药品审批流程+受理号含义+评审时间

药品审批流程
药品审批受理号
2005年以后受理号分为四部分,第一部分是申请的基本信息(四位字母),第二部份是年份(第五、六位),第三部分是流水号(第七至十一位),第四部分(十二、十三位)是受理单位标识。药品、辅料注册申请受理号共十三位,药包材注册申请受理号共十二位。
前面的四位字母意思分别是
第一位:C表示国产,J表示进口
第二位:X表示新药,Y表示已有国家标准(即仿制药)
第三位:H表示化学药品,Z表示中药,S表示生物制品,F表示辅料
第四位:L表示申请临床,S表示申请上市(即生产),B表示补充申请,Z表示再注册,F 表示分包装
如“CXHL0600001甘”表示国产新药化学药品临床申请,是2006年受理的该类型第一个注册申请,甘肃省局上报;“JYHF0600001桂”表示进口已有国家标准化学药品分包装申请,是2006年受理的该类型第一个注册申请,广西区局上报。
2005年以前受理号的大体意思是:
X:表示申请国产注册或补充(新药)
Y:表示申请国产注册或补充(已有国家标准,即仿制药)
FX:申请仿制(西药)
FZ:申请仿制(中药)
FX、FZ前面加B,即BFX或BFZ即为补充申请的意思
BFX/BFZ:申请仿制补充(西药/中药)
:申请新药证书及生产(化药)
CXL:申请新药临床研究(化药)
CXZ:申请新药试生产转正式生产(化药)
CXS、CXL、CXZ或CX后面加B即为补充的意思CZS:申请新药证书及生产(中药)CZL:申请新药临床研究(中药)
CZZ:申请新药试生产转正式生产(中药)
CZS、CZL、CZZ或CZ后面加B即为补充的意思
J:为申请进口注册或补充
B:申请进口药品补充
A:申请进口药品注册证
AS:申请进口药品注册证(生物制品)
AZ:申请进口药品注册证(中药)
H:申请进口药品注册证换发
HS:申请进口药品注册证换发(生物制品)
HZ:申请进口药品注册证换发(中药)
CSS:申请生物制品试生产转正式生产
CSL:申请生物制品临床研究
CSZ:申请生物制品试生产转正式生产
CSS、CSL、CSZ、CS后面加B即为补充的意思
药品审评时间
本文讨论的药品审评时间,指申报临床或生产的药品品种从进入药审中心到得出审评结论的时间,这其中包含了每个受理号等待审评的时间,以及技术审评所需花费的时间。
技术审评时间在《药品注册管理办法》中有规定,申报新药临床试验和生产的技术审评工作时间分别为90日、150日,若按工作日来计,分别约为4个月、7个月;160日的仿制药申请工作时间则约为7-8个月。所以,药品审评的大部分时间实际上都花费在等待时间上,该时间则视申报数量和药品审评中心的审评效率等因素来定。
选取了2011-2014年获得临床和生产批准的1.1、3.1类新药以及2013-2014年获得批准的6类仿制药的具体数据,来看一下药品审评时间的具体情况。
1.1类新药的平均审评时间(未在国内外上市销售的)
申报临床的平均审评时间为14个月,总体趋于稳定;申报生产的平均审评时间为29个月(以获得生产批件为准),2014 年审评时间增加到了42个月,远超于总体平均水平。
3.1类新药的平均审评时间(已在国外上市销售且未在国内上市销售的药品)
申报临床的平均审评时间为27个月,逐年持续增加,总体水平是1.1 类新药审评时间的近2倍;申报上市的平均审评时间为34个月,总体也呈增加趋势,且2014 年的平均审评时间与1.1类新药的审评时间一致,为42个月。
6类仿制药的平均审评时间
申报临床和申报上市的平均审评时间分别为29个月和25个月,总体均呈小幅上升趋势。
进口药品的评审时间
申请临床的审评时间为6-10个月;申请上市的审评时间则跨度较大,快则20 个月之内,慢则近24个月甚至62个月。从总历时来看,平均历时59个月,约为5年。
2020年第1批国家药品标准批件公告(
2020年第1批国家药品标准批件公告(2020)2020年1月20日 修订批件 2 辛伐他汀分散片 片剂 XGB2020-001 WS1-(X-006)-2010Z-2020 各省、自治区、直辖市食品药品监督管理局 各省、自治区、直辖市食品药品监督管理局 2020年1月20日 修订颁布件 3 藿香正气胶囊 胶囊剂 ZGB2020-9 WS3-B-2266-96-20-2020 各省、自治区、直辖市食品药品监督管理局 各省、自治区、直辖市食品药品监督管理局 2020年1月21日 修订颁布件
4 妇科养血颗粒 颗粒剂 ZGB2020-15 WS-5001-(B-0001)-2014Z-2020 各省、自治区、直辖市食品药品监督管理局各省、自治区、直辖市食品药品监督管理局2020年1月21日 修订颁布件 5 xx补肾胶囊 胶囊剂 ZGB2020-13 WS-1046(ZD-0436)-2002-2012Z-2020 各省、自治区、直辖市食品药品监督管理局各省、自治区、直辖市食品药品监督管理局2020年1月21日 修订颁布件 6 复方蛤青胶囊 胶囊剂
ZGB2020-16 YBZ18112005-2009Z-2020 各省、自治区、直辖市食品药品监督管理局各省、自治区、直辖市食品药品监督管理局2020年1月21日 修订颁布件 7 xx养血糖浆 糖浆剂 ZGB2020-17 WS3-B-2339-97-2020 各省、自治区、直辖市食品药品监督管理局各省、自治区、直辖市食品药品监督管理局2020年1月21日 修订颁布件 8 xx 丸剂 ZGB2020-8 YBZ15012005-2014Z-2020 各省、自治区、直辖市食品药品监督管理局
新药临床申请审批流程图
