尤启东药物化学药物1--5
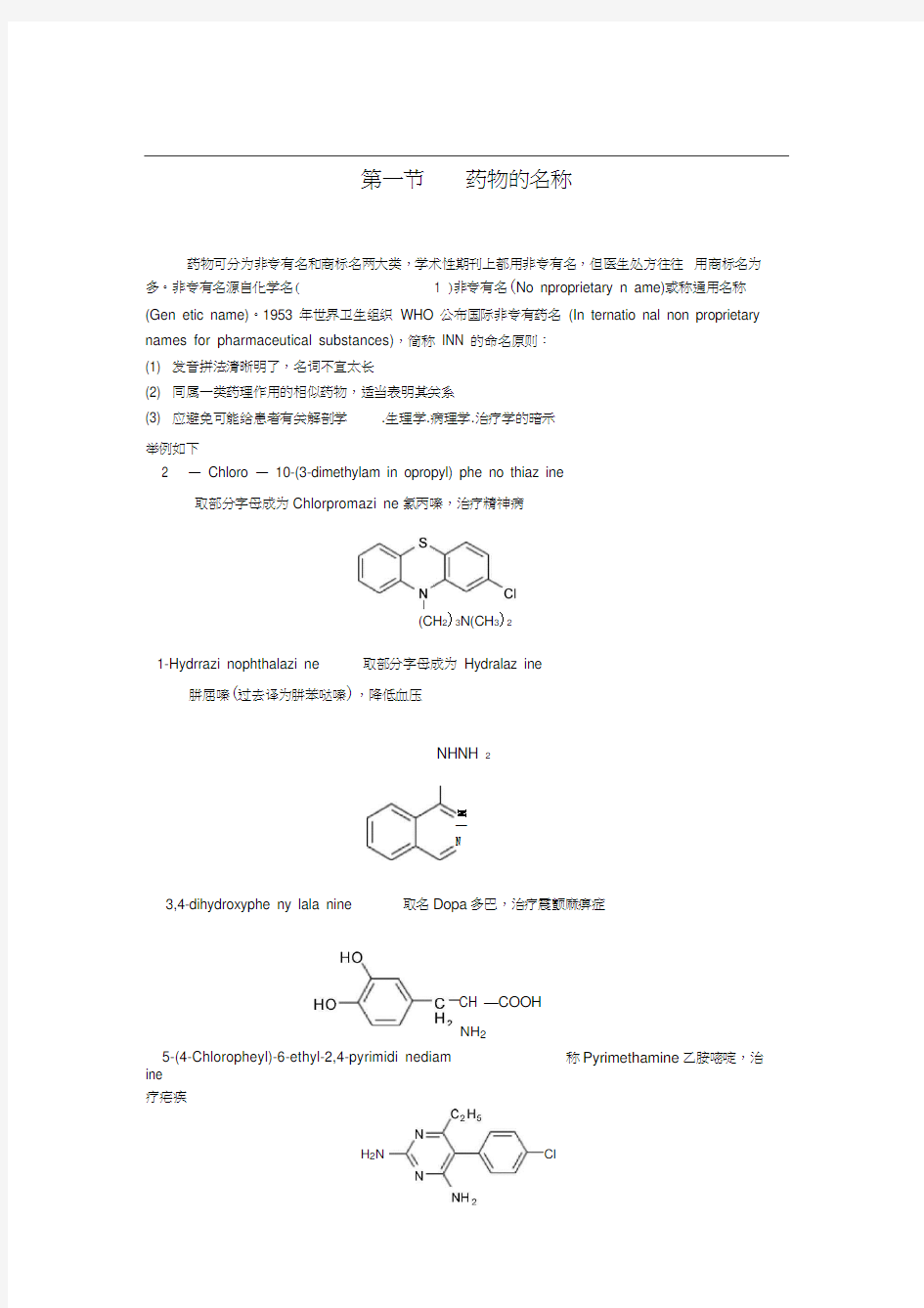

第一节药物的名称
药物可分为非专有名和商标名两大类,学术性期刊上都用非专有名,但医生处方往往用商标名为多。非专有名源自化学名( 1 )非专有名(No nproprietary n ame)或称通用名称(Gen etic name)。1953 年世界卫生组织WHO 公布国际非专有药名(In ternatio nal non proprietary names for pharmaceutical substances),简称INN 的命名原则:
(1) 发音拼法清晰明了,名词不宜太长
(2) 同属一类药理作用的相似药物,适当表明其关系
(3) 应避免可能给患者有关解剖学.生理学.病理学.治疗学的暗示
举例如下
2 —Chloro —10-(3-dimethylam in opropyl) phe no thiaz ine
取部分字母成为Chlorpromazi ne氯丙嗪,治疗精神病
1-Hydrrazi nophthalazi ne 取部分字母成为Hydralaz ine
肼屈嗪(过去译为肼苯哒嗪),降低血压
NHNH 2
3,4-dihydroxyphe ny lala nine 取名Dopa多巴,治疗震颤麻痹症
NH2
5-(4-Chloropheyl)-6-ethyl-2,4-pyrimidi nediam ine
疗疟疾称Pyrimethamine乙胺嘧啶,治
(CH2)3N(CH3)2
N
—
N
CH —COOH H2N Cl
药理作用相似药物
Cimetidi ne
Ran itidi ne
Famotid ine
西咪替丁
雷尼替丁
法莫替丁
局部麻醉药
Coca ine可卡因
Proca ine普鲁卡因
Lidoca ine利多卡因
头孢类抗生素
Cefaclor头孢龙罗
Cefradi ne头孢柱定
Ceftizoxime头孢唑肟
商标名称各公司注册商标的名称, 不能冒用
非专有名称译名商标名称
Pyrimetham ine乙胺嘧啶Daraprim
Chlorpromaz ine氯丙嗪Prozil
Chloramphe
nicol
氯霉素Chlormyceti
n
Hydralaz ine肼屈嗪Apresoli ne
Methado ne美沙酮Dolophi ne
中文译名要求贯彻药政条理,名称要简洁?明确.科学?不准用代号,可以音译,也可意译,也可音意合译。若外文名称音节较少,结构较复杂,尽量音译。
Cortiso ne 可的松Lidocai ne 利多卡因
Morphi ne 吗啡Dexamethaso ne 地塞米松
Dopa 多巴
若外文名称显示化学基团,对照译简短,化学名称意译
Proglumide 丙谷胺
Pyrimethami ne 乙胺嘧啶若外文名称音译较简明,用音译
Spir ono lact one
An ethol trith one
Gua nethidi ne 胍乙啶(降压)外文名称显示部分结构,用意音结合
Dopamine多巴胺
Phe ny toin 苯妥因(治癫痫)
H
Chlorpromaz ine 氯丙嗪
相似药理作用的药物,其译名也应表示关系例如青霉素类字尾都用西林
