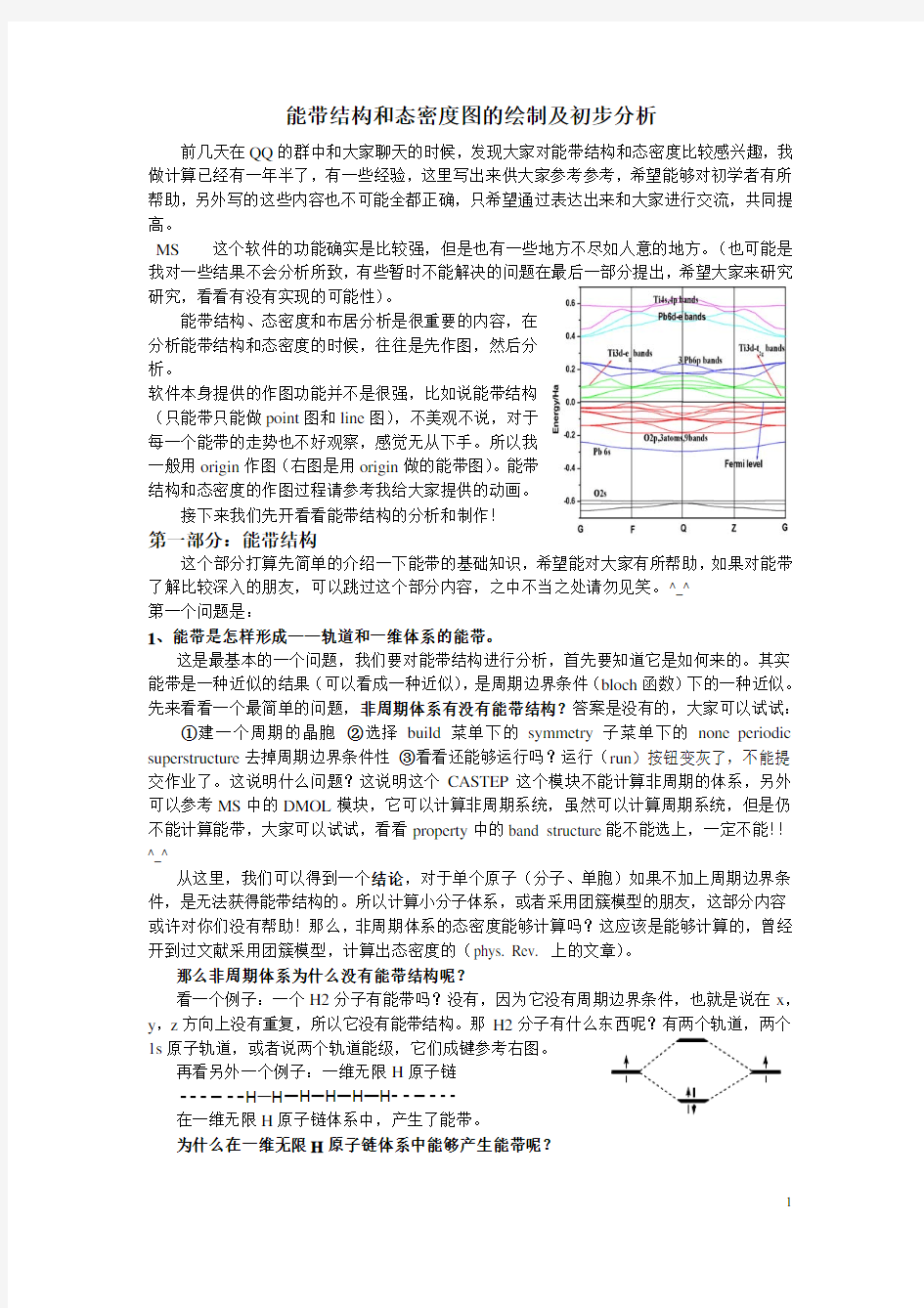
一般用origin作图(右图是用origin做的能带图)。能带
结构和态密度的作图过程请参考我给大家提供的动画。
接下来我们先开看看能带结构的分析和制作!
第一部分:能带结构
这个部分打算先简单的介绍一下能带的基础知识,希望能对大家有所帮助,如果对能带了解比较深入的朋友,可以跳过这个部分内容,之中不当之处请勿见笑。^_^
第一个问题是:
1、能带是怎样形成——轨道和一维体系的能带。
这是最基本的一个问题,我们要对能带结构进行分析,首先要知道它是如何来的。其实能带是一种近似的结果(可以看成一种近似),是周期边界条件(bloch函数)下的一种近似。先来看看一个最简单的问题,非周期体系有没有能带结构?答案是没有的,大家可以试试:
①建一个周期的晶胞②选择build菜单下的symmetry子菜单下的none periodic superstructure去掉周期边界条件性③看看还能够运行吗?运行(run)按钮变灰了,不能提交作业了。这说明什么问题?这说明这个CASTEP这个模块不能计算非周期的体系,另外可以参考MS中的DMOL模块,它可以计算非周期系统,虽然可以计算周期系统,但是仍不能计算能带,大家可以试试,看看property中的band structure能不能选上,一定不能!!^_^
从这里,我们可以得到一个结论,对于单个原子(分子、单胞)如果不加上周期边界条件,是无法获得能带结构的。所以计算小分子体系,或者采用团簇模型的朋友,这部分内容或许对你们没有帮助!那么,非周期体系的态密度能够计算吗?这应该是能够计算的,曾经开到过文献采用团簇模型,计算出态密度的(phys. Rev. 上的文章)。
那么非周期体系为什么没有能带结构呢?
看一个例子:一个H2分子有能带吗?没有,因为它没有周期边界条件,也就是说在x,y,z方向上没有重复,所以它没有能带结构。那H2分子有什么东西呢?有两个轨道,两个
1s原子轨道,或者说两个轨道能级,它们成键参考右图。
再看另外一个例子:一维无限H原子链
H H H H H H
在一维无限H原子链体系中,产生了能带。
为什么在一维无限H原子链体系中能够产生能带呢?
因为,每一个H 原子有一个1s 轨道,由于在X 轴方向(H 原子周期排列的方向)引入周期边界条件,所以这个体系有无数(阿佛加得罗)个H1s 的轨道能级,这些具有相同能量的能级轨道处于简并的状态。如果两个相邻的H 原子之间距离较大,不能够成键,那么这无数个简并的能级将排成一条水平的直线,这条直线很长,无法画下来,那么我们只有压缩它,将他压缩到一个区间([0,a π
]),这样每一个能级用一个点表示,由于点较多,看起
来好像形成了一条线,这样能带就形成了。
如果用函数的语言来描述,周期排列我们采用bloch 函数表示,我们解这些函数,就得到了一些k 矢量,在一维体系中k 矢量表示平移操作,k 的取值见下图,由于H1s 轨道能级有无数个,所以k 平移操作矢量就有无数个(注意k 是量
子化的),所以将它们压缩到[0,a π
]这个区间,就成了能带
结构中的横坐标,另外这个矢量也可以指向-X 方向,所以在[-
a π,0]这个区间能带的图像和[0,a π
]对称。
当H 原子之间的距离逐渐接近,它们的原子轨道要进行组合,形成一个成键分子轨道和一个反键分子轨道,那么原来能带是一条水平的直线,现在就要开始发生弯曲了(两个分子轨道能量不一样,导致能带发生弯曲),所以[0,a π
]这个区间,能带开始有带宽(散度),
随着H 原子的距离逐渐接近,可以预料,成键分子轨道和反键分子轨道的分裂越大,能带的带宽(最高能级-最低能级)越大。所以,相邻轨道之间的重叠越大,成键程度越大,带宽就越大。
另外值得一提的是,k 矢量还可以表示节点的数目。当k =0的时候表示什么呢?表示节点数为0,没有节点,所以k =0表示的H1s 轨道组合(组成分子轨道的原子轨道都带+号)具有最大的成键,能量最低。随着k 值的增大,节点数逐渐增多,体系的能量上升,最后k =a π
时,H1s 轨道组合成的分子轨道能量最高(原子轨道为+-+-……交替)。所以H 原子链的能带结构是一条向上弯曲的曲线(能带),k =0能量最低,k =a π
时能量最高。
这里要特别注意,并不是k =0的时候节点数都为0,比如(+-,+-,+-,……)这样的p 轨道,如果它们沿着X 轴周期排列,那么k =0的时候将具有最大的节点数,这时候形成的分子轨道将是能量最大的,随着k 的逐渐增加,节点数逐渐减小,所以这时候能带将向下伸展,这与H 原子链的情况刚好相反。
大家都知道,这样的H 原子链是不可能稳定的,最后都要变成H2分子,能带要消失,这是一个什么样的过程呢?在这个一维周期体系中轨道能级的数目假设为N (无限大),那么这个体系的电子数是多少呢?答案是N ,那么这些电子在这个能带上是如何分布的呢?当然是按照能量从低到高的顺序来填充的了,这样由于每一个轨道能够容纳2个电子,而这个能带只有较低能量的部分被填充(能带半充满),所以这时候要产生畸变(Peierls 畸变,即固体物理中的姜-泰勒效应),H 原子之间要产生相对振动(虚模,能量不稳定),以降低体系的能量,这样,H2分子就形成了,而能带也由于H2原子的形成破坏了周期条件,当这些H2分子不再沿着X 轴方向形成周期排列的时候,能带也就消失了(变成非周期体系)。
结论:一个原子的一个原子轨道在一维周期条件下将产生一条能带,能带的带宽取决于这些原子轨道的在周期方向上的成键强度,强度越大,带宽越大,成键越弱,带宽越小,如
果周期方向上没有成键,能带将是一条直线。另外能带是向上伸展还是向下伸展取决于原子轨道的特性,或者说是体系的拓补性质。
接下来我们看看,布里渊区里面的高对称点(G ,X ,F ,M 等)是怎么来的。
2、 布里渊区的高对称点
前面讲了一维周期条件下的布里渊区的能带是一条线,如果加上二维(X ,Y )的周期边界条件,这些能带又会变成什么样呢?答案是一个面,由原来的线组成一个面。因为在一维情况下我们用一条能带来表示k 矢量(对称操作)和能级的关系,可以用E (k )来表示,这构成第一布里渊区(即k 的取值范围[0,a π
])。对于二维周期体系,我们需要两个平移矢
量k x 和k y ,所以能带可以用E (k x ,k y )来表示,当k x =0时,变成E (0,k y ),得到一条能带(线,y 方向上与一维周期情况的能带类似);当k y =0时,变成E (k x ,0),得到一条能带(线)。由于k x 和k y 是矢量,它们可以组合成另外一个矢
量,这个矢量不是沿着X 轴,也不是沿着Y 轴,实际上沿着该
矢量仍是能够得到一个能带(线)的,这样的矢量有很多,所
有的这些能带(线)将构成一个面。如果我们在做能带结构图
的时候,将能带结构按照二维的面画出来是很困难的,而三维
的情况更加困难,因为对称操作有很多,k 矢量的取值有很多,