现在有三种申报方法 一.申报1.1类药进口(进口药) (1)方法:a.等药品在国外上市后,不在国内生产。申请分包进口(类似于代理)。 b.等药品在国外上市后,完善国内厂房技术,在国内生产。 (2)费用:临床实验37.6万,生产59.39万。 (3)难点:a.需要等药物在国外上市后才可以进行,延长周期。 b.如果在国内生产,需要完善国内厂房技术,延长周期、增加费用。 c.如果国外生产,分包进入国内时可以申请免除临床实验,但是不能确定可以申请成 功。 d.需要所有的临床资料,包括所有的实验数据。 二.申报1.1类新药临床试验(国产药) (1)方法:直接在国内申报1类新药,相关药理毒理研究资料由国外进行,提供相关证明,然后工艺研究、质量研究跟稳定性研究在国内进行,等国内药厂达到生产要求后,将药品在国内生产,按照国内药来报。 (2).费用:申请临床实验19.2万,申请生产43.2万。 (3)难点:a.需要等国内工厂具备相关生产条件后才可以进行申报。 b.无法申请避免临床。 三.申报国际多中心临床试验 (1)基本流程+费用+时间:主要临床基地伦理委员会审(费用...,时间...)+临床审批(费用37.6万,时间205天)+药品清关(费用...+时间...)(时间相对较短) (2)方法:申报程序与国内1.1类化学新药临床试验申报大致相同,但是需要更多的临床资料和
证明性文件。 (3)申请地点:国家药监局北京市西城区宣武门西大街28号大成广场3门一层(总局办公大楼 西侧),邮编100053 (4)流程图: 注:斜线前为一般审批时限,斜线后为特殊审批时限,均为工作日。 (5)申报资料列表: (一)概要部分
药品注册流程及所需资料
(二)已有国家标准的药品 《药品注册申请表》 1.综述资料 资料编号1、药品名称 资料编号2、证明性文件。 资料编号4、对主要研究结果的总结及评价。 资料编号5、药品说明书样稿、起草说明及最新参考文献。 资料编号6、包装、标签设计样稿。 2.药学研究资料 资料编号7、药学研究资料综述。 资料编号8、药材来源及鉴定依据。 资料编号12、生产工艺的研究资料及文献资料,辅料来源及质量标准。 资料编号15、药品标准草案及起草说明,并提供药品标准物质及有关资料。 资料编号16、样品检验报告书。 资料编号17、药物稳定性研究的试验资料及文献资料。 资料编号18、直接接触药品的包装材料和容器的选择依据及质量标准。 以上申报材料具体要求详见《药品注册管理办法》附件一。 (一)申报资料的一般要求: 1、申报资料按《药品注册管理办法》(国家食品药品监督管理局令第17号)附件一规定的资料顺序编号,按编号分别装订,申报资料首页为申报资料目录。 2、申报资料应使用A4纸打印,内容完整、清楚,不得涂改。 3、资料封面应包含以下信息:药品名称、资料项目编号、项目名称、申请机构联系人姓名、电话、地址,试验资料完成机构名称、主要完成人、参加人、电话、原始资料保存地点。并须加盖各机构公章。 4、资料按套装入档案袋,档案袋封面注明:申请分类、注册分类、药品名称、本袋所属第X套第X袋每套共X袋、原件/复印件、申请机构、联系人、电话。 5、注册申请报送2套完整申请资料(其中至少1套为原件)和1套综述资料(可为复印件),各袋均应包含1份申请表。 6、《药品注册申请表》:从国家食品药品监督管理局网站(https://www.360docs.net/doc/9a15044711.html,)下载,按要求填写后打印并保存,用于提交的申请表电子文件与书面申请表的数据核对码必须一致,并一并提交。 7、临床试验总结资料封面应有临床试验组长单位盖章,各临床试验分总结应有试验单位盖章。 (二)申报资料的具体要求: 1、《药品注册申请表》:临床试验完成后申请生产或仅申请新药证书,应重新填写《药品注册申请表》 该表是申请人提出药品注册申请的基本文件,同时也是药监部门对该申请进行审批的依据,其填写必须准确、规范,并符合填表说明的要求。
药品注册标准附件
附件4: 药品补充申请注册事项及申报资料要求 一、注册事项 (一)国家食品药品监督管理局审批的补充申请事项: 1.持有新药证书的药品生产企业申请该药品的批准文号。 2.使用药品商品名称。 3.增加中药的功能主治、天然药物适应症或者化学药品、生物制品国内已有批准的适应症。 4.变更用法用量或者变更适用人群范围但不改变给药途径。 5.变更药品规格。 6.变更药品处方中已有药用要求的辅料。 7.改变影响药品质量的生产工艺。 8.修改药品注册标准。 9.替代或减去国家药品标准处方中的毒性药材或处于濒危状态的药材。 10.进口药品、国内生产的注射剂、眼用制剂、气雾剂、粉雾剂、喷雾剂变更直接接触药品的包装材料或者容器;使用新型直接接触药品的包装材料或者容器。 11.申请药品组合包装。 12.新药的技术转让。 13.修订或增加中药、天然药物说明书中药理毒理、临床试验、药代动力学等项目。 14.改变进口药品注册证的登记项目,如药品名称、制药厂商名称、注册地址、药品有效期、包装规格等。 15.改变进口药品的产地。 16.改变进口药品的国外包装厂。 17.进口药品在中国国内分包装。 18.其他。 (二)省级食品药品监督管理部门批准国家食品药品监督管理局备案或国家食品药品监督管理局直接备案的进口药品补充申请事项: 19.改变国内药品生产企业名称。
20.国内药品生产企业内部改变药品生产场地。 21.变更直接接触药品的包装材料或者容器(除上述第10事项外)。 22.改变国内生产药品的有效期。 23.改变进口药品制剂所用原料药的产地。 24.变更进口药品外观,但不改变药品标准的。 25.根据国家药品标准或者国家食品药品监督管理局的要求修改进口药品说明书。 26.补充完善进口药品说明书安全性内容。 27.按规定变更进口药品包装标签。 28.改变进口药品注册代理机构。 29.其他。 (三)省级食品药品监督管理部门备案的补充申请事项: 30.根据国家药品标准或者国家食品药品监督管理局的要求修改国内生产药品说明书。 31.补充完善国内生产药品说明书安全性内容。 32.按规定变更国内生产药品包装标签。 33.变更国内生产药品的包装规格。 34.改变国内生产药品制剂的原料药产地。 35.变更国内生产药品外观,但不改变药品标准的。 36.其他。 二、申报资料项目及其说明 1.药品批准证明文件及其附件的复印件: 包括与申请事项有关的本品各种批准文件,如药品注册批件、补充申请批件、商品名批准文件、药品标准颁布件、药品标准修订批件和统一换发药品批准文号的文件、《新药证书》、《进口药品注册证》、《医药产品注册证》等。附件包括上述批件的附件,如药品标准、说明书、标签样稿及其他附件。 2.证明性文件: (1)申请人是药品生产企业的,应当提供《药品生产许可证》及其变更记录页、营业执照、《药品生产质量管理规范》认证证书复印件。申请人不是药品生产企业的,应当提供其机构合法登记证明文件的复印件。