carfecillin卡非西林
hetacillin海地西林
螺内酯(利尿)
菌三流(利胆)
------- N
O
mezlocillin 美洛西林
译名应避免暗示治疗用途,例如消炎镇痛药
acetaminophen 扑热息痛
应称醋氨酚
B 受体阻滞剂原译名
propranolol 心得安 alprenolol 心得舒 practolol 心得宁 oxprenolol 心得平 pindolol 心得静
近年国外发展的系列药物增多,很难一一意译,药典委员会主张译音
propranolol 普萘洛尔 alprenolol 阿普洛尔 practolol 普托洛尔 pindolol 吲哚洛尔 oxprenolol 氧烯洛
尔 acebutolol 醋丁洛尔 atenolol 阿替洛尔 metoprolol 美托洛尔 nadolol 纳多洛尔 timolol 噻吗洛尔 原来已有意译的药名也统一为系列名称
ampicillin 原译氨苄青霉素,现译氨苄西林 oxacillin 原译苯唑青霉素,现译苯唑西林 cimetidine 原译
甲氰咪胍,现译西咪替丁
benoxylate 扌卜热痛
H 3C H 3C "'
indomethasine 消炎痛
应称吲哚美辛
CH 3O
HO
NH —CO — CH 3
benzydamine 炎痛静
NHCOCH 3
CH 2Ph
N — CH 2)
CH 2—O
CH
N CO
第二节酶反应与酶抑制剂
2. 1酶反应
酶是生物体内化学反应的催化剂,象细菌这样简单的生物,细胞内大约有3000种酶, 分别催化3000种不同化学变化。
在生命活动中,有时在瞬间要消耗巨大能量。动物在原野奔腾追逐,禽鸟在高空展翅翱翔,伴随着肌肉迅速收缩,需要代谢反应迅速进行,将储存的能量高速释放,酶在这里起提高反应速率的催化作用。如果没有酶的帮助,我们一次进餐得化上50年的时间才能消化
完毕,而在酶的帮助下,要不了几个小时,肚子又饿了。糖或脂肪氧化会产生能量,把糖或脂肪放在空气里燃烧,氧化作用很快完成,热量一下释放出来。这样的反应如果在体内进行,放热产生高温,会损伤机体。因此,在生命活动中,氧化作用必须缓慢进行,以便最有效地利用能量;同时热量必须温和地放出,以便长期保持一定的体温。所以代谢分解必须和燃烧有所不同,代谢分解每步反应由不同的酶催化。我们摄取食物后消化,由一系列酶催化分解,
酶由体内各个部分分泌。唾液和胰腺分泌淀粉酶,化解淀粉;胃分泌蛋白酶;肠分泌蛋白酶
和糜蛋白酶,水解蛋白质。肠还释放脂酶,水解脂肪,于是食物从大分子分级分解成小分子。当食物变成单糖,氨基酸,脂酸后,便在肠壁吸收,渗入到血流,运输到各种组织。
当食物代谢分解释放的能量有盈余时,酶的催化作用又促使产生一些化学物质,把能量暂时储存起来,而当需用时,又可迅速释出。例如三磷酸腺苷(ATP)即为能量储存物质,分子内有3个磷酸基,在分解而裂去一个磷酸基时,可释出33千焦能量,变为二磷酸腺苷
(ADP )。同样,二磷酸腺苷再水解,脱去一个磷酸基,变成单磷酸腺苷,又可释出33千焦能量,但单磷酸腺苷再水解脱去最后一个磷酸基时,只释出12.5千焦能量。
A —P —P —P+H2O - A —P —P + P —OH+33Kg
A-P—P+H2O - A —P +P —OH +33Kg