所以一个可行的办法就是让k 的取值沿着一定的路径走,最后
回到起点。如右图(二维情况)。
这样,我们只要选择一些较高的对称点,就可以确定这个路径。比如二维的布里渊区是一个面,这个面上每一点与原点(G 点或Γ点)的连线都构成一个k 矢量,有一个k 矢量就有一个能级对应(E (k ))。所以,二维的能带结构是这个布里渊区上的一个平面(面积),如上图,按照Γ——>X ——>M ——>Γ这个路径走,就可以得到一个可以大致反映布里渊区上的能带平面的一个近似图,这就是二维的能带结构。具体的能带图的展开见下图。
三维的能带展开见下图:
前面讲了一维H 原子链的能带结构,提到这个体系具有一个能带,并且是向上弯曲的一个能带。这个能带是这样来的,H 原子链里面的基本单位(单体,单胞)是一个H 原子,每一个原子有一个原子轨道,即每一个基本单位有一个能级轨道,加上周期条件以后,这一系列轨道能就变为一个能带了。假如,现在以两个H 原子作为一个基本单位呢?能带结构又是如何的呢?这就是能带重叠的问题
3、 能带的折叠
如上图,将2个氢原子作为一个基本单位,这时候能带结构是什么样子的呢?很容易理解,根据前面的知识,单胞的原子轨道的数目决定了能带的数目,所以这样划分体系将有将有两个能带。
但是这个体系与前面1个H 原子周期链是一样的,只不过人为地进行了划分,能带结构就变了吗?是的,能带确实变了,那么能带将怎样变化呢?下图分别是1个H 原子为单胞和2个H 原子为单胞的能带结构。
第一个图可以清楚的看到,能带底部是成键的,能带的顶部是反键的,中间是非键的(成键与反键相当),能带向上伸展(弯曲)。在第二个图,原来的中部的非键轨道分别变成了两个能带的反键和成键轨道(相同颜色表示可以重叠成键,不同颜色表示中间有一个节点)。实际上,这两个图是有关系的,能够反映相同的内容,首先,布里渊区从原来的[0,
a π]变为[0,a 2π
],这也是可以理解的,原来的能带长度要变成原来的一半(因为周期方向上的单胞数减少一半,原来有N 个单胞,以2个H 原子为一个单胞后,单胞数变为一半,所以布里
渊区要减半)。其次,原来的能带在[0,a π
]展开,现在由于布里渊区减半,能带不能在[0,a 2π
]
这个小区间画出来,所以能带结构将产生折叠,由原来的一个能带变为2个能带,并且是以k =a 2π
为对称轴,将原来[a 2π,a π
]区间的能带折叠过去,所以就得到了2H 原子为单胞的
能带结构。如果我们将单胞取3个原子,或者取4个原子,体系的能带将如何变化?体系的能带分别变为3条和4条能带,并且是2次或3次折叠原来的能带。
另外,上面的例子我们也可以从另外一个角度考虑问题,即2个H 原子组成一个H2分子,形成成键分子轨道和反键分子轨道,把这两个分子轨道看成一个“原子”的两个“原子轨道”,给这个“原子”加上周期条件以形成能带,这
样也得到2个能带。这种想法可以进行适当的扩充,
比如我们研究一个晶体,首先我们可以把单胞找出来,
然后将单胞的轨道能级画出来,考虑它们之间的成键,
最后得到一组分子轨道,以这组分子轨道为基础,给
它们加上周期条件,形成能带。这样的想法什么时候
有用呢?在组合成分子轨道以后,这些分子轨道并不是
不变的,有可能和相邻晶胞的分子轨道再次组合,这
种情况在过渡金属氧化物中非常普遍,所以在考虑成
键的时候除了单胞内成键情况以外,还需要考虑晶胞间分子轨道的组合。
能带的折叠实际上是从不同的角度考虑问题而已,其实能带结构的本质还是一样的。如果我们理解了这点,那么我们在分析能带的时候,就不会笨到建立一个很大的超胞去研究其能带结构。所以我们要研究一个体系的能带结构时候,尽量选最小的单胞,以减少能带的数目。
不幸的是,有时候我们往往要计算掺杂的情况而不得不建立一个很大的单胞(超胞),而掺杂的比例又与超胞的大小有关。如果要做1/8掺杂,那么起码要将8个晶胞做成一个单胞(超胞),并将其中一个原子替换掉,从而实现掺杂。这样能带的数目将变为原来的8倍,这样我们计算出来的能带将是一团曲线交缠在一起,再也无法分开。
大家可以试试建立一个超胞,然后计算能带,看看能带结构如何!
所以能带结构的分析对于比较大的单胞(超胞)或者具有较多原子轨道的单胞将变得很麻烦。而且MS 中画出的能带结构很难看,如果我们要用origin 作图的画,又要反复的copy +paste 数据,所以建议在计算具有较多原子轨道(分子轨道)的体系时,尽量不用能带结构分析问题(虽然它很有用^_^)。用态密度和PDOS 就好多了。
另外,简单的提一下Fermi 能级的概念,有很多书都将最高占据分子轨道(HOMO )的顶部作为Fermi 能级,所以我也采用这种观点,不知道大家是怎么理解的?
下面我简单讲讲我做的体系的能带结构,具体做法参加我提供的动画演示。
4、3维体系的能带——PbTiO3的能带结构
要分析能带,首先要知道每个能带是属于那个轨道的,所以首先要画出单胞的轨道能级图,下图是钛酸铅的轨道能级示意图,不是很准确(因为有一些轨道成键没有连线,仅给价带和导带部分连线了)。
在轨道能级图里面先将各个原子的轨道能级标出来(横轴的能量是参考了PDOS 加上的,没有也没有关系^_^)。接着考虑成键,主要是中心Ti 原子和O 原子成键(模型参考下图),有O 原子的2p 轨道组成价带,2s 轨道的能量最低Pb 的4s 轨道比O2p 轨道能量低,比O2s 轨道高。另外Ti 原子处于TiO6八面体中心,所以3d 能级要分裂成3下(3t 2g 态,3d xy ,3d xz ,3d
yz 单胞1单胞2
二次组合
轨道)和2上(e g 态,即3d x 2-y 2,3d z 2轨道),这些轨道构成导带。Pb 的6d ,Ti 的4s ,4p 轨道在Ti3d 轨道上面。
有了这些信息我们就可以绘制能带,并可以用不同的颜色表示不同轨道形成的能带,具体的绘制过程见动画。
接下来看看能带能够说明什么问题。
第一点:O2s 能带有带宽,表明其参与成键(参与成
键的有Ti 的3d z 2轨道,Ti4s ,4p z 轨道,还有Pb 原子轨道
的部分贡献,能带结构看不出来,但是态密度可以看出
来)。
第二点:Pb 的6s 能带带宽比较大,说明Pb6s 轨道参
与成键,由于Pb 与Ti 原子很远(3.45A )所以只能是与
O2p 轨道成键(能量相近,并且有带宽,从后面态密度分
析也可以得到同样的结论)
第三点:O2p 能带组成价带,体系中有3个O 原子,
共有9条O2p 能带,其中每3个是一组(G 点是3重简并
的)。
第四点:Ti3d 的5个能级分裂成2组,3下2上,组成导带。
第五点:Pb 的6p 和6d 能带在Ti 的3的能带上面,而且Pb 的6的也要分裂,还有一些能带图中未能表示出来,另外Ti4s ,4p 能带在最上面,且带宽比较大,说明它们参与了与O2p 轨道成键。
当然,能带结构还能给出很多信息,比如带隙等,由于水平有限,所以只能看出一些简单的内容!希望对能带结构比较了解的朋友能够把这部分内容更加完善,大家共同探讨和学习!