国家药品标准
药品管理法:国务院药品监督管理部门颁布的《中华人民共和国药典》和药品标准为国家药品标准。 第一百三十六条国家药品标准,是指国家食品药品监督管理局颁布的《中华人民共和国药典》、药品注册标准和其他药品标准,其内容包括质量指标、检验方法以及生产工艺等技术要求。 药品注册标准,是指国家食品药品监督管理局批准给申请人特定药品的标准,生产该药品的药品生产企业必须执行该注册标准。 药品注册标准不得低于中国药典的规定 1.药典标准 2.卫生部中药成方制剂一至二十一册 3.卫生部化学、生化、抗生素药品第一分册 4.卫生部药品标准(二部)一册至六册; 5.卫生部药品标准藏药第一册、蒙药分册、维吾尔药分册; 6.新药转正标准1至88册(正不断更新) 7.国家药品标准化学药品地标升国标一至十六册; 8.国家中成药标准汇编内科心系分册、内科肝胆分册、内科脾胃分册、内科气血津液分册、内科肺系(一耳鼻喉皮肤科分册、经络肢体脑系分册;)、(二)分册、内科肾系分册、外科妇科分册、骨伤科分册、口腔肿瘤儿科分册、眼科 9.国家注册标准(针对某一企业的标准,但同样是国家药品标准) 10.进口药品标准 1级标准:就是《中国药典》(中华人民共和国药典),是药典委员会制定,每5 年修订一次。2010年新版中国药典分3部,一部中药标准,二部化学药标准,三部生物制剂标准。另外2010年版第一增补本自2012年10月1日起施行。2级标准:是局颁标准(国家药监局)或部颁标准(卫生部),药品标准开头字母WS是卫生部批准的,药品标准开头字母 YB是国家药监局批准的。3级标准:基本已经废除,一般是指各省、自治区、直辖市制定的中药炮制或中药饮片标准。在药
药品补充申请注册事项及申报资料要求
附件4:药品补充申请注册事项及申报资料要求一、注册事项 (一)国家食品药品监督管理局审批的补充申请事项: 1、持有新药证书的药品生产企业申请该药品的批准文号。 2、使用药品商品名称。 3、增加中药的功能主治或者化学药品、生物制品国内已有批准的适应症。 4、变更用法用量或者变更适用人群范围但不改变给药途径。 5、变更药品规格。 6、变更药品处方中已有药用要求的辅料。 7、改变影响药品质量的生产工艺。 8、修改药品注册标准。 9、替代或减去国家药品标准处方中的毒性药材或处于濒危状态的药材。 10、进口药品、国内生产的注射剂、眼用制剂、气雾剂、粉雾剂、喷雾剂变更直接接触药品的包装材料或者容器;使用新型直接接触药品的包装材料或者容器。 11、申请药品组合包装。 12、新药的技术转让。 13、修订或增加中药、天然药物说明书中药理毒理、临床试验、药代动力学等项目。 14、改变进口药品注册证的登记项目,如药品名称、制药厂商名称、注册地址、药品有效期、包装规格等。 15、改变进口药品的产地。 16、改变进口药品的国外包装厂。 17、进口药品在中国国内分包装。 18、其它 (二)省级食品药品监督管理部门批准国家食品药品监督管理局备案或国家食品药品监督管理局直接备案的进口药品补充申请事项: 19、改变国内药品生产企业名称。 20、国内药品生产企业内部改变药品生产场地。 21、变更直接接触药品的包装材料或者容器(除上述第10事项外)。 22、改变国内生产药品的有效期。 23、改变进口药品制剂所用原料药的产地。 24、变更进口药品外观,但不改变药品标准的。 25、根据国家药品标准或者国家食品药品监督管理局的要求修改进口药品说明书。 26、补充完善进口药品说明书安全性内容。 27、按规定变更进口药品包装标签。
新药申报与审批程序
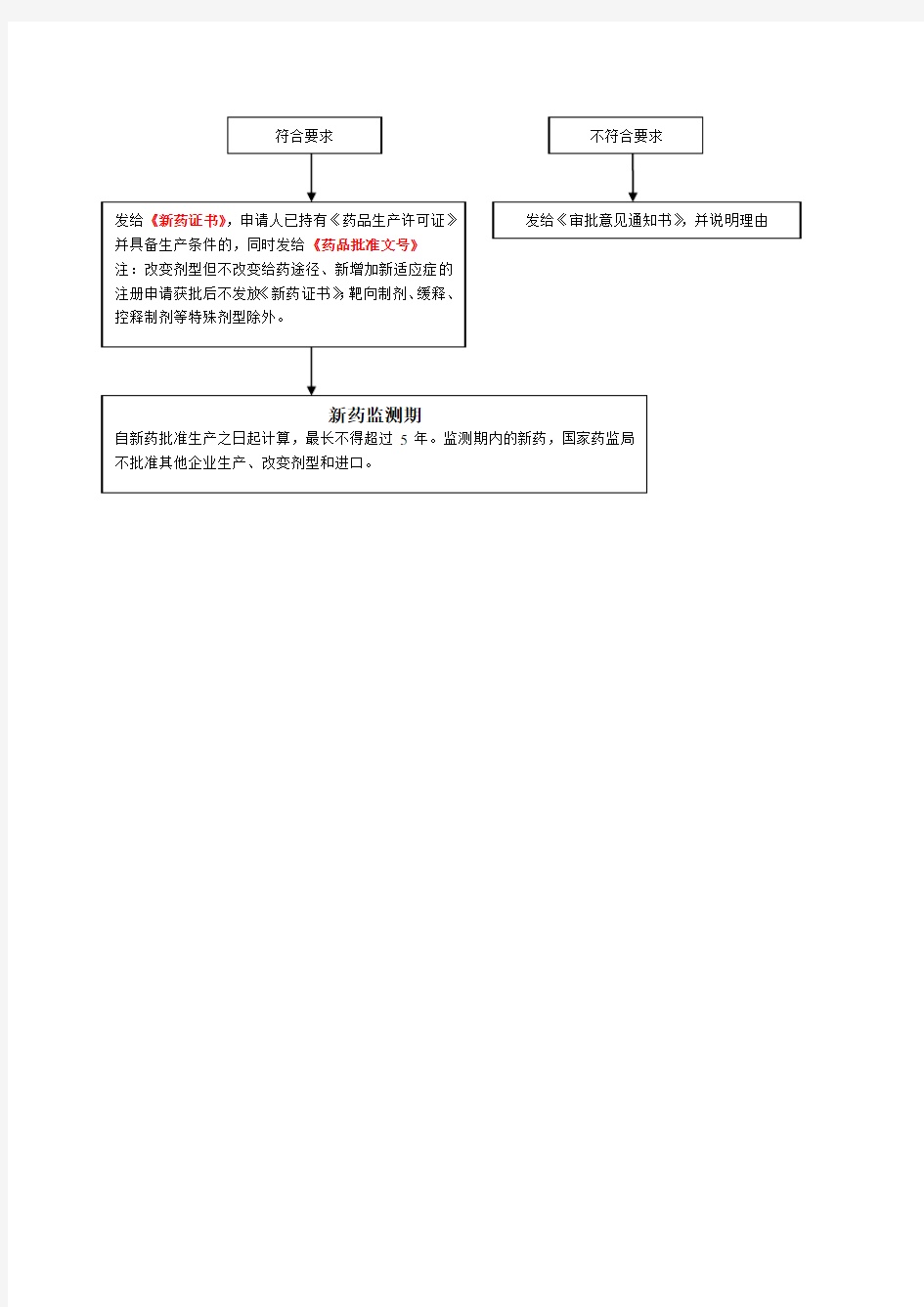
按照《新药审批办法》第五章的规定:“新药的申报与审批分为临床研究和生产上市两个阶段。初审由省级药品监督管理部门负责,县市由国家药品监督管理局负责。” 现结合图1新药申报与审批基本程序示意图介绍该程 1.申报单位填写新药临床研究(或生产)申请表,连同申报的技术资料和样品报省、自治区、直辖市药品监督管理部门。省级药品监督管理部门进行初审,即对新药的各项原始资料是否齐全进行审查;同时,派员对试制条件进行实地考察,填写考察报告表。对已报齐所有应报资料的,正式通知申报单位收审;同时将样品和技术资料转省、自治区、直辖市药品检验所审核。对应报资料不全的,予以退审,将申请表和资料退回申报单位并提出退审理由。 2.省、自治区、直辖市药品检验所按新药审批各项技术要求完成对申报资料的审查和样品的检验。 药检所的审核系指对新药的药学(包括药理、毒理)研究资料进行审查和对样品进行实验检验;不包括为申报单位进行新的检测方法的研究。药检所审核完毕后,提出质量标准和对药学(包括药理、毒理)方面的综合审查意见,送省级药品监督管理部门。 3.省级药品监督管理部门初审通过同意上报的,在新药临床研究(或生产)申请表签署意见,连同申报的技术资料一式5份报国家药品监督管理局注册司进行形式审查。 3’.新生物制品和按《新药审批办法》第二十六条所列新药,由申报单位填写申请表,连同申报的技术资料一式五份直接报国家药品监督管理局注册司。样品检验和质量标准复核由中国药品生物制品检定所负责。4.国家药品监督管理局注册司经形式审查合格的,向申报单位发出收取审评费的通知。同时交药品审评中心安排技术审查、审评委员会审评及必要的复核等工作。 4.形式审查不合格的,予以退审。 5.技术审评通过后,将建议批准的或退审的审评报告及意见,报国家药品监督管理局药品注册司。