A—P+H2O ---------------------- - A+ P—OH +12.5Kg
2.2青霉素
许多药物的作用机理,在于抑制酶的催化作用,从而干扰生命活动。这种药物的结构,可能与酶反应的底物的结构相似。青霉素和头孢菌素的抑制细菌作用,是由于干扰细菌合成
细胞壁。细菌依赖着细胞壁保护其细胞,细胞壁由一些多糖链和多肽链交织而成。在交织构
成过程中,一条包括由D-丙氨酰-D-丙氨酸二肽的链加到一条有几个甘氨酸组成的肽链上,这加成反应由转肽酶与羧肽酶所催化。青霉素和头孢菌素的构象和D-丙氨酰-D-丙
氨酸的构象十分相象,因之青霉素正好适应D-丙氨酰-D-丙氨酸在酶上的作用部位。而
药物与酶分子作用部位的结合,意味着占有这部位而排斥了丙氨酸二肽的结合,这样就干扰
了肽链的交织,从而阻止了细胞壁的构成,于是危及细菌的生长。这作用称为代谢拮抗
(Biological antagonism ),丙氨酸肽链是代谢物,青霉素是拮抗物。拮抗物( Antaganist )
与代谢物(Metabilite )间存在一定构象关系。青霉素和头孢菌素的半合成类似物中,也存在类似构效关系,只有构象与前述青霉素或D-丙氨酰-D-丙氨酸相似的,才有抑菌作用。
R — CON S CH3
~CH3
COOH
CH 2
COOH
2
D--丙氨酰丙氨酸
头孢菌素类化合物环内第三位存有双键,该双键如迁移至第二位,便失去抑菌活性, 因为双键位置的
变异, 也同时意味着构象的差别。 在头孢菌素类化合物中, 羧基占有环上的 假平伏键。在这构象中,酰氨基的氧原子与羧基的碳原子相距较近。在没有抗菌活性的△ —头孢菌素类化合物中, 羧基处于环上的假直立键。 在这构象中,酰胺基的氧原子与羧基的 碳原子相距较远。前者构象与丙氨酸二肽相近, 因此也可在酶上同一受体部位作用,
后者的
构象就有偏离,就不适应多肽的作用所在,因此不能与酶很稳固结合而产生抑菌作用。
△ 2—头抱菌素类 (无抗菌活性)
2. 3抗艾滋病毒药物
艾滋病即获得性免疫缺陷综合症
(Acquired immune deficiency syndrome ),病毒是 RNA
病毒,以RNA 为模板,合成DNA ,通过将有关核苷酸制成磷酸酯而成为长链。因为所用原
料包括脱氧胸腺嘧啶核苷,将其 3位OH 基改为叠氮基成为齐多夫定(zidovudine,AZT )与 脱氧胸腺嘧啶核苷结构相近,都能与逆转录酶结合,但叠氮基不能磷酸化,不能形成 DNA
长链。艾滋病患者服用齐多夫定后体重增加,免疫功能改善,免疫
T 细胞增加,高剂量组
患者体内找不到病毒。齐多夫定虽不能根治艾滋病,但可降低该病死亡率,可减少并发症的 发作及严重性。
脱氧胸腺嘧啶核苷
一些类似的药物也可抑制艾滋病毒繁殖,如
Didanosine,DDI 可延缓艾滋病毒进程,延
长患者寿命;Zalcitabine(ddC)与齐多夫定合用,可见体内免疫
T 细胞增多,dioxolane-T 以
O 代替CH 成非糖结构,代谢降解减慢,对艾滋病毒明显抑制;还有
stavudine (D4T )
dida nosine
ddI
青霉素类药物
头孢菌素类 (有抗菌活性)
齐多夫定
zalcitab
ine
RCO —
R'
HO...