下面我主要讲一下态密度的一些基本内容和初步的分析以及态密度的绘制。 第二部分:态密度
首先来看一下态密度的意义,前面提到当单胞(超胞)很大的时候,含有的原子轨道(分子轨道)的数目就很多,采用能带分析的时候,必须处理很多能带。在单个分子中,我们可以挑选出一个或一小组能决定分子的几何构型,反应性能等的轨道作为前线轨道或者价轨道,但是,在多到几乎无法可数的晶体轨道(组成能带的能级)中,就没有可能找到某一个能级来决定一种几何构型或反应性能。
举个例子来说,如果做表面成键或者吸附,首先要建立按照一定的晶面(如[110]面)取向的表面构型,然后就是考虑吸附上去的原子对这个表面的影响情况,如果计算能带,当
表面吸附的覆盖度很小时,这时候需要建的表面就很大,能带的数目自然就增加很大数目,而影响到表面结构和性质的只要那么几条能带(吸附原子和被吸附原子的能带),如何从这么多的能带中找到它们呢,这显然时吃力不讨好的事情(我觉得这时候用能带来解释不现实),那么我们怎么考虑吸附质吸附上去以后,表面的结构和性质呢。我个人认为,只能靠计算得到的态密度来分析了,特别时偏态密度(态密度投影或称PDOS),比较被吸附原子和吸附原子在吸附前或者吸附后的偏态密度应该能够很好的说明情况,而且如果能够加上布居分析,局部的结构和性质应该可以弄清楚。比如做TiO2[110]面吸附乙酸,我们可以计算TiO2晶体的态密度,计算TiO2[110],和吸附后的TiO2[110]的态密度,这样,比较前两者,就知道从晶体变到表面以后,吸附原子的成键变化情况,比较后两者,又可以知道吸附前和吸附后,表面的结构怎么变化(成键等),性质发生什么变化。所以我个人认为,在计算表面的结构性质的时候,应该更注意局部变化的情况。
那么态密度的物理意义是什么呢?
1、态密度的意义
态密度的物理意义应该是比较简单的。大家都知道,能带结构的纵坐标是能量,假如在这个坐标轴上取ΔE这一个很小能量范围(比如0.005000~0.005001eV这个范围),那么这个能量间隔范围内又多少个能级,或者说又多少个原子轨道(分子轨道)呢?别忘了,能带是怎么来的,是无数个能级“压缩”而成的,而且能带是量子化的,所以在这个能量范围必然又一定数量的能级(轨道)存在。所以从这里开始,不再谈论单个的原子轨道或能级了,因为这没有什么意义。而是我们将这一能量范围内的能级作为一组来考虑,所以态密度的概念就得出来了,即E+dE这个能量范围内的能级数,如果E+dE这个能量范围内轨道(能级数)越多越密集,态的密度越大,所以说态密度的概念是很贴切的。
而且要注意态密度是根据能带得来的,两者有一定的对应关系,比如在E+dE这个能量间隔没有能带,那么有态密度吗?这个答案是很显然的,没有能带,没有能级,哪能有态密度呢?所以这时候态密度图在这个能量范围内将是0。从这里可以得到一个结论,能带按照纵坐标轴投影过去,就得到态密度,所以态密度为0的地方,能带图上一定没有能带经过。
在态密度中还有一个问题就是,有的峰很高,有的峰很低,这是为什么呢?为什么会产生这种情况?这个很容易理解,比如一条水平的能带经过E+dE这个区间,那么这个区间的能级数是不是最大的呢?当然是,这个能带的所有能级都处于这个能量范围内,所以态的数目最大,在态密度图中表现为一个很尖锐的峰。如果能带的带宽较大,这说明态密度图中它很平缓,并且跨过的能量区间越大。一句话,能带越平,态密度峰越尖锐,能带越宽,态密度越平缓,当然离域性越强了。
这里有两个问题:第一个问题:能带越平,是否成键越弱,或者不成键?即态密度峰越尖锐,成键越弱或不成键呢?第二个问题:能带带宽越大,即态密度峰跨度越大越平缓,成键是否越强?
这两个问题,我查了一些书,但是没有答案,我个人的理解如下。对于第一个问题,能带越平,不表示不成键或成键弱,为什么呢,比如两个原子轨道形成很强的σ键,那么根据分子轨道理论,将形成一个成键分子轨道和一个反键分子轨道,成键越强,两者分离越大,形成这两个分子轨道以后,在这个基础上在形成能带结构,也就是在X,Y,Z方向上加上周期条件,假如在这3个周期方向单胞之间的距离比较远,那么两个分子轨道不能产生2次组合,所以得到的能带只能是两条水平直线,所以我个人认为,能带平缓,或者态密度峰尖锐不能说明这个轨道不成键或者成键比较弱。
第二个问题,我认为是这样的,带宽越大,成键越强,因为带宽越大,态密度峰跨度越大,离域性越强,虽然不一定跟那个原子成键,但是其上面的电子离域了,肯定要成键,离域越强,成键越强。
上面两个问题是我自己思考的答案,当然不一定对,希望大家能够探讨探讨!
接下来,想说说用态密度如何分析成键的问题。
2、 态密度与成键
下面的图是能级,能带以及态密度的关系,两个原子的原子轨道组合以后,得到两个分子轨道,在周期边界条件下,这两个分子轨道形成两个能带,根据能带的宽度和斜率,可以得到态密度的近似图。
上图右边的态密度图表示的是总的态密度,不是PDOS ,总态密度与PDOS 的关系如何呢?要解决这个问题,还得回到能带结构,在能带结构中,同一个能量E 画一条水平横线,与能带相交,交点有可以能很多个,并且有可能和几条能带相交,与这条直线相交的若干个点对应于态密度图上能量为E 的一个点,也就是说这个点包含了若干个能带的贡献,所以得到总的态密度图,如果非要区分这些交点中属于那些轨道,则总态密度就得按照一定的原则子划分到不同原子上,就得到了PDOS 。总之,总态密度是所有能带的贡献,而如果要对这些贡献划分为某一个原子的贡献,则产生PDOS 。
再回到上面的图,上图中左边为分子轨道,这个分子轨道可以用波函数2211χχψc c +=来描述,其中1χ和2χ表示两个原子轨道,那么它们组成的每一个分子轨道应该都有两个原子轨道的贡献。但是对于成键轨道,主要以电负性大的原子的原子轨道为主(比如O2p 轨道),即成键分子轨道主要是电负性大的原子的原子轨道,当中掺入了部分另外一个原子轨道的贡献;而反键分子轨道主要是由电负性小的原子的原子轨道构成,并且有电负性大的原子轨道的贡献。也就是所这两个能带不是某一个原子轨道独自形成的的,每一个能带都是一个原子轨道为基础,掺入了其他轨道的贡献,当然这个性质在能带结构中是无法反映的,你不知道其他原子的原子轨道对某一个能带的贡献。那么,什么能够反映这个性质呢?答案是态密度,更准确一些讲是PDOS 。下面看看如何用PDOS 表示这个性质!
如下图:
这个图有两部分态密度组成,能量较低的部分态密度相当于低能的能带产生,高能部分的态密度是高能能带产生(参考上面的轨道-能带-态密度图)。这样低能部分的态密度对应于成键分子轨道,它由两个部分组成(红色部分+黑色部分=总态密度),其中面积大的部分态密度是由电负性大的原子的原子轨道产生,而面积小的部分是另外一个原子轨道对这个分子轨道的贡献(对低能能带的贡献),而对于高能部分的态密度刚好相反,所以根据PDOS可以判断成键的情况。也就是说态密度发生“共振”是成键的一个明显标志。
对于态密度,另外还有一些东西要注意的:
第一:对态密度曲线的积分等于电子数,比如体系由10个电子,10个电子肯定是按照能量从低到高的顺序排列,那么对态密度进行积分,当电子数达到10的时候,这个地方就是fermi能级(积分曲线可以用origin做)。
第二:偏态密度积分至fermi能级得到某一个原子某一个轨道的电子填充的数目。
第三:如果成键作用加强,那么成键分子轨道要下移,反键分子轨道要上移,导致态密度要发生移动,一个向下移动,一个向上移动,而能带则变宽。
大家如果要验证态密度的移动,可以计算一些亚晶格的态密度,然后组合,看看移动的方向。比如钛酸铅,去掉Ti原子和Pb原子,得到O3的亚晶格,计算它的态密度;去掉O3和Ti计算Pb原子的亚晶格,最后用同样方法计算Ti原子的亚晶格的态密度。根据算出的结果和PbTiO3的态密度比较,会发现很明显的特征,起码态密度的移动很明显,而且轨道要分裂(如Ti分裂成3下2上结构),等等!
从上面的讨论和分析可以清楚的知道,PDOS具有很大的用处,不但可以分析成键,也可以分析电子在何处(对不同PDOS积分)!