注册变更国家局
药品补充申请注册事项及申报资料要求 一、注册事项 (一)国家食品药品监督管理局审批的补充申请事项: 1.持有新药证书的药品生产企业申请该药品的批准文号。 2.使用药品商品名称。 3.增加中药的功能主治、天然药物适应症或者化学药品、生物制品国内已有批准的适应症。 4.变更用法用量或者变更适用人群范围但不改变给药途径。 5.变更药品规格。 6.变更药品处方中已有药用要求的辅料。 7.改变影响药品质量的生产工艺。 8.修改药品注册标准。 9.替代或减去国家药品标准处方中的毒性药材或处于濒危状态的药材。 10.进口药品、国内生产的注射剂、眼用制剂、气雾剂、粉雾剂、喷雾剂变更直接接触药品的包装材料或者容器;使用新型直接接触药品的包装材料或者容器。 11.申请药品组合包装。 12.新药的技术转让。 13.修订或增加中药、天然药物说明书中药理毒理、临床试验、药代动力学等项目。 14.改变进口药品注册证的登记项目,如药品名称、制药厂商名称、注册地址、药品有效期、包装规格等。 15.改变进口药品的产地。 16.改变进口药品的国外包装厂。 17.进口药品在中国国内分包装。 18.其他。 (二)省级食品药品监督管理部门批准国家食品药品监督管理局备案或国家食品药品监督管理局直接备案的进口药品补充申请事项: 19.改变国内药品生产企业名称。 20.国内药品生产企业内部改变药品生产场地。 21.变更直接接触药品的包装材料或者容器(除上述第10事项外)。 22.改变国内生产药品的有效期。 23.改变进口药品制剂所用原料药的产地。 24.变更进口药品外观,但不改变药品标准的。 25.根据国家药品标准或者国家食品药品监督管理局的要求修改进口药品说明书。
新药准入审批制度流程
新药准入审批制度 一、新药审批会议制度 1、药事管理委员会每年度召开1--2次会议,审批新药申请和老药淘汰,参加会议人数须超过应到人数的一半以上。 2、药剂科将初选合格的申请表整理汇总,编制"药事管理委员会讨论药品目录",标明每个品种的商品名、化学名、剂型、规格、报销属性、申请科室、主要用途、生产厂家、参考价格、批准文号、现有同类产品等属性。 3、参会人员每人1份目录,听取申请科室委员对药品有关内容介绍,经提出讨论意见后,在目录上无记名投票。 4、得票超过参会人数半数者为批准购入和淘汰药品,但原则上应控制每次增加和淘汰品种超过30﹪个。 5、药事管理委员会可根据申请情况确定是否请申请人到会答辩。 二、新药申请及审批程序 (一)临床科室申请 1、凡申请购入医院从未使用过,或因各种原因停用半年以上的药品,均应由科室申请,新药还包括不同剂型或不同规格的品种。 2、专科用药须由相应的专科申请,一般情况下西药由西医科室申请,中药由中医科室申请,专科用药由专科科室申请。 3、申请医师须具备主治医师以上职称,申请购入的药品须经全科讨论,科主任签字后附有关资料交药剂科。所附资料包括由生产厂家提供的新药证书、药品说明书、GMP证书、药检报告、药价批单、临床研究报告以及其他用以证明该药优势的文件资料等。
4、申请表中各项内容应填写完整,剂型、类别栏目中应注明是否为何地医保药品及国产、进口、合资等属性。 5、科室主任应对所申请药品用量负责,购入后造成积压浪费由申请科室承担责任,药剂科定期报告用药情况。 (二)医务部审核并签署意见 1、医务部审核申请表及所附资料,内容不全的申请无效。 2、符合下列条件的品种方可提交药事管理委员会审批: (1)本院尚未购入使用的新成分的药品。 (2)注射剂必须是GMP产品。 (3)已有同成分进口产品,因价格因素可申请一种国产药并存(国产药品价格须便宜30%以上)。 (4)现有品种为自费药品时,可申请同种医保用药品种(例如:注射剂自费-口服剂型医保;进口药自费-国产药医保)。 (5)同成分品种,可在质量相同的情况下,以价格低取代价格高的品种。 (6)不同科室申请由不同厂家生产的同一成分药品时,医务部和药剂科共同只认可一份申请。 3、按上述条件初审合格后,医务部主任签署意见。 (三)药剂科审查资料并编制"药事管理委员会讨论药品目录" 1、药剂科和监审科对新药证书、药检报告、药价批单、药品说明书、GMP证书等资料的有效性进行审核,如有疑义可退回申请。 2、按照申请科室汇总,编制"药事管理委员会讨论药品目录",内容包括每个申请品种的商品名、化学名、剂型、规格、报销属性、申请科室、主要用途、生产厂家、参考价格、批准文号、现有同类产品等属性、。
药品注册分类及注册流程
目录 1. 化学药品注册分类 2. 境内申请人新药申报流程 3. 化学药品申报资料要求 4. 化学药品临床试验要求
化学药品注册分类 1.未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2.改变给药途径且尚未在国内外上市销售的制剂。 3.已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4.改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5.改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6.已有国家药品标准的原料药或者制剂。 以上,属注册分类1~5类按新药申报程序申请注册,6类按仿制药申请程序申请注册。
境内申请人,新药申报流程(以上注册分类中1~5类申报流程) 准备报临床申报资料(具体资料项目要求附后) 向省食品药品监督管理局报送申请资料 拿到受理号,相关进度, 便可以从SFDA网站上 查询https://www.360docs.net/doc/9a15044711.html, 省局,受理,5工作日内组织对药物研制情况及原始资料进行现场核查;30工作日内完成现场核查,将初审意见,《药品注册研制现场核查报告》,申报资料送交 国家食品药品监督管理局药品审评中心(CDE) 审评进度,审评人员名 单及联系方式可以从 CDE网站查询。 https://www.360docs.net/doc/9a15044711.html, CDE对申报资料进行技术审评(90工作日) 如果必要,CDE将要求申请人补充资料