CH 3
OH
HO.
CH
3
N 3
HOH
stavudi ne
dtT
艾滋病毒RNA的外衣是蛋白质。病毒蛋白由大分子的前体蛋白水解而成,水解的断裂点是苯丙氨酸与脯氨酸间的肽键和酪氨酸与脯氨酸间的酰胺键。
模仿蛋白质的片断结构,带有苯丙氨酰脯氨酸的结构改造的化合物成为indinavir,n elfi navir,rit on
avir,raqui navir 是艾滋病毒蛋白水解酶抑制剂,应用后迅速降低病毒浓度。
2. 4血管紧张素转化酶抑制剂
肾脏分泌有一种酶称肾素(Rennin),又称肾高血压蛋白酶,产生后分泌至血循环中,使血浆中一种球蛋白分解,产生血管紧张素( angiotensin),这种蛋白称血管紧张素原(angiotensinogen),来自肝脏。裂去羧端的肽段后,生成10个氨基酸组成的血管紧张素I, 后者又经在肺脏产生的血管紧张素转化酶( angiotensin convertingenzyme )的催化作用,再裂去羧端的两个氨基酸,成为八肽的血管紧张素H。肾素,血管紧张素,醛固酮合成一个系统,对维持生命活动起重要作用。醛固酮调节电解质的平衡与血压的上下,由血管紧张素n
诱导分泌。血管紧张素n又通过收缩小动脉以升高血压,从而起到调节血压的作用。当体内钠离子浓度下降时,肾素与血管紧张素水平就升高;当钠离子过量时其水平又降低。肾素,血管紧张素系统保证体内一定的钠离子浓度。如果体内调节失灵,产生过量血管紧张素与醛
固酮时,又可导致高血压。
肾素
NH2 —天冬—精—缬—酪—异亮—组—脯—苯丙—组—亮一|—缬—异亮—组……
血管紧张素原
转化酶
NH2 —天冬—精—缬—酪—异亮—组—脯—苯丙—| —组—亮一COOH
血管紧张素I
NH2 —天冬—精—缬—酪—异亮—组—脯—苯丙—COOH
血管紧张素n
血管紧张素n是强效的升高血压物。血管紧张素I本身没有升高血压作用。如果抑制血管紧张素转化酶从而限制血管紧张素n的产生,自应有降低血压作用。1965年,Ferrira
等发现拉丁美洲所产毒蛇毒汁中提取的物质可阻止狗产生实验性肾型高血压,其有效成分是
一组由9 —13个氨基酸组成的多肽物质,对血压的效应正是由于抑制了血管紧张素转化酶。这组多肽相互间有着结构上的相似性,其中替普罗肽(teprotide,SQ20881 )的氨基酸顺序为:HOH
H C H C
dioxola ne-
T
NH 2—焦谷色一脯一精一脯一谷胺一异亮一脯一脯一
它对好几种动物模型都有降压作用。
当时血管紧张素转化酶的结构尚未阐明,
但知其为一种羧肽酶, 与胰羧肽酶相似,1973
年早就发现D -苄基丁二酸是羧肽酶的竞争性抑制剂。
血管紧张素转化酶是二肽羧肽酶,
其
催化作用水解裂去 2个氨基酸。酶上的阳离子部位与酶中心的锌离子间距离比羧肽酶 A 的
相应距离多一个氨基酸残基。鉴于脯氨酸对替普罗肽所起的作用,便合成了
2-D -甲基丁
二酰脯氨酸(SQ13, 297)与2-D —甲基戊二酰脯氨酸, 实验结果抑制血管紧张素转化酶作 用远比D -苄基丁二酸强。在其分子中,羧基是与酶系统的锌离子作用的基团,
将其换为氨
基、酰氨基、胍基等,作用并不增强,但如换为巯基,可生成难以解离的硫醇锌盐,与酶的 结合更为牢固,抑制作用更强,称卡托普利(
Captopril,SQ14,225 ),又称巯甲丙脯酸。
CH 2C 6H 5
HOOC —CH2—CH 一COOH
SQ 13,297
卡托普利分子中的巯基,易与体内一些蛋白作用,从而产生皮疹等副作用。因而在卡 托普利的基础上
又开发了伊那普利( enalapril ),赖诺普利(lisinopril )等新的血管紧张素 转化酶抑制剂,作为心血管系统药物。
COOH
CH 3
I
HOOC —CH 2—CH —CO — N
CH 3
HS — CH 2—CH —CO —N
COOH
卡托普利
COOH