下面看看我计算的钛酸铅的态密度:
上面的左图是Ti-O原子之间的成键分析,可以发现O2p轨道和Ti3d轨道有明显的“共振”,另外Ti4p在O2p有一部分贡献;Ti4s,4p,3d对O2s都有态密度贡献(考虑到对称性匹配,它们应该成σ键)
上面的右图可以发现,Pb的6s和O2p有态密度共振,也成键;另外Pb6d和Pb6d在O2p态密度处有明显的峰(有贡献),所以O2p与Pb6s,6d也是成键的。
虽然,态密度能够分析成键的情况,并且能够积分出电子的数目从而确定电子在原子轨道上的分布情况。但是对于成键的强弱很难以定量说明,也就是说,成键强,到底有多强,成键弱,到底有多弱这样的问题用态密度比较难以回答。一个办法就是积分PDOS的贡献峰,但是,这样做也不见得可行,比如大家看看上面O2p的贡献峰,O2p在Pb6s的贡献峰积分,就可以知道它与Pb6s轨道成键时对Pb6s轨道的贡献。但是由于Ti3d和Pb6p态密度
峰重叠了,O2p对这两个轨道的贡献峰是一个总的贡献,区分不开对Ti3d和Pb6p的贡献。
态密度对成键的定量化描述不是很强,所以我们往往要进行布居分析,原子布居和键布居,根据布居的数值来判定成键的强弱。
态密度的基本内容就这些,至于怎么作图,可以用origin做,大家看看我提供的动画,动画中有部分内容是积分曲线的制作。
第三:MS中Castep和Dmol模块的一些问题
最后,想讲使用MS中的一些问题提出来,由于我一直没有解决,在这里提出,大家看看能不能实现。
第一个问题就是MS中能不能做晶体轨道重叠布居图(COOP图)?
前面提到用DOS和PDOS 分析成键,对于其强弱的变化比较难以描述,所以要借助布居分析,而晶体轨道重叠布居图,就是重叠布居权重的态密度。我们还是那1维H原子链来说明问题,如下图:
在这个体系中,能带下面是成键的,中间是非键的,上部是反键的。当电子填充在能带下部的时候,填充越多成键越强,当下半部分能带电子填满以后,在往上填充,电子就会跑到反键轨道上,使得成键逐渐减弱。COOP图能够很直观的反映出成键的情况,是一个分析成键情况的有力工具,可是MS中好像做不出来,但是ADF有这个功能,所以在这个地方提一下,大家看看MS中有没有实现的可能性。
第二个问题就是重叠布居的问题。重叠布居是键强弱的一个度量,重叠布居本身是有很多轨道贡献的叠加,这里提出这个问题是MS中能够把没有原子轨道对重叠布居的贡献烈出来吗?
另外还有一些问题,以后慢慢在论坛或者群里面在问好了。^_^
另外,强烈推荐一本书,上面的一些观点都是从这本书中找到更为详细的解释。
书名:固体与表面
作者:[美国] R.霍夫曼著
郭洪煪李静译
王作新郑冲校
出版社:北京工业出版社
我的联系方式:carlon@https://www.360docs.net/doc/8d8901031.html,或carlon2000@https://www.360docs.net/doc/8d8901031.html,
界面缺陷态密度与衬底电阻率取值对硅异质结光伏电池性能的影响
界面缺陷态密度与衬底电阻率取值 对硅异质结光伏电池性能的影响? 周骏1,2, 邸明东2, 孙铁囤3,孙永堂2,汪昊2 (1. 宁波大学理学院光学与光电子技术研究所, 浙江 宁波315211) (2. 江苏大学机械工程学院光信息科学与技术系, 江苏 镇江 212013) (3. 常州亿晶光电科技有限公司, 江苏 常州 213223) 在不同的异质结前界面缺陷态密度(D it1)和异质结背界面缺陷态密度(D it2)条件下,对P 型单 晶硅(c-Si(p))为衬底的硅异质结太阳能电池(TCO / a–Si: H (n +) / c–Si (p) / a–Si: H (p +) / TCO ) 的衬底电阻率R 与电池性能的关系进行数值研究。结果表明:衬底电阻率R 的取值不仅决定于异 质结前界面缺陷态,也与异质结背界面缺陷态有关,即前界面缺陷态密度D it1决定衬底电阻率的 最优值R op ,且R op 随着D it1的增大而增大; R>R op 时, 背界面缺陷态密度D it2对衬底电阻率的可取值 范围具有较大影响,D it2越大可取衬底电阻率的范围越小。 关键词:SHJ 太阳能电池;c–Si (p)衬底电阻率;c–Si (p)/(a–Si: H )界面缺陷;AFORS_HET PACC: 7340L, 8630J, 6185 1 引 言 对于以c-Si(p)为衬底的硅异质结(SHJ)太阳能电池,异质结界面特性对电 池的性能有显著影响[1-2],如衬底电阻率与异质结界面c–Si 耗尽区厚度的关系, 以及由此引起的硅异质结太阳能电池性能的变化[3]等。然而,对于c-Si(p)衬底 电阻率与硅异质结太阳能电池性能的关系,目前研究的还不够深入,长期以来都 是将R =1.0Ω为衬底的最佳电阻率,而将R =1.0~25.0cm 作Ωcm 视为可用的衬底 电阻率[4-5]。最近,文献[6]研究不同前异质结界面缺陷态密度情况下衬底电阻率 与电池性能的关系,指出衬底电阻率最优值R op 的取值将随着前界面缺陷态密度 D it1的降低而减少,突破了人们一直以来认为R =1.0Ωcm 底的最佳电阻率的 观点。但是,文献[6]设计的电池结构有一些缺点,如采用的铝背面场要在高于 800c 的温度下制备,对SHJ 太阳能电池的能量转换效率的提高产生一定限制。此 外,文献[6]对衬底电阻率与背异质结界面缺陷态密度的关系和对电池性能的影 响没有研究。实际上,氢化非晶硅(a–Si:H )和氢化微晶硅(μc-Si:H )因其低温 为衬0 ?国家自然科学基金(批准号:60977048)资助课题,浙江省“钱江人才”项目(2007R10015)、宁波市重点实验 室基金项目(2007A22006),江苏大学与常州亿晶光电科技有限公司联合研发项目和宁波大学王宽成幸福基 金资助课题。 通讯作者:周骏(1958-),男,教授,安徽马鞍山人,主要从事光电子功能材料与器件制备研究。 E-mail :ejzhou@https://www.360docs.net/doc/8d8901031.html,
标准正态分布的密度函数样本
幻灯片1 正态分布 第二章 第七节 一、标准正态分布的密度函数 二、标准正态分布的概率计算 三、一般正态分布的密度函数 四、正态分布的概率计算幻灯片2 正态分布的重要性正态分布是概率论中最重要的分布, 这能够由 以下情形加以说明: ⑴ 正态分布是自然界及工程技术中最常见的分布之一, 大量的随机现象都是服从或近似服从正态分布的.能够证明, 如果一个随机指标受到诸多因素的影响, 但其中任何一个因素都不起决定性作用, 则该随机指标一定服从或近似服从正态分布. 这些性质是其它 ⑵ 正态分布有许多良好的性质, 许多分布所不具备的. ⑶ 正态分布能够作为许多分布的近似分布.幻灯片3 -标准正态分布下面我们介绍一种最重要的正态分布 一、标准正态分布的密度函数若连续型随机变量X 的密度函数为定义 则称X 服从标准正态分布,
记为标准正态分布是一种特别重要的它的密度函数经常被使用, 分布。 幻灯片4 密度函数的验证 则有 ( 2) 根据反常积分的运算有能够推出 幻灯片5 标准正态分布的密度函数的性质若随机变量 , X 的密度函数为 则密度函数的性质为: 的图像称为标准正态( 高斯) 曲线幻灯片6 随机变量 由于 由图像可知, 阴影面积为概率值。对同一长度的区间 , 若这区间越靠近 其对应的曲边梯形面积越大。标准正态分布的分布规律时”中间多, 两头少” . 幻灯片7 二、标准正态分布的概率计算 1、分布函数分布函数为幻灯片8 2、标准正态分布表书末附有标准正态分布函数数值表, 有了它, 能够解决标准正态分布的概率计算.表中给的是x > 0时,①(x)的值. 幻灯片9 如果由公式得令则幻灯片10
初学VASP中电子态密度计算设置参考
初学VASP中电子态密度计算基本设置参考主要分成三步:一、结构优化;二、静态自洽计算;三、非自洽计算以Al-FCC为例子 第一步结构优化 输入文件(INCAR, POTCAR, POSCAR, KPOINT) INCAR文件 System=Al ISTART=0 ISMEAR=1 SIGMA=0.2 ISPIN=2 GGA=91; VOSKOWN=1; EDIFF=0.1E-05; EDIFFG=-0.01 IBRION=2 NSW=50 ISIF=2 (OR 3) NPAR=10 POTCAR 文件直接在势库中拷贝 POSCAR文件 Al 4.05 1.0 0.0 0.0 0.0 1.0 0.0
0.0 0.0 1.0 4 Direct 0.0 0.0 0.0 0.5 0.5 0.0 0.5 0.0 0.5 0.0 0.5 0.5 KPOINT 文件 Automatic generation Mohkorst Pack 15 15 15 0.0 0.0 0.0 第二步静态自洽计算 INCAR:PREC = Medium,ISTART = 0,ICHARG = 2,ISMEAR = -5输入文件(INCAR, POTCAR, POSCAR, KPOINT) INCAR文件 System=Al ISTART=0 ISMEAR=1 SIGMA=0.2 ISPIN=2