药品审批
某药业研制一个已在国内上市销售的进口化学药制剂,其原料药未进口,注册时需要提交哪些研究资料?试提出加快药品审批进度的建议。 从题目“某药业研制一个已在国内上市销售的进口化学药制剂,其原料药未进口,注册时需要提交哪些研究资料?”可知该药品在注册分类里属于第6类——已有国家药品标准的原料药或制剂。在注册时需要申报的资料项目包括综述资料、药学研究资料、药理毒理研究资料以及临床试验资料。 首先,每个注册药品都要提交综述资料,其中包括:药品名称、证明性文件、立题目的与依据、对主要研究结果的总结及评价、药品说明书样稿、起草说明及最新参考文献包装和标签设计样稿。 制剂处方及工艺的研究资料及文献资料(单独申请注册药物制剂,必须提供原料药的合法来源证明文件,一式2 份,分别放入资料项目2 的资料和资料项目13 号的资料中。使用国产原料药的申请人,应当提供该原料药的药品批准证明文件、检验报告书、药品标准、原料药生产企业的营业执照、《药品生产许可证》、《药品生产质量管理规范》认证证书、与该原料药生产企业签订的供货协议、销售发票等的复印件。使用进口原料药的,应当提供与该原料药生产企业或国内合法的销售代理商签订的供货协议、《进口药品注册证》或者《医药产品注册证》、口岸药品检验所检验报告书、药品标准复印件等。药品注册过程中,研制制剂所用的进口原料药未取得《进口药品注册证》或者《医药产品注册证》的,必须经国家食品药品监督管理局批准。)提供资料包括确证化学结构或者或组份的试验资料及文献资料、质量研究工作的试验资料及文献资料、药品标准草案及起草说明、样品的检验报告书、辅料的来源及质量标准、药物稳定性研究的试验资料及文献资料和直接接触药品的包装材料和容器的选择依据及质量标准。 药理毒理资料包括药理毒理研究资料项下的16项药理毒理研究资料综述,以及说明项下第17项——局部用药除按所属注册分类及项目报送相应资料外,应当报送资料项目21,必要时应当进行局部吸收试验。资料项目21为过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等特殊安全性试验资料和文献资料。 关于临床试验资料部分,除国内外相关的临床试验资料综述以外,对于注册
新药注册特殊审批管理规定
新药注册特殊审批管理规定 第一条为鼓励研究创制新药,有效控制风险,根据《药品注册管理办法》,制定本规定。 第二条根据《药品注册管理办法》第四十五条的规定,国家食品药品监督管理局对符合下列情形的新药注册申请实行特殊审批: (一)未在国上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂,新发现的药材及其制剂; (二)未在国外获准上市的化学原料药及其制剂、生物制品; (三)治疗艾滋病、恶性肿瘤、罕见病等疾病且具有明显临床治疗优势的新药; (四)治疗尚无有效治疗手段的疾病的新药。 主治病证未在国家批准的中成药【功能主治】中收载的新药,可以视为尚无有效治疗手段的疾病的新药。 属于(一)、(二)项情形的,药品注册申请人(以下简称申请人)可以在提交新药临床试验申请时提出特殊审批的申请。 属于(三)、(四)项情形的,申请人在申报生产时方可提出特殊审批的申请。 第三条国家食品药品监督管理局根据申请人的申请,对经审查确定符合本规定第二条情形的注册申请,在注册过程中予以优先办理,并加强与申请人的沟通交流。 第四条申请人申请特殊审批,应填写《新药注册特殊审批申请表》(附件1),并提交相关资料。 《新药注册特殊审批申请表》和相关资料应单独立卷,与《药品注册管理办法》规定的申报资料一并报送药品注册受理部门。 第五条药品注册受理部门受理后,应将特殊审批申请的相关资料随注册申报资料一并送交国家食品药品监督管理局药品审评中心。 第六条国家食品药品监督管理局药品审评中心负责对特殊审批申请组织审查确定,并将审查结果告知申请人,同时在国家食品药品监督管理局药品审评中心上予以公布。 (一)属于本规定第二条(一)、(二)项情形的,国家食品药品监督管理局药品审评中心应在收到特殊审批申请后5日进行审查确定; (二)属于本规定第二条(三)、(四)项情形的,国家食品药品监督管理局药品审评中心应在收到特殊审批申请后20日组织专家会议进行审查确定。 特殊审批申请的审查确定时间包含在《药品注册管理办法》规定的技术审评工作时间。 第七条国家食品药品监督管理局药品审评中心对获准实行特殊审批的注册申请,按照相应的技术审评程序及要求开展工作。负责现场核查、检验的部门对获准实行特殊审批的注册申请予以优先办理。 第八条获准实行特殊审批的注册申请,申请人除可以按照国家食品药品监督管理局药品审评中心的要求补充资料外,还可以对下列情形补充新的技术资料: (一)新发现的重大安全性信息; (二)根据审评会议要求准备的资料; (三)沟通交流所需的资料。
国家药品标准物质管理办法
国家药品标准物质管理办法 第一条为进一步规范国家药品标准物质的管理,根据《中华人民共和国药品管理法》、《药品注册管理办法》及《体外诊断试剂注册管理办法(试行)》的相关规定,制定本办法。 第二条国家药品标准物质的标定、审核、批准以及国家药品标准物质的对外供应,应遵守本办法。 第三条国家药品标准物质是指供国家药品标准中物理和化学测试及生物方法试验用,具有确定特性量值,用于校准设备、评价测量方法或者给供试药品赋值的材料或物质。 国家药品标准物质分为生物标准品、生物参考品、化学对照品、对照药材/对照提取物、医疗器械对照材料和标准品、体外诊断试剂标准品、药用辅料对照品及药包材对照物质等。 第四条中国食品药品检定研究院(以下简称中检院)负责对标定的药品标准物质从原材料选择、制备方法、标定方法、标定结果、定值准确性、量值溯源、稳定性、分装与包装条件、成本等进行全面审核,并作出可否作为国家药品标准物质的结论。 第五条中检院设立标准物质管理处,负责国家药品标准物质工作的综合、组织、协调和管理,中检院设立国家药品标准物质委员会,负责国家药品标准物质全面技术审核,并负责相关技术指导规范的审定。
