乙炔与格氏试剂的反应机理

乙炔与格氏试剂反应原理
塔尔图,爱沙尼亚,塔尔图大学,有机化学研究所
摘要
乙炔和溴化苯镁的反应在乙醚中进行并添加了少量三乙胺且没有催化剂条件下的反应动力学。该反应原理由Grignard等人提出,在溴化镁乙炔与Grignard试剂反应中得到补充说明。各自的速率常数已确定。三乙胺在不同程度上催化了这些反应,其中溴化镁乙炔对其催化作用最为敏感。催化剂可能的作用途径已被讨论,并强调了亲核援助的重要作用。?1999 Elsevier Science S.A.保留所有权。
关键词:格氏试剂;乙炔;动力学机制;溶剂效应
.1.介绍
乙炔与格氏试剂反应,提供了一个溴化镁乙炔和双溴镁乙炔的混合物,它可以作为一个乙炔化合物[1]的合成的中间物。由于反应进行缓慢,而且产率是由反应条件预设的,所以需要反应机理方面的知识来控制这一进程。
R C≡CMgX型格氏试剂最初是由lotsitch [2]用氢置换法制备的。格氏试剂二价镁本身也是由Iotsitch通过将乙炔通入一种可挥发的乙溴化镁溶液[3]准备的。
Grignard 等人[4]建议了一种合成双溴镁乙炔的两步反应机制:
Kleinfeller 和Lohmann [5] 研究了乙烷在乙炔和乙溴化镁的反应中的变化的动力学。双溴镁乙炔的合成的第一步反应及其进一步的歧化作用被确认了。然而,作者没能遵循足够长的时间来观察溴化镁和双溴镁产物的平衡。Jones等人[6]也质疑了平衡的出现并且建议了以下的反应机制:
在一部更早的作品[7]中,我们遵循了乙炔和溴化苯镁的反应机制,同时消耗乙炔并有苯的变化。测量是在乙醚和四氢呋喃中添加三乙胺并没有催化剂的情况下进行的。乙醚中的溴化镁苯溶液中极少量的三乙胺被发现能够很大程度地加快反应并极大地改变这一进程的运动特点。三乙胺在四氢呋喃溶液中微弱的作用表明供体溶剂合物的区别(讨论可见参考文献[8])。然而,结果的讨论基于Grignard等人提出的原理(见上)。
在此论文中,我们报告了这一反应的再次调查的结果。用了相似的实验方法。看来Grignard
的原理不得不用溴化镁乙炔与Grignard试剂反应来补充说明。
2.实验
2.1.材料
所有的操作都必须仔细地用净化的试剂和溶液在干燥的氩中进行。溴化苯镁是用传统的方式[9]来准备和分析的。
2.2.动力学测量
溴化苯镁和乙炔的反应是在20°C恒温的100毫升玻璃容器中进行的。反应池中的混合物上方有磁搅拌器、注射试剂的入口和蒸汽的采样。
反应容器由纯净的氩彻底净化。然后,用50毫升的溴化镁苯的乙醚溶液(0.9–1.1 毫升),5毫升的纯净甲苯(GLC分析的国内标准)和一定量的三乙胺(达5.6毫克)。
反应混合物被回流5分钟来取代惰性气体,然后迅速地用硅鼻中隔换掉冷凝器。到达热平衡后,乙炔气流被打开。有一个不断记录气体消耗值的自动气量计为反应池提供恒压乙炔。开始,烧瓶中的起始乙炔体积被记录下来,然后,乙炔到达饱和溶液的消耗量及其进一步的的消耗被记录下来。测量的被吸收的乙炔的体积减少到正常的情况并被转化为摩尔。为了方便动力学计算中数据的使用,消耗乙炔的摩尔量指的是反应混合物的体积(mol l-1)
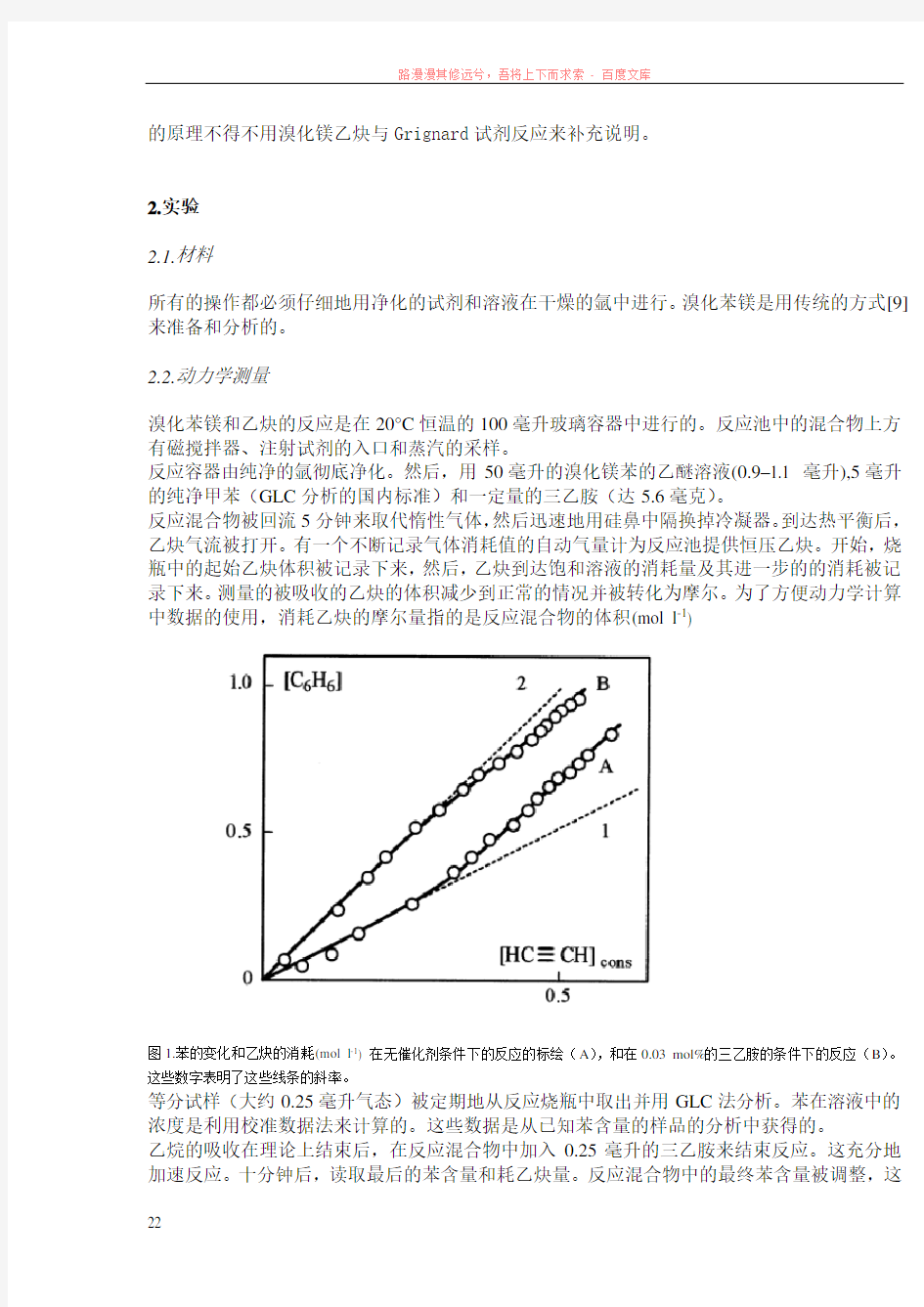
图1.苯的变化和乙炔的消耗(mol l-1) 在无催化剂条件下的反应的标绘(A),和在0.03 mol%的三乙胺的条件下的反应(B)。这些数字表明了这些线条的斜率。