GGA=91; VOSKOWN=1; EDIFF=0.1E-05; EDIFFG=-0.01 #IBRION=2 #NSW=50 #ISIF=2 (OR 3) NPAR=10 POTCAR 文件直接在势库中拷贝 POSCAR文件 Al 4.05 1.0 0.0 0.0 0.0 1.0 0.0 0.0 0.0 1.0 4 Selective Dynamic Direct 0.0 0.0 0.0 T T T 0.5 0.5 0.0 T T T 0.5 0.0 0.5 T T T 0.0 0.5 0.5 T T T KPOINT 文件 Automatic generation
半导体物理与器件公式以及全参数
半导体物理与器件公式以及参数 KT =0.0259ev N c =2.8?1019N v =1.04?1019 SI 材料的禁带宽度为:1.12ev. 硅材料的n i =1.5?1010 Ge 材料的n i =2.4?1013 GaAs 材料的n i =1.8?106 介电弛豫时间函数:瞬间给半导体某一表面增加某种载流子,最终达到电中性的时间,ρ(t )=ρ(0)e ?(t /τd ),其中τd =?σ,最终通过证明这个时间与普通载流子的寿命时间相比十分的短暂,由此就可以证明准电中性的条件。 E F 热平衡状态下半导体的费米能级,E Fi 本征半导体的费米能级,重新定义的E Fn 是存在过剩载流子时的准费米能级。 准费米能级:半导体中存在过剩载流子,则半导体就不会处于热平衡状态,费米能级就会发生变化,定义准费米能级。 n 0+?n =n i exp (E Fn ?E Fi kT )p 0+?p =n i exp [?(E Fp ?E Fi )kT ] 用这两组公式求解问题。 通过计算可知,电子的准费米能级高于E Fi ,空穴的准费米能级低于E Fi ,对于多子来讲,由于载流子浓度变化不大,所以准费米能级基本靠近热平衡态下的费米能级,但是对于少子来讲,少子浓度发生了很大的变化,所以费米能级有相对比较大的变化,由于注入过剩载流子,所以导致各自的准费米能级都靠近各自的价带。
过剩载流子的寿命: 半导体材料:半导体材料多是单晶材料,单晶材料的电学特性不仅和化学组成相关而且还与原子排列有关系。半导体基本分为两类,元素半导体材料和化合物半导体材料。 GaAs主要用于光学器件或者是高速器件。 固体的类型:无定型(个别原子或分子尺度内有序)、单晶(许多原子或分子的尺度上有序)、多晶(整个范围内都有很好的周期性),单晶的区域成为晶粒,晶界将各个晶粒分开,并且晶界会导致半导体材料的电学特性衰退。 空间晶格:晶格是指晶体中这种原子的周期性排列,晶胞就是可以复制出整个晶体的一小部分晶体,晶胞的结构可能会有很多种。原胞就是可以通过重复排列形成晶体的最小晶胞。三维晶体中每一个等效的格点都可以采用矢量表示为r=pa?+qb?+sc?,其中矢量a?,b?,c?称为晶格常数。晶体中三种结构,简立方、体心立方、面心立方。 原子体密度=每晶胞的原子数每晶胞的体积
利用Excel的NORMSDIST计算正态分布函数表1
利用Excel的NORMSDIST计算正态分布函数表1
利用Excel的NORMSDIST函数建立正态 分布表 董大钧,乔莉 沈阳理工大学应用技术学院、信息与控制分院, 辽宁抚顺113122 摘要:利用Excel办公软件特有的NORMSDIST函数可以很准确方便的建立正态分布表、查找某分位数点的正态分布概率值,极大的提高了数理统计的效率。该函数可返回指定平均值和标准偏差的正态分布函数,将其引入到统计及数据分析处理过程中,代替原有的手工查找正态分布表,除具有直观、形象、易用等特点外,更增加了动态功能,极大提高了工作效率及准确性。 关键词:Excel;正态分布;函数;统计 引言 正态分布是应用最广泛的连续概率分布,生产与科学实验中很多随机变量的概率分布都可以近似地用正态分布来描述。例如,在生产条件不变的情况下,某种产品的张力、抗压强度、口径、长度等指标;同一种生物体的身长、体重等指标;同一种种子的重量;测量同一物体的误差;弹着点沿某一方向的偏差;某个地区的年降水量;以及理想气体分子的速度分量等等。一般来说,如果一个量是由许多微小的独立随机因素影响的结果,那么就可以认为这个量具有正态分
布。从理论上看,正态分布具有很多良好的性质,许多概率分布可以用它来近似;还有一些常用的概率分布是由它直接导出的,例如对数正态分布、t分布、F分布等。在科学研究及数理统计计算过程中,人们往往要通过某本概率统计教材附录中的正态分布表去查找,非常麻烦。若手头有计算机,并安装有Excel软件,就可以利用Excel的NORMSDIST( x )函数进行计算某分位数点的正态分布概率值,或建立一个正态分布表,准确又方便。 1 正态分布及其应用 正态分布(normal distribution)又名高斯分布(Gaussian distribution),是一个在数学、物理及工程等领域都非常重要的概率分布,在统计学的许多方面有着重大的影响力。若随机变量X服从一个数学期望为μ、标准方差为σ2的高斯分布,记为N(μ,σ2)。则其概率密度函数为正态分布的期望值μ决定了其位置,其标准差σ决定了分布的幅度。因其曲线呈钟形,因此人们又经常称之为钟形曲线。我们通常所说的标准正态分布是μ = 0,σ = 1的正态分布。服从正态分布的随机变量的概率规律为取与μ邻近的值的概
一十种概率密度函数
一十种概率密度函数 function zhifangtu(x,m) %画数据的直方图,x表示要画的随机数,m表示所要画的条数%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%% a=min(x); b=max(x); l=length(x); h=(b-a)/m; %量化x x=x/h; x=ceil(x); w=zeros(1,m); for i=1:l for j=1:m if (x(i)==j) %x(i)落在j的区间上,则w(j)加1 w(j)=w(j)+1; else continue end end end w=w/(h*l); z=a:h:(b-h); bar(z,w); title('直方图') function y=junyun(n) %0-1的均匀分布,n代表数据量,一般要大于1024 %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%% y=ones(1,n); x=ones(1,n); m=100000; x0=mod(ceil(m*rand(1,1)),m); x0=floor(x0/2); x0=2*x0+1; u=11; x(1)=x0; for i=1:n-1 x(i+1)=u*x(i)+0; x(i+1)=mod(x(i+1),m); x(i)=x(i)/m; end %x(n)单位化
x(n)=x(n)/m; y=x; function y=zhishu(m,n) %指数分布,m表示指数分布的参数,m不能为0.n表示数据量,n一般要大于1024 %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%% x=junyun(n); for i=1;n if (x(i)==0) x(i)=0.0001; else continue; end end u=log(x); y=-(1/m)*u; function y=ruili(m,n) %瑞利分布,m是瑞利分布的参数,n代表数据量,n一般要大于1024 %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%% x=junyun(n); for i=1:n if (x(i)==0) x(i)=0.0001; else continue; end end u=(-2)*log(x); y=m*sqrt(u); function y=weibuer(a,b,n) %韦布尔分布,a,b表示参数,b不能为0.n表示数据量,一般要大于1024 %a=1时,是指数分布 %a=2时,是瑞利分布%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%% x=junyun(n); for i=1:n if (x(i)==0) x(i)=0.0001; else continue; end
标准正态分布的密度函数
正态分布 第二章 第七节 一、标准正态分布的密度函数 二、标准正态分布的概率计算 三、一般正态分布的密度函数 四、正态分布的概率计算 幻灯片2 正态分布的重要性正态分布是概率论中最重要的分布, 这可以由 以下情形加以说明: ⑴正态分布是自然界及工程技术中最常见的分布 之一, 大量的随机现象都是服从或近似服从正态分布的. 可以证明, 如果一个随机指标受到诸多因素的影响, 但其中任何一个因素都不起决定性作用, 则该随机指标 一定服从或近似服从正态分布. 这些性质是其它 ⑵正态分布有许多良好的性质, 许多分布所不具备的. ⑶正态分布可以作为许多分布的近似分布. 幻灯片3 -标准正态分布 下面我们介绍一种最重要的正态分布 一、标准正态分布的密度函数 若连续型随机变量X的密度函数为 定义 则称X服从标准正态分布, 记为 标准正态分布是一种特别重要的 它的密度函数经常被使用, 分布。 幻灯片4 密度函数的验证 则有 (2)根据反常积分的运算有 可以推出 幻灯片5 标准正态分布的密度函数的性质