第六条标准物质管理处负责组织药品注册申请与药品标准修订中新增的药品标准物质原料的申报与受理,申报的药品标准物质原料及其研究资料,应符合《国家药品标准物质申报、备案办法》及相关类别药品标准物质原料的要求。 第七条国家药品标准物质的原材料应满足国家药品标准的要求,由标准物质管理处负责组织相关品种原料的制备或采购。对于特殊来源要求的药品标准物质,可通过采取自行制备、向有关机构或特定人群收集的方式,获得符合国家药品标准要求的原材料。对于药品标准物质原材料供应不足或难以获得的品种,应由标准物质管理处汇总后报送相关单位,并提请国家食品药品监督管理局协调解决。 第八条国家药品标准物质的标定及定值方法,应遵照国家药品标准、中检院发布的《国家药品标准物质技术规范》以及具体类别标准物质定值方法的要求。 第九条标准物质管理处可以组织有能力的药品检验所、研究机构和生产企业等单位协作标定国家药品标准物质。 第十条参照国际惯例,国家药品标准物质不规定有效期,但应在规定的储存和使用条件下,定期进行特性量值的稳定性核查。 第十一条国家药品标准物质更换批号、停止使用及撤销的品种,需经国家药品标准物质委员会批准后,标准物质管理处及时向社会公布。
药品审批流程+受理号含义+评审时间
药品审批流程
2005年以后受理号分为四部分,第一部分是申请的基本信息(四位字母),第二部份是年份(第五、六位),第三部分是流水号(第七至十一位),第四部分(十二、十三位)是受理单位标识。药品、辅料注册申请受理号共十三位,药包材注册申请受理号共十二位。 前面的四位字母意思分别是 第一位:C表示国产,J表示进口 第二位:X表示新药,Y表示已有国家标准(即仿制药) 第三位:H表示化学药品,Z表示中药,S表示生物制品,F表示辅料 第四位:L表示申请临床,S表示申请上市(即生产),B表示补充申请,Z表示再注册,F 表示分包装 如“CXHL0600001甘”表示国产新药化学药品临床申请,是2006年受理的该类型第一个注册申请,甘肃省局上报;“JYHF0600001桂”表示进口已有国家标准化学药品分包装申请,是2006年受理的该类型第一个注册申请,广西区局上报。 2005年以前受理号的大体意思是: X:表示申请国产注册或补充(新药) Y:表示申请国产注册或补充(已有国家标准,即仿制药) FX:申请仿制(西药) FZ:申请仿制(中药) FX、FZ前面加B,即BFX或BFZ即为补充申请的意思 BFX/BFZ:申请仿制补充(西药/中药) :申请新药证书及生产(化药) CXL:申请新药临床研究(化药) CXZ:申请新药试生产转正式生产(化药) CXS、CXL、CXZ或CX后面加B即为补充的意思CZS:申请新药证书及生产(中药)CZL:申请新药临床研究(中药) CZZ:申请新药试生产转正式生产(中药) CZS、CZL、CZZ或CZ后面加B即为补充的意思 J:为申请进口注册或补充 B:申请进口药品补充 A:申请进口药品注册证 AS:申请进口药品注册证(生物制品) AZ:申请进口药品注册证(中药) H:申请进口药品注册证换发 HS:申请进口药品注册证换发(生物制品) HZ:申请进口药品注册证换发(中药) CSS:申请生物制品试生产转正式生产 CSL:申请生物制品临床研究 CSZ:申请生物制品试生产转正式生产 CSS、CSL、CSZ、CS后面加B即为补充的意思
药品注册申报流程
药品申报流程流程 一、国外已上市品种: 1 直接进口制剂 以国外公司名义申请制剂的进口注册证,注册工作由国外公司在国内的办事处或委托的代理机构办理。国内公司仅作为其经销商。此类情况仅是商业层面上的合作,国内公司不以自己名义进行药品注册申报,当然也可作为国外公司的代理机构,受其委托,代为办理相关注册事宜。 申请进口药品注册证,按化学药品注册分类3提交申报资料,如已在国内进行多中心临床试验,可直接申请药品进口注册证,自提交申报资料受理之日起计算,其总的流程约为12个月,主要经过的部门有:国家药监局受理中心、药品审评中心、中检所、国家药监局药品注册司化药处、陕西药监局注册处。 进口药品注册(无需临床试验)的申报流程图 如所申请品种未在中国进行临床试验,则需按化学药品注册分类3要求进行临床前申报,申请药物临床试验批件,流程及所经部门与申请药品进口注册证相
同。 根据化学药品注册分类3相关临床要求实施临床试验后,按分类3提交申请生产申报资料,申请药品进口注册证。流程及所经部门与申请药品进口注册证相同。 进口药品注册(临床批件)的申办流程图 进口药品注册(完成临床药品进口注册证)的申办流程图
2 进口制剂分装 以国外公司名义申请大包装制剂进口,然后国内分装公司以自己名义按进口制剂分装相关规定申请制剂分装。申请流程及所经部门与直接进口制剂相同。3. 进口原料药,在国内加工制剂 原料药由国外公司申请进口注册证,.制剂由国内加工企业按国内品种申请药品注册证及生产批文,具体注册类别根据国内上市状态按注册管理办法相关规定划分(一般为3类或6类)。 原料药的进品注册申请按进口药品注册(临床批件)申请流程进行申报。制剂按国产药品化学药品注册分类3或注册分类6进行申报。具体流程及所经部门参照新药临床试验申报流程(1-5类)、新药生产申请流程(1-5类)及仿制药申报流程(6类)进行。 4. 3类新药(即仿国外) 按化学药品注册分类3的申报流程进行申报,目前我公司新药“吡非尼酮”
药品标准