等分试样(大约0.25毫升气态)被定期地从反应烧瓶中取出并用GLC法分析。苯在溶液中的浓度是利用校准数据法来计算的。这些数据是从已知苯含量的样品的分析中获得的。
乙烷的吸收在理论上结束后,在反应混合物中加入0.25毫升的三乙胺来结束反应。这充分地加速反应。十分钟后,读取最后的苯含量和耗乙炔量。反应混合物中的最终苯含量被调整,这
是由于最初的溶液中的苯浓度被当作最初的活跃格氏试剂的浓度。基本镁的酸量滴定的确定浓度在最初的试剂中超过活跃格氏浓度4–6%。
3.结果与讨论
调查乙炔与溴化苯镁在乙醚中的反应。在这一反应中,乙炔的消耗和苯的变化都被记录了下来。动力学测量也是在少量三乙胺中进行的(达9.6×10-4 mol每摩尔格氏试剂)。
研究发现,在恒定乙炔溶液浓度下,苯的变化是这个反应的第一步。乙炔的消耗似乎是更为复杂的动力学,这一过程的运动顺序在反应中不断变化着。在这种情况下,苯的变化的摩尔量和乙炔的消耗的标绘对于反应原理来说是有告知作用的。
由图一(绘图A)可见,在反应的开始阶段且没有催化剂的情况下,1摩尔苯的产生伴随着1摩尔乙炔的消耗。随着反应的进行,到了反应末尾时,2摩尔苯的产生伴随着1摩尔乙炔的消耗。除了反应的巨大加速,三乙胺的添加也改变了原理图((图.1, B)。在反应的前半阶段,消耗1摩尔乙炔产生了整整2摩尔苯。而且,绘制的图略低于直线。
表格1
乙炔与溴化镁苯在乙醚中20°C时各自的反应常数(k×104, l mol-1s-1)。
a三乙胺相对于格氏试剂的摩尔速率
b乙炔的消耗
c苯的变化
在无催化剂情况下观测到的动力学由于某种原因可以由Grignard等人提出的原理解释(见第一部分),但有三乙胺的情况与此方案不能兼容。因此,包含由Jones等人首次提议的溴化镁乙炔与格氏试剂反应是明智的。
因此,以下的反应方案是接受了验证的:
对于建议的反应方案,可由以下微分方程写出:
[A]cons代表消耗乙炔的摩尔量,被指作反应溶液的体积。[A]、[B]和[G]分别为乙炔、苯和活跃溴化苯镁的浓度。符号I 1和I 2分别表示Iotsitch 络合物,溴化镁乙炔和双溴化镁乙炔。
在这一反应中,由于乙炔在反应混合物中的浓度是恒定的(0.18 M at 20°C),微分方程可表示如下:
在这一过程的任一时间,由化学计量学我们可以得出:
因此:
且:
[B]∞代表反应末尾时苯的浓度(测定可见第二部分)
从微分方程的积分计算得到的速率常数被置于表格1中.乙炔的观测值k1与苯的变化值吻合较好,再加上数据一致,这可以得出一些有趣的结论(如下)并且确认被提出的机械方案的正确性。
三乙胺对不同反应的催化效果不同。并且,一些速率常数与催化剂浓度呈非线性关系。表格1中的速率常数是由以下方程式处理的:
TEA表示三乙胺
乙炔与溴化苯镁反应的速率常数(k1)及其与双溴化镁乙炔的速率常数(k3)随着胺浓度的增加而进行非线性增长(n﹦1, 表格2)。显然,用一种比乙醚更强的原料物质可以提高束缚于镁原子的有机半族的亲核性,从而加快其与弱酸性乙炔的反应。但是,溴化镁乙炔与溴化苯镁
(k2, n﹦1.85)或与溴化镁乙炔本(k4, n﹦1.16)身的反应的线性关系是奇特的。出人意料的结果表明
表格2
在有三乙胺参与的情况下的反映步骤的催化常数
两种试剂的良好溶剂化作用。亲核溶剂化作用在基础试剂上提高速率的作用是不证自明的,相似的溴化镁乙炔的溶剂化作用应该会降低乙炔试剂的酸度进而降低反应速率。对这个争论的一个解释强调了用炔属进行亲核援助[10]的重要性。Dessy等人[11,12]建议的四中心点过渡状态意味着对碳素酸阴离子部分对镁原子的一次亲核攻击。