,X的密度函数为 则密度函数的性质为: 的图像称为标准正态(高斯)曲线。 幻灯片6 随机变量 由于 由图像可知,阴影面积为概率值。 对同一长度的区间 ,若这区间越靠近 其对应的曲边梯形面积越大。 标准正态分布的分布规律时“中间多,两头少”. 幻灯片7 二、标准正态分布的概率计算 1、分布函数 分布函数为 幻灯片8 2、标准正态分布表 书末附有标准正态分布函数数值表,有了它,可以解决标准正态分布的概率计算. 表中给的是x > 0时, Φ(x)的值. 幻灯片9 如果 由公式得 令 则 幻灯片10 例1 解 幻灯片11 由标准正态分布的查表计算可以求得, 当X~N(0,1)时, 这说明,X 的取值几乎全部集中在[-3,3]区间内,超出这个范围的可能性仅占不到0.3%. 幻灯片12 三、一般正态分布的密度函数 如果连续型随机变量X的密度函数为 (其中 为参数) 的正态分布,记为 则随机变量X服从参数为 所确定的曲线叫 作正态(高斯)曲线. 幻灯片13
DOS态密度
态密度(Density of States,简称DOS) 在DOS结果图里可以查瞧就就是导体还就就是绝缘体还就就是半导体,请问怎么瞧。理论就就是什么?或者哪位老师可以告诉我这方面得知识可以通过学习什么获得。不胜感激。 查瞧就就是导体还就就是绝缘体还就就是半导体,最好还就就是用能带图DOS得话瞧费米能级两侧得能量差 谢希德。复旦版得《固体能带论》一书中有,请参阅!另外到网上或者学校得数据库找找“第一性原理”方面得论文,里面通常会有一些计算分析。下面有一篇可以下载得:ZnO得第一性原理计算 hoffman得《固体与表面》对态密度得理解还就就是很有好处得。 下面这个就就是在版里找得,多瞧瞧吧: 如何分析第一原理得计算结果 用第一原理计算软件开展得工作,分析结果主要就就是从以下三个方面进行定性/定量得讨论:1 ?、电荷密度图(charge density); 2、能带结构(EnergyBand Structure);?3、态密度(Density ofStates,简称DOS)。??电荷密度图就就是以图得形式出现在文章中,非常直观,因此对于一般得入门级研究人员来讲不会有任何得疑问。唯一需要注意得就就就是这种分析得种种衍生形式,比如差分电荷密图(def-ormationchargedensity)与二次差分图(difference chargedensity)等等,加自旋极化得工作还可能有自旋极化电荷密度图(spin-polarizedc harge density)。所谓“差分”就就是指原子组成体系(团簇)之后电荷得重新分布,“二次”就就是指同一个体系化学成分或者几何构型改变之后电荷得重新分布,因此通过这种差分图可以很直观地瞧出体系中个原子得成键情况。通过电荷聚集(accumulation)/损失(depl etion)得具体空间分布,瞧成键得极性强弱;通过某格点附近得电荷分布形状判断成键得轨道(这个主要就就是对d轨道得分析,对于s或者p轨道得形状分析我还没有见过)。分析总电荷密度图得方法类似,不过相对而言,这种图所携带得信息量较小。?能带结构分析现在在各个领域得第一原理计算工作中用得非常普遍了。但就就是因为能带这个概念本身得抽象性,对于能带得分析就就是让初学者最感头痛得地方。关于能带理论本身,我在这篇文章中不想涉及,这里只考虑已得到得能带,如何能从里面瞧出有用得信息。首先当然可以瞧出这个体系就就是金属、半导体还就就是绝缘体。判断得标准就就是瞧费米能级与导带(也即在高对称点附近近似成开口向上得抛物线形状得能带)就就是否相交,若相交,则为金属,否则为半导体或者绝缘体。对于本征半导体,还可以瞧出就就是直接能隙还就就是间接能隙:如果导带得最低点与价带得最高点在同一个k点处,则为直接能隙,否则为间接能隙。在具体工作中,情况要复杂得多,而且各种领域中感兴趣得方面彼此相差很大,分析不可能像上述分析一样直观与普适。不过仍然可以总结出一些经验性得规律来。主要有以下几点: 1) 因为目前得计算大多采用超单胞(supercell)得形式,在一个单胞里有几十个原
DOS态密度
态密度(Density of States,简称DOS) 在DOS结果图里可以查看是导体还是绝缘体还是半导体,请问怎么看。理论是什么?或者哪位老师可以告诉我这方面的知识可以通过学习什么获得。不胜感激。 查看是导体还是绝缘体还是半导体,最好还是用能带图 DOS的话看费米能级两侧的能量差 谢希德。复旦版的《固体能带论》一书中有,请参阅!另外到网上或者学校的数据库找找“第一性原理”方面的论文,里面通常会有一些计算分析。 下面有一篇可以下载的: ZnO的第一性原理计算 hoffman的《固体与表面》对态密度的理解还是很有好处的。 下面这个是在版里找的,多看看吧: 如何分析第一原理的计算结果 ? ?? ?用第一原理计算软件开展的工作,分析结果主要是从以下三个方面进行定性/定量的讨论: ??1、电荷密度图(charge density); ??2、能带结构(Energy Band Structure); ??3、态密度(Density of States,简称DOS)。 ? ? ? ???电荷密度图是以图的形式出现在文章中,非常直观,因此对于一般的入门级研究人员来讲不会有任何的疑问。唯一需要注意的就是这种分析的种种衍生形式,比如差分电荷密图(def-ormation charge density)和二次差分图(difference charge density)等等,加自旋极化的工作还可能有自旋极化电荷密度图(spin-polarized charge density)。所谓“差分”是指原子组成体系(团簇)之后电荷的重新分布,“二次”是指同一个体系化学成分或者几何构型改变之后电荷的重新分布,因此通过这种差分图可以很直观地看出体系中个原子的成键情况。通过电荷聚集(accumulation)/损失(depletion)的具体空间分布,看成键的极性强弱;通过某格点附近的电荷分布形状判断成键的轨道(这个主要是对d轨道的分析,对于s或者p轨道的形状分析我还没有见过)。分析总电荷密度图的方法类似,不过相对而言,这种图所携带的信息量较小。 ? ?? ?能带结构分析现在在各个领域的第一原理计算工作中用得非常普遍了。但是因为能带这个概念本身的抽象性,对于能带的分析是让初学者最感头痛的地方。关于能带理论本身,我在这篇文章中不想涉及,这里只考虑已得到的能带,如何能从里面看出有用的信息。首先
数学分布(泊松分布、二项分布、正态分布、均匀分布、指数分布)生存分析贝叶斯概率公式全概率公式(新)
数学期望:随机变量最基本的数学特征之一。它反映随机变量平均取值的大小。又称期望或均值。它是简单算术平均的一种推广。例如某城市有10万个家庭,没有孩子的家庭有1000个,有一个孩子的家庭有9万个,有两个孩子的家庭有6000个,有3个孩子的家庭有3000个,则此城市中任一个家庭中孩子的数目是一个随机变量,记为X,它可取值0,1,2,3,其中取0的概率为0.01,取1的概率为0.9,取2的概率为0.06,取3的概率为0.03,它的数学期望为0×0.01+1×0.9+2×0.06+3×0.03等于1.11,即此城市一个家庭平均有小孩1.11个,用数学式子表示为:E(X)=1.11。 也就是说,我们用数学的方法分析了这个概率性的问题,对于每一个家庭,最有可能它家的孩子为1.11个。 可以简单的理解为求一个概率性事件的平均状况。 各种数学分布的方差是: 1、一个完全符合分布的样本 2、这个样本的方差 概率密度的概念是:某种事物发生的概率占总概率(1)的比例,越大就说明密度越大。比如某地某次考试的成绩近似服从均值为80的正态分布,即平均分是80分,由正态分布的图形知x=80时的函数值最大,即随机变量在80附近取值最密集,也即考试成绩在80分左右的人最多。 下图为概率密度函数图(F(x)应为f(x),表示概率密度):
离散型分布:二项分布、泊松分布 连续型分布:指数分布、正态分布、X2分布、t分布、F分布 抽样分布 抽样分布只与自由度,即样本含量(抽样样本含量)有关 二项分布(binomial distribution):例子抛硬币 1、重复试验(n个相同试验,每次试验两种结果,每种结果概率恒定———— 伯努利试验) 2、
16种常见概率分布概率密度函数、意义及其应用
目录 1. 均匀分布 (1) 2. 正态分布(高斯分布) (2) 3. 指数分布 (2) 4. Beta分布(:分布) (2) 5. Gamm 分布 (3) 6. 倒Gamm分布 (4) 7. 威布尔分布(Weibull分布、韦伯分布、韦布尔分布) (5) 8. Pareto 分布 (6) 9. Cauchy分布(柯西分布、柯西-洛伦兹分布) (7) 2 10. 分布(卡方分布) (7) 8 11. t分布................................................ 9 12. F分布 ............................................... 10 13. 二项分布............................................ 10 14. 泊松分布(Poisson 分布)............................. 11 15. 对数正态分布........................................