第三节药品标准 一、药品标准概述 1.药品标准是指对药品的质量指标、生产工艺和检验方法所作的技术要求和规定,内容包括药品的名称、成分或处方的组成;含量及其检查、检验方法;制剂的辅料;允许的杂质及其限量要求以及药品的作用、用途、用法、用量;注意事项;贮藏方法等。中药材、中成药、化学原料药及其制剂,生物制品等应根据各自的特点设置不同的项目。 2.国家药品标准《药品管理法》规定,国务院药品监督管理部门颁布的《中国药典》和药品标准为国家药品标准。 国家药品标准包括《中国药典》及增补本,经国家食品药品监督管理局批准的药品注册标准和颁布的其他药品标准,以及与药品质量指标、生产工艺和检验方法相关的技术指导原则和规范。 二、药品标准的分类 依据《药品管理法》规定,我国的药品标准分为国家药品标准和炮制规范。 1.国家药品标准分类《中国药典》、国家食品药品监督管理局颁布的药品标准和药品注册标准。 (1)《中国药典》由国家药典委员会编纂,国家食品药品监督管理局颁布。《中国药典》是国家药品标准的核心,是国家为保证药品质量、保护人民用药安全有效而制定的法典。 《中国药典》于1953年编纂出版第一版以后,相继于1963年、1977年分别编纂出版。从1985年起每5年修订颁布新版药典,现行版为2010年版《中国药典》。 2010年版《中国药典》是新中国成立以来第九版药典,本版药典收载品种总计4567个,与2005年版《中国药典》相比新增品种1386个;一部收载药材及饮片、植物油脂和提取物、成方和单味制剂共2165个,其中新增1019个,修订634个;二部收载化学药品、抗生素、生化药品、放射性药品及药用辅料共2271个,其中新增330个,修订1500个;三部收载生物制品131个品种,其中新增37个,修订94个;药典附录新增47个,修订154个。药用辅料标准新增130多种。 (2)国家食品药品监督管理局颁布的药品标准这类药品标准是指未列入《中国药典》而由国家食品药品监督管理局颁布的药品标准,以及与药品质量指标、生产工艺和检验方法相关的技术指导原则和规范。 (3)药品注册标准是指国家食品药品监督管理局批准经申请人特定药品的标准,生产该药品的生产企业必须执行该注册标准。 根据《标准化法》规定和国际惯例,国家标准是市场准入的最低标准,原则上行业标准高于国家标准,企业标准应高于行业标准。所以,药品注册标准不得
新药审批流程与审批时间
中国创新药咨询与服务先锋CRO 新药审批流程及审批时间
一.国家、省级药品监督管理局审批职责 国家药品监督管理局主管全国药品注册管理工作。国家药品监督管理局药品注册司(以下称药品注册司)代表国家药品监督管理局受理药品注册申请、下达审评任务、批准药品注册、办理发证事宜,并对药品注册工作实施监督管理。各省、自治区、直辖市药品监督管理部门受理辖区内新药、仿制药品的申请,并按规定完成初审工作(国家另有规定的除外)。 二.技术审评职责 国家药品监督管理局药品审评中心(以下称药品审评中心)是国家药品监督管理局药品注册的技术审评机构,在药品注册司的领导下进行新药、进口药品、仿制药品的技术审评工作,提出审评综合意见,上报药品注册司审核。国家药品监督管理局药品审评专家负责提出新药(含新生物制品)、进口药品、仿制药品技术审评咨询意见。审评会由国家药品监督管理局从国家药品审评专家库中随机遴选专家召开。中国药品生物制品检定所负责一类新药和新生物制品的实验室技术复核。省级药品检验所负责辖区内的二至五类新药、仿制药品实验室技术复核。口岸药品检验所负责进口药品的实验室技术复核。 三.初审程序 经省级药品监督管理部门初审的药品申请人应填写申请表,连同要求申报的技术资料和样品报省级药品监督管理部门。省级药品监督管理部门应根据有关审批管理规定,对申报资料进行审查。申报资料不全的,退还申请人;申报齐全的,填写正式受理通知书,同时安排省级药品检验所进行实验室技术复核;在申请人交纳审评费后,进行以下审核工作:对原始资料的真实性、完整性、规范性进行审核;对研制现场进行考察;组织技术审评并提出初审意见。符合要求者,由省级药品监督管理部门报药品注册司,不符合要求的补充资料或作出退审决定。凡初审时要求补充资料的,申请人应在完成相应工作后将补充资料报省级药品监督管理部门审核,通过者方可报药品注册司。省级药品监督管理部门新药的初审审评方式结合本地实际,并参照本工作程序自行制定,报药品注册司备案。
关于在药品包装标签上印制说明书二维码的指导意见
关于在药品包装标签上印制说明书二维码的指导意见 我局为进一步落实国家药品监督管理局《药品说明书和标签管理规定》的有关要求,根据国家相关法律法规,结合我省实际,制定此指导意见。 一、适用范围 (一)药品说明书文字内容多,因客观原因导致纸张尺寸小,说明书字号小于5号标准字号时,企业应以药品说明书二维码方式在药品包装上印刷,并以药品补充申请形式向省局提出备案; (二)药品说明书字号大于等于5号标准字号,企业可根据需要在药品包装上印制药品说明书二维码,并以药品补充申请形式向省局提出备案。 二、申报形式及资料 (一)根据国家药品标准或国家药监局的要求修改国内生产药品说明书和按规定变更国内生产药品包装标签的备案事项,申报资料如下: 1、药品批准证明文件及其附件:包括与申请事项有关的本品各种批准文件,如药品注册批件、补充申请批件、商品名称批准文件、药品标准颁布件、药品标准修订批准和统一制发药品批准文号的文件、《新药证书》、《进口药品注册证》、《医药产品注册证》等。
附件包括上述批件的附件,如药品质量标准、说明书、标签样稿及其他附件。 2、修订的药品说明书样稿,并附详细修订说明。 3、修订药品标签样稿,在左下角预留二维码印制位置,并附详细修订说明。 4、提供药品处方、生产工艺、质量标准。 5、生产药品制剂所用原料药的来源。改变原料药来源的,应当提供批准证明文件。 6、提供新的国家药品标准或者国家药品监督管理局要求修改说明书的文件。 7、提供新修订说明书的w o r d版,字号不得小于5号。 (二)补充完善国内生产药品说明书安全性内容和按规定变更国内生产药品包装标签的备案事项,提供如下资料: 1、药品批准证明文件及其附件:包括与申请事项有关的本品各种批准文件,如药品注册批件、补充申请批件、商品名称批准文件、药品标准颁布件、药品标准修订批准和统一制发药品批准文号的文件、《新药证书》、《进口药品注册证》、《医药产品注册证》等。附件包括上述批件的附件,如药品质量标准、说明书、标签样稿及其他附件。 2、修订的药品说明书样稿,并附详细修订说明。 3、修订药品标签样稿,在左下角预留二维码印制位置,并附详细修订说明。