亲核援助的加速效果明显地超过了酸度降低的降速效果。至于在溴化镁乙炔的双分子歧化反应中,三乙胺在速率常数为k4时的效果偏低于其在速率常数为K2时的效果这个原因并不是很清楚。显然,增长的位阻会导致原料物质溶剂化作用能力的降低。同样地,有机团的庞大会阻止原料物质和镁中心的络合。以很强的Br?nsted为基础的三乙胺对于溴化苯镁似乎是一种比乙醚更好的基础物质,但不是对于由烷基团组成的比甲基更大的格氏试剂[8,13]。我们的实验结果表明乙炔基类比苯基需要更大的空间,但是这是可疑的。相反地,假如溴化镁乙炔被更强地溶剂化了,镁原子的亲电性就会降低,因此可推测亲核援助的敏感性会降低。
致谢
此作由爱沙尼亚基金会支持(批准号3058)
参考文献
[1] L. Brandsma, H.D. Verkruijsse, Synthesis of Acetylenes, Allenes
and Cumulenes, Elsevier, Amsterdam, 1981.
[2] J. Iotsitch, Bull. Soc. Chim. Fr. 28 (3) (1902) 922.
[3] J. Iotsitch, Bull. Soc. Chim. Fr. 30 (3) (1903) 210.
[4] V. Grignard, L. Lapayre, T. Faki, Bull. Soc. Chim. Fr. 43 (4)
(1928) 931.[5] H. Kleinfeller, H. Lohmann, Ber. Deut. Chem. Ges. 71 (1938) 2608.
[6] E.R.H. Jones, L. Skattebo ¨ l, M.C. Whiting, J. Chem. Soc. (1956) 4765.
[7] (a) M. Lopp, E. Otsa, V. Pa ¨ llin, A. Tuulmets, Org. React. Tartu
13 (1976) 506. (b) M. Lopp, E. Otsa, V. Pa ¨ llin, A. Tuulmets,
Chem. Abstr. 87 (1977) 117278.
[8] (a) A. Tuulmets, Reakts. Sposobn. Org. Soedin. 11 (1974) 81. (b)
A. Tuulmets, Chem. Abstr. 82 (1975) 42803.
[9] B.J. Wake?eld, Organomagnesium Methods in Organic Synthe-
sis, Academic Press, New York, 1995.
[10] C.K. Ingold, Structure and Mechanism in Organic Chemistry,
Cornell University Press, London, 1969.
[11] R.E. Dessy, J.H. Wotiz, C.A. Hollingsworth, J. Am. Chem. Soc.
79 (1957) 358.
[12] R.E. Dessy, Y. Okuzumi, A. Chen, J. Am. Chem. Soc. 84 (1962) 2899.
[13] A. Tuulmets, D. Panov, J. Organomet. Chem. 575 (1999) 182.