1. 均匀分布 均匀分布X ~U(a,b)是无信息的,可作为无信息变量的先验分布。
2. 正态分布(高斯分布) 当影响一个变量的因素众多,且影响微弱、都不占据主导地位时,这个变量 很可能服从正态分布,记作 X~N (」f 2)。正态分布为方差已知的正态分布 N (*2)的参数」的共轭先验分布。 1 空 f (x ): —— e 2- J2 兀 o' E(X), Var(X) _ c 2 3. 指数分布 指数分布X ~Exp ( )是指要等到一个随机事件发生,需要经历多久时间。其 中,.0为尺度参数。指数分布的无记忆性: Plx s t|X = P{X t}。 f (X )二 y o i E(X) 一 4. Beta 分布(一:分布) f (X )二 E(X) Var(X)= (b-a)2 12 Var(X)二 1 ~2
利用Excel的NORMSDIST计算正态分布函数表1
利用Excel的NORMSDIST函数建立正态 分布表 董大钧,乔莉 沈阳理工大学应用技术学院、信息与控制分院,辽宁抚顺113122 摘要:利用Excel办公软件特有的NORMSDIST函数可以很准确方便的建立正态分布表、查找某分位数点的正态分布概率值,极大的提高了数理统计的效率。该函数可返回指定平均值和标准偏差的正态分布函数,将其引入到统计及数据分析处理过程中,代替原有的手工查找正态分布表,除具有直观、形象、易用等特点外,更增加了动态功能,极大提高了工作效率及准确性。 关键词:Excel;正态分布;函数;统计 引言 正态分布是应用最广泛的连续概率分布,生产与科学实验中很多随机变量的概率分布都可以近似地用正态分布来描述。例如,在生产条件不变的情况下,某种产品的张力、抗压强度、口径、长度等指标;同一种生物体的身长、体重等指标;同一种种子的重量;测量同一物体的误差;弹着点沿某一方向的偏差;某个地区的年降水量;以及理想气体分子的速度分量等等。一般来说,如果一个量是由许多微小的独立随机因素影响的结果,那么就可以认为这个量具有正态分布。从理论上看,正态分布具有很多良好的性质,许多概率分布可以用它来近似;还有一些常用的概率分布是由它直接导出的,例如对数正态分布、t分布、F分布等。在科学研究及数理统计计算过程中,人们往往要通过某本概率统计教材附录中的正态分布表去查找,非常麻烦。若手头有计算机,并安装有Excel软件,就可以利用Excel的NORMSDIST( x )函数进行计算某分位数点的正态分布概率值,或建立一个正态分布表,准确又方便。 1 正态分布及其应用 正态分布(normal distribution)又名高斯分布(Gaussian distribution),是一个在数学、物理及工程等领域都非常重要的概率分布,在统计学的许多方面有着重大的影响力。若随机变量X服从一个数学期望为μ、标准方差为σ2的高斯分布,记为N(μ,σ2 )。则其概率密度
能带结构分析、态密度和电荷密度的分析
电荷密度图、能带结构、态密度的分析 能带图的横坐标是在模型对称性基础上取的K点。为什么要取K点呢?因为晶体的周期性使得薛定谔方程的解也具有了周期性。按照对称性取K点,可以保证以最小的计算量获得最全的能量特征解。能带图横坐标是K点,其实就是倒格空间中的几何点。纵坐标是能量。那么能带图应该就是表示了研究体系中,各个具有对称性位置的点的能量。我们所得到的体系总能量,应该就是整个体系各个点能量的加和。 主要是从以下三个方面进行定性/定量的讨论: 1、电荷密度图(charge density); 2、能带结构(Energy Band Structure); 3、态密度(Density of States,简称DOS)。 电荷密度图是以图的形式出现在文章中,非常直观,因此对于一般的入门级研究人员来讲不会有任何的疑问。唯一需要注意的就是这种分析的种种衍生形式,比如差分电荷密图(def-ormation charge density)和二次差分图(difference charge density)等等,加自旋极化的工作还可能有自旋极化电荷密度图(spin-polarized charge density)。所谓“差分”是指原子组成体系(团簇)之后电荷的重新分布,“二次”是指同一个体系化学成分或者几何构型改变之后电荷的重新分布,因此通过这种差分图可以很直观地看出体系中个原子的成键情况。通过电荷聚集(accumulation)/损失(depletion)的具体空间分布,看成键的极性强弱;通过某格点附近的电荷分布形状判断成键的轨道(这个主要是对d轨道的分析,对于s或者p轨道的形状分析我还没有见过)。分析总电荷密度图的方法类似,不过相对而言,这种图所携带的信息量较小。 成键前后电荷转移的电荷密度差。此时电荷密度差定义为:delta_RHO = RHO_sc - RHO_atom 其中RHO_sc 为自洽的面电荷密度,而RHO_atom 为相应的非自洽的面电荷密度,是由理想的原子周围电荷分布堆彻得到的,即为原子电荷密度的叠加(a superposition of atomic charge densities)。需要特别注意的,应保持前后两次计算(自洽和非自洽)中的FFT-mesh 一致。因为,只有维数一样,我们才能对两个RHO作相应的矩阵相减。 能带结构分析现在在各个领域的第一原理计算工作中用得非常普遍了。首先当然可以看出这个体系是金属、半导体还是绝缘体。对于本征半导体,还可
正态分布概率公式(部分)
Generated by Foxit PDF Creator ? Foxit Software https://www.360docs.net/doc/8d8901031.html, For evaluation only.
图 62正态分布概率密度函数的曲线 正态曲线可用方程式表示。 n 当 →∞时,可由二项分布概率函数方程推导出正态 分布曲线的方程:
fx= (61 ) () .6
式中: x—所研究的变数; fx —某一定值 x出现的函数值,一般称为概率 () 密度函数 (由于间断性分布已转变成连续性分布,因而我们只能计算变量落在某 一区间的概率, 不能计算变量取某一值, 即某一点时的概率, 所以用 “概率密度” 一词以与概率相区分),相当于曲线 x值的纵轴高度; p—常数,等于 31 .4 19……; e— 常数,等于 2788……; μ 为总体参数,是所研究总体 5 .12 的平均数, 不同的正态总体具有不同的 μ , 但对某一定总体的 μ 是一个常数; δ 也为总体参数, 表示所研究总体的标准差, 不同的正态总体具有不同的 δ , 但对某一定总体的 δ 是一个常数。 上述公式表示随机变数 x的分布叫作正态分布, 记作 N μ ,δ2 ), “具 ( 读作 2 平均数为 μ,方差为 δ 的正态分布”。正态分布概率密度函数的曲线叫正态 曲线,形状见图 62。 (二)正态分布的特性
1、正态分布曲线是以 x μ 为对称轴,向左右两侧作对称分布。因 =
的
数值无论正负, 只要其绝对值相等, 代入公式 61 ) ( .6 所得的 fx 是相等的, () 即在平均数 μ 的左方或右方,只要距离相等,其 fx 就相等,因此其分布是 () 对称的。在正态分布下,算术平均数、中位数、众数三者合一位于 μ 点上。
统计学三大分布及正态分布的关系
统计学三大分布与正态分布的关系 [1] 张柏林 41060045 理实1002班 摘要:本文首先将介绍2χ分布,t 分布,F 分布和正态分布的定义及基本性质, 然后用理论说明2χ分布,t 分布,F 分布与正态分布的关系,并且利用数学软件MATLAB 来验证之. 1.三大分布函数[2] 1.12χ分布 2()n χ分布是一种连续型随机变量的概率分布。这个分布是由别奈梅(Benayme)、赫尔默特(Helmert)、皮尔逊分别于1858年、1876年、1900年所发现,它是由正态分布派生出来的,主要用于列联表检验。 定义:若随机变量12n ,,X X …X 相互独立,且都来自正态总体01N (,) ,则称统计量222 212n =+X X χ++…X 为服从自由度为n 的2χ分布, 记为22~()n χχ. 2χ分布的概率密度函数为 122210(;),2()200n x n x e x n f x n x --?≥??=Γ???? ,2χ分布的密度函数图形是一个只取非负值的偏态分布,如下图.
卡方分布具有如下基本性质: 性质1:22(()),(())2E n n D n n χχ==; 性质2:若221122(),()X n X n χχ==,12,X X 相互独立,则21212~()X X n n χ++; 性质3:2 n χ→∞→时,( n )正态分布; 性质4:设)(~2 2n α χχ,对给定的实数),10(<<αα称满足条 件:αχχα χα ==>?+∞ ) (2 22)()}({n dx x f n P 的点)(2 n α χ为)(2n χ分布的水平α的上侧分位数. 简称为上侧α分位数. 对不同的α与n , 分位数的值已经编制成表供查 用. 2()n χ分布的上α分位数 1.2t 分布 t 分布也称为学生分布,是由英国统计学家戈赛特在1908年“student ”的笔名 首次发表的,这个分布在数理统计中也占有重要的位置. 定义:设2 ~0~X N χ(,1),Y (n ),,X Y 相互独立,,则称统计量/T Y n = 服从自由度为n 的t 分布,记为~()T t n .