药品注册资料整理要求及注册程序
药品注册申请程序及资料整理要求 申报资料整理要求 申报资料完成后课题组应向注册部提供1套原件,2+1套资料袋。其中1套原件交省局(注册部负责更换资料),注册申请表、研制情况申报表各5份(公司公章由注册部统一盖), 应同时提供注册申请表的电子版,注意申请表应为RVT格式。 补正资料通知下达后,需补正的资料请交1套给注册部,其余6+1套资料课题组自行补正,寄送资料1周前注册部通知课题组提交全部资料(包括纸质资料和全部资料的pdf文件),由注册部负责替换、存档。 补充申请:国家审批的,提供资料3套,申请表5份。省局审批的,提供资料1套,申请表5份。 化学药品6类:申报时应提供药品注册申请表、药品研制情况申报表、药品生产现场检查申请表(生产时间应安排在递送资料2周后)各5份。应提供单独的生产工艺规程,以便进行生产现场检查。生物等效性试验完成后,填写研制情况申报表(仅需填写临床信息),提交5套临床资料(其中3套要求完整图谱)。 申报生产时,要对临床批件中所提意见进行答复,答复意见放在资料最上方。 6类药的化学名称应与所仿制的品种完全一致(以申报时国家局网站公布的名称为准)。 注射剂不需29、30号资料。其他制剂需准备生物等效性临床方案。 复方固体制剂:主要活性成分按等比例增减的,辅料种类不改变,按照不同规格进行申报;主要活性成分不等比例的,按照不同品种进行申报。 注册申请表的填写: 一个规格填写一份注册申请表,研制情况申请表也是一个规格一份表。不同规格可只准备同一套资料。 原料为化药3类,制剂为化药6类的,申请表中原料的申报阶段为生产。 如需减免临床,资料3中应提供依据。 如需特殊审批,应填写特殊审批申请表,并按照《药品注册管理办法》中相关规定准备单独成卷资料。申报化学1类药和肿瘤、艾滋治疗药物均需申请特殊审批。 申请表15项应将同期申报的原料和其它规格的情况填写清楚。如申报品种为改变酸根、碱基的,前期已申报同类品种的,也应在15项填写。 16项包材应按照YBB标准名称填写完整。 19项所用辅料有型号规格区分的,应在表格中注明 28项中,生产地址应与生产许可证中登记的地址完全一致。 33项应填写所有委托试验的研究机构,请勿遗漏。 研制情况申请报表的填写:(请务必区分临床前用表和临床后用表) 表中研究地点应填写具体地址(**市**路**号**幢); 药学研究注明样品检验机构(单独一行)
国家食品药品监督管理局国家药品标准
国家食品药品监督管理局 国家药品标准 -B-3464-98-2009 WS 3 追风透骨丸 Zhuifeng Tougu Wan 【处方】制川乌白芷制草乌香附(制) 甘草白术(炒)没药(制)麻黄 川芎乳香(制)秦艽地龙 当归茯苓赤小豆羌活 天麻赤芍细辛防风 天南星(制)桂枝甘松 【性状】本品为红褐色的水蜜丸,除去包衣后显褐棕色至黑棕色;气微香,味苦。 【鉴别】(1)取本品,置显微镜下观察:不规则分枝状团块无色,遇水合氯醛液渐溶化;菌丝无色或淡棕色,直径4~6μm(茯苓)。气孔特异,侧面观保卫细胞呈哑铃状(麻黄)。种皮柵状细胞成片,红色,侧观细胞狭长,长约50μm,细胞壁上部有细纵沟纹,胞腔明显,内含红棕色物;表面观呈类多角形,胞腔小,孔沟细密(赤小豆)。草酸钙簇晶直径7~38μm,散在或存在于薄壁细胞中,常数个纵向排列成行(赤芍)。纤维成束,周围薄壁细胞含草酸钙方晶,形成晶纤维(甘草)。分泌细胞呈类圆形,直径35~72μm,内含淡黄棕色至红棕色分泌物,其周围5~8个薄壁细胞作放射状环列(香附)。斜纹肌纤维无色,散在或相互绞结,弯曲或稍平直,直径4~26μm (地龙)。 (2)取本品15g,研细,加1%盐酸溶液50ml,加热回流1小时,滤过,滤液用乙醚振摇提取2次,每次40ml,酸水液备用,合并乙醚提取液,低温蒸干,残渣加无水乙醇2ml使溶解,作为供试品溶液。另取羌活对照药材0.4g,同法制成对照药材溶液。照薄层色谱法(附录Ⅵ B)试验,吸取上述两种溶液各2μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以正己烷-乙酸乙酯(1:1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。 (3)取[鉴别](2)项下备用的酸水液,用浓氨试液调节pH值至10,用三氯甲烷振摇提取3次,每次30ml,合并三氯甲烷液,蒸干,残渣加无水乙醇1ml使溶解,作为供试品溶液。另取盐酸麻黄碱对照品,加无水乙醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(附录ⅥB)试验,吸取供试品溶液10μl、对照品溶液2μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-浓氨试液(4:1:0.1)为展开剂,展开,取出,晾干,喷以茚三酮试液,于105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。 (4)取本品5g,研细,加乙醚30ml,密塞,超声处理10分钟,取出,放冷,滤过,,滤液挥干,残渣加乙醇1ml使溶解,作为供试品溶液。另取川芎对照药材、当归对照药材各0.5g,分别加乙醚20ml,密塞,超声处理10分钟,取出,放冷,滤过,,滤液挥干,残渣分别加乙醇1ml使溶解,作为对照药材溶液。照薄层色谱法(附录ⅥB)试验,吸取上述三种溶液各5μl,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯(6:1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)