期末ref清华大学固体物理王燕
填空三十分 格波和平面波的区别 八道简答四十分 固体物理中的绝热近似是什么意思 从能带理论解释为什么存在导体、半导体、绝缘体 波矢空间和倒格空间有什么关系?为什么说波矢空间可以看作准连续 三道计算各十分 1.画正三角形晶格的倒格子 h-bar^2 7 1 2.已知E(k)=————(— - coska + — cos2ka) ma^2 8 8 求能带宽度和能带顶底的有效质量 3.已知omiga=cq^2,求频谱? 07邹建平考题 一填空(30分) 固体物理中常用_________边界条件 理想状态中导热能力最好的是___________ 热膨胀系数与格林爱森常数的关系 金属导电载体?半导体导电载体? 考虑了散射后的运动方程 1.准自由电子近似,把_____作为0级波函数,把_______作为微扰项;紧束缚近似把____作为0级波函数,把____作为微扰项 2.一维单原子链,如果已知色散关系w=c*q^2,求G(w) 3. 3.准经典近似下,速度v=_____ ,有效质量的表达式________ 4.一维单原子链,晶格常数a,N个原子,求紧束缚近似下的E(k)=______ 5.低温条件,晶格比热和____成正比,电子比热和_____成正比 6.半导体中,载流子在外场力作用下是__运动,考虑了散射后的运动方程__,低场条件下的迁移率__ 7.一维N原子链,一个能带中有多少能级?容纳多少电子? 二简答(30分) 1.晶体膨胀时,费米能级如何变化,温度升高时,费米能级如何变化? 2.波矢空间与倒格空间有何关系?为什么说波矢空间内的状态点是准连续的? 3.布里渊区边界上的能级 4.什么叫简正振动模式,简正振动数目,格波数目,格波振动模式数目是否是一回事? 5.极性,非极性晶体晶格散射机制,驰豫时间与温度的关系 6.温度很低时时,对于无限长的晶体,是热超导材料还是热绝缘材料? 三(15') 1.一维单原子链的态密度
能带,态密度图分析
能带结构和态密度图的绘制及初步分析 前几天在QQ的群中和大家聊天的时候,发现大家对能带结构和态密度比较感兴趣,我做计算已经有一年半了,有一些经验,这里写出来供大家参考参考,希望能够对初学者有所帮助,另外写的这些内容也不可能全都正确,只希望通过表达出来和大家进行交流,共同提高。 MS这个软件的功能确实是比较强,但是也有一些地方不尽如人意的地方。(也可能是我对一些结果不会分析所致,有些暂时不能解决的问题在最后一部分提出,希望大家来研究 研究,看看有没有实现的可能性)。 能带结构、态密度和布居分析是很重要的内容,在 分析能带结构和态密度的时候,往往是先作图,然后分 析。 软件本身提供的作图功能并不是很强,比如说能带结构 (只能带只能做point图和line图),不美观不说,对于 每一个能带的走势也不好观察,感觉无从下手。所以我 一般用origin作图(右图是用origin做的能带图)。能带 结构和态密度的作图过程请参考我给大家提供的动画。 接下来我们先开看看能带结构的分析和制作! 第一部分:能带结构 这个部分打算先简单的介绍一下能带的基础知识,希望能对大家有所帮助,如果对能带了解比较深入的朋友,可以跳过这个部分内容,之中不当之处请勿见笑。^_^ 第一个问题是: 1、能带是怎样形成——轨道和一维体系的能带。 这是最基本的一个问题,我们要对能带结构进行分析,首先要知道它是如何来的。其实能带是一种近似的结果(可以看成一种近似),是周期边界条件(bloch函数)下的一种近似。先来看看一个最简单的问题,非周期体系有没有能带结构?答案是没有的,大家可以试试: ①建一个周期的晶胞②选择build菜单下的symmetry子菜单下的none periodic superstructure去掉周期边界条件性③看看还能够运行吗?运行(run)按钮变灰了,不能提交作业了。这说明什么问题?这说明这个CASTEP这个模块不能计算非周期的体系,另外可以参考MS中的DMOL模块,它可以计算非周期系统,虽然可以计算周期系统,但是仍不能计算能带,大家可以试试,看看property中的band structure能不能选上,一定不能!!^_^ 从这里,我们可以得到一个结论,对于单个原子(分子、单胞)如果不加上周期边界条件,是无法获得能带结构的。所以计算小分子体系,或者采用团簇模型的朋友,这部分内容或许对你们没有帮助!那么,非周期体系的态密度能够计算吗?这应该是能够计算的,曾经开到过文献采用团簇模型,计算出态密度的(phys. Rev. 上的文章)。 那么非周期体系为什么没有能带结构呢? 看一个例子:一个H2分子有能带吗?没有,因为它没有周期边界条件,也就是说在x,y,z方向上没有重复,所以它没有能带结构。那H2分子有什么东西呢?有两个轨道,两个 1s原子轨道,或者说两个轨道能级,它们成键参考右图。 再看另外一个例子:一维无限H原子链 H H H H H H 在一维无限H原子链体系中,产生了能带。 为什么在一维无限H原子链体系中能够产生能带呢?
正态分布概率公式(部分)
图 6-2 正态分布概率密度函数的曲线 正态曲线可用方程式表示。当n→∞时,可由二项分布概率函数方程推导出正态分布曲线的方程: f(x)= (6.16 ) 式中: x —所研究的变数; f(x) —某一定值 x 出现的函数值,一般称为概率密度函数(由于间断性分布已转变成连续性分布,因而我们只能计算变量落在某一区间的概率,不能计算变量取某一值,即某一点时的概率,所以用“概率密度”一词以与概率相区分),相当于曲线 x 值的纵轴高度; p —常数,等于 3.14 159 ……; e —常数,等于 2.71828 ……;μ为总体参数,是所研究总体的平均数,不同的正态总体具有不同的μ,但对某一定总体的μ是一个常数;δ也为总体参数,表示所研究总体的标准差,不同的正态总体具有不同的δ,但对某一定总体的δ是一个常数。 上述公式表示随机变数 x 的分布叫作正态分布,记作 N( μ , δ2 ) ,读作“具平均数为μ,方差为δ 2 的正态分布”。正态分布概率密度函数的曲线叫正态曲线,形状见图 6-2 。 (二)正态分布的特性 1 、正态分布曲线是以 x= μ为对称轴,向左右两侧作对称分布。因的数值无论正负,只要其绝对值相等,代入公式( 6.16 )所得的 f(x) 是相等的,即在平均数μ的左方或右方,只要距离相等,其 f(x) 就相等,因此其分布是对称的。在正态分布下,算术平均数、中位数、众数三者合一位于μ点上。
2 、正态分布曲线有一个高峰。随机变数 x 的取值范围为( - ∞,+ ∞ ),在( - ∞ ,μ)正态曲线随 x 的增大而上升,;当 x= μ时, f(x) 最大;在(μ,+ ∞ )曲线随 x 的增大而下降。 3 、正态曲线在︱x-μ︱=1 δ处有拐点。曲线向左右两侧伸展,当x →± ∞ 时,f(x) →0 ,但 f(x) 值恒不等于零,曲线是以 x 轴为渐进线,所以曲线全距从 -∞到+ ∞。 4 、正态曲线是由μ和δ两个参数来确定的,其中μ确定曲线在 x 轴上的位置 [ 图 6-3] ,δ确定它的变异程度 [ 图 6-4] 。μ和δ不同时,就会有不同的曲线位置和变异程度。所以,正态分布曲线不只是一条曲线,而是一系列曲线。任何一条特定的正态曲线只有在其μ和δ确定以后才能确定。 5 、正态分布曲线是二项分布的极限曲线,二项分布的总概率等于 1 ,正态分布与 x 轴之间的总概率(所研究总体的全部变量出现的概率总和)或总面积也应该是等于 1 。而变量 x 出现在任两个定值 x1到x2(x1≠x2)之间的概率,等于这两个定值之间的面积占总面积的成数或百分比。正态曲线的任何两个定值间的概率或面积,完全由曲线的μ和δ确定。常用的理论面积或概率如下: 区间μ ± 1 δ面积或概率 =0.6826 μ ± 2 δ =0.9545 μ ± 3 δ=0.9973 μ± 1.960δ=0.9500 μ ±2.576 δ =0.9900
四面体法测态密度
Wuhan, Hubei, 430074, P. R. China 中华人民共和国 湖北 武汉 《计算材料学》课程设计 指导老师:江建军教授 电子科学与技术系 2004年6月
Wuhan, Hubei, 430074, P. R. China 中华人民共和国 湖北 武汉 四面体法测状态密度 夏玄赵超荣孙金中程捷兰天吕璐 赵珊刘志英岳雄伟向伟席明鹏 (华中科技大学电子科学与技术系,武汉 430074) 摘要:以面心立方(fcc)晶体的s能带为例, 利用紧束缚法计算其能带,利用四面体法计算其态密度。即将第一布里渊区分成一些微小的四面体,计算每一个四面体对空间一点的态密度的贡献而进行态密度计算的方法。分析当分割四面体个数变化时态密度曲线的拟合程度。 关键词:紧束缚法能带密度函数四面体法第一布里渊区态密度态密度曲线 The Calculation of Dos with Tetrahedron Method Abstract: Take the energy band of S orbital of fcc crystal as an example, we measure it by Tight-Binding Theory and then calculate its density of state (Dos) with Tetrahedron Method. With this method, firstly the first Brillouin Zone is divided into some tiny tetrahedrons, and each tetrahedron's contributing to Dos is calculated, then the total Dos can be known. Finally, we use different number of tetrahedrons to get different Dos lines and analyze the congruity of them with other calculation methods. Key words: Tight-Binding Theory; Dos function; Tetrahedron Method; Density of state; Dos line. 1 引言 在研究非晶半导体电子性质的努力中,许多研究工作者试图将固体中各种各样的无序状态进行分类和理想化。典型的种类是在结构,组分,网络形状,定量和拓扑学等的无序状态。这样的分类通常依据特定的理论结构。为了回避问题的复杂性,抓住问题的实质,关键在于研究理想化的模型系统,从而设想将整个空间机构化分成许多基本单元——四面体单元的组合,对每个四面体单独考虑最后叠加。四面体法作为一种空间机构分析与综合的工具, 有这样的特点:这就是它对于使用者数学方面的要求并不高;它与几何学的结合给人以直观、形象的感受, 使得该方法易于被人理解、接收; 它涉及到的矢量知识都是最基本的矢量运算。