蛋白质分析相关数据库及网站
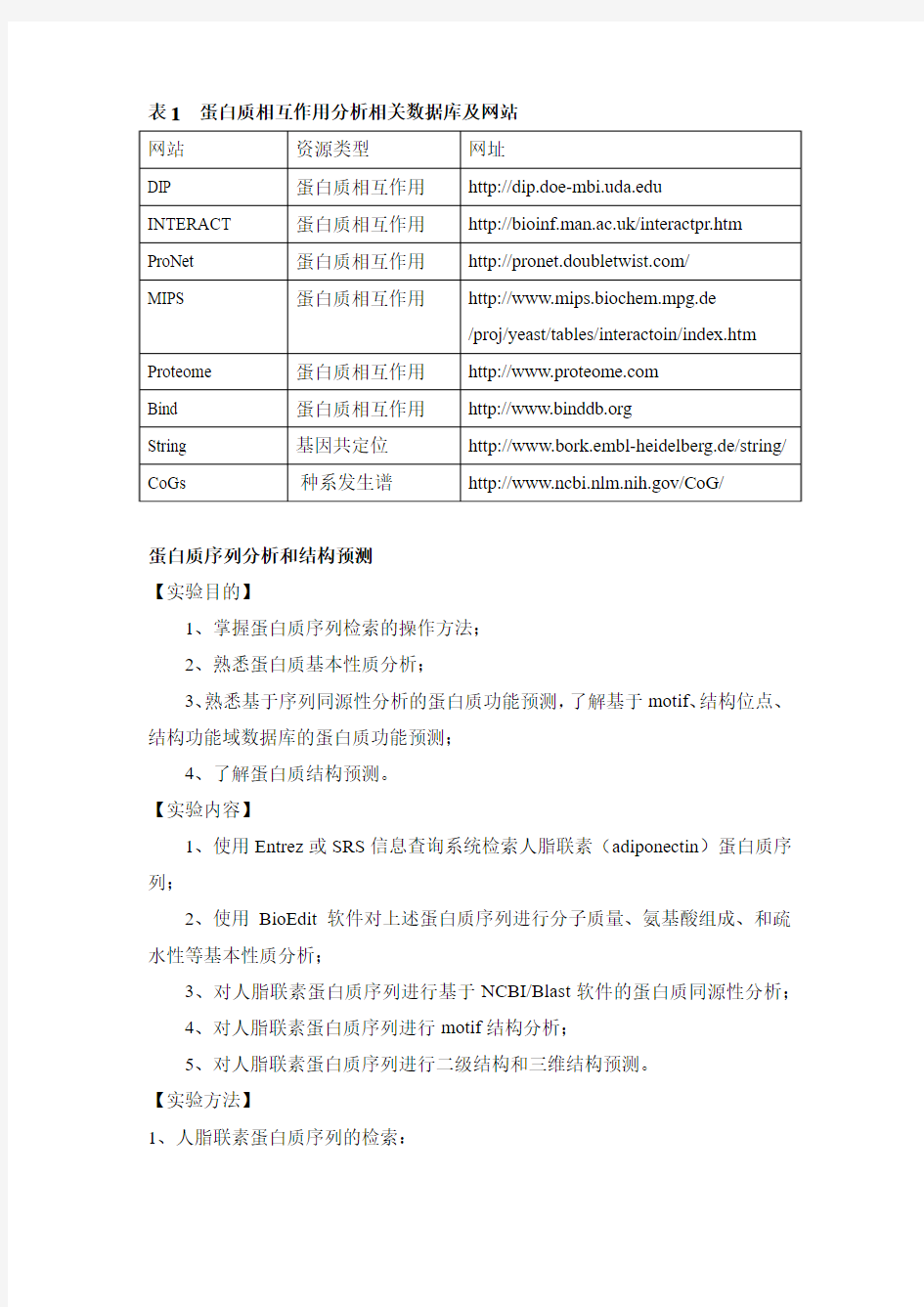

表1蛋白质相互作用分析相关数据库及网站
蛋白质序列分析和结构预测
【实验目的】
1、掌握蛋白质序列检索的操作方法;
2、熟悉蛋白质基本性质分析;
3、熟悉基于序列同源性分析的蛋白质功能预测,了解基于motif、结构位点、结构功能域数据库的蛋白质功能预测;
4、了解蛋白质结构预测。
【实验内容】
1、使用Entrez或SRS信息查询系统检索人脂联素(adiponectin)蛋白质序列;
2、使用BioEdit软件对上述蛋白质序列进行分子质量、氨基酸组成、和疏水性等基本性质分析;
3、对人脂联素蛋白质序列进行基于NCBI/Blast软件的蛋白质同源性分析;
4、对人脂联素蛋白质序列进行motif结构分析;
5、对人脂联素蛋白质序列进行二级结构和三维结构预测。
【实验方法】
1、人脂联素蛋白质序列的检索:
(1)调用Internet浏览器并在其地址栏输入Entrez网址(https://www.360docs.net/doc/9a4099134.html,/Entrez);
(2)在Search后的选择栏中选择protein;
(3)在输入栏输入homo sapiens adiponectin;
(4)点击go后显示序列接受号及序列名称;
(5)点击序列接受号NP_004788 (adiponectin precursor;adipose most abundant gene transcript 1 [Homo sapiens])后显示序列详细信息;
(6)将序列转为FASTA格式保存(参考上述步骤使用SRS信息查询系统检索人脂联素蛋白质序列);
2、使用BioEdit软件对人脂联素蛋白质序列进行分子质量、氨基酸组成和疏水性等基本性质分析:
打开BioEdit软件→将人脂联素蛋白质序列的FASTA格式序列输入分析框→点击左侧序列说明框中的序列说明→点击sequence栏→选择protein→点击Amino Acid Composition→查看该蛋白质分子质量和氨基酸组成;或者选择protein后,点击Kyte & Doolittle Mean Hydrophobicity Profile→查看该蛋白质分子疏水性水平;
3、人脂联素蛋白质序列的蛋白质同源性分析:
(1)进入NCBI/Blast网页;
(2)选择Protein-protein BLAST (blastp);
(3)将FASTA格式序列贴入输入栏;
(4)点击BLAST;
(5)查看与之同源的蛋白质;
4、人脂联素蛋白质序列的motif结构分析:
(1)进入http://hits.isb-sib.ch/cgi-bin/PFSCAN网页;
(2)将人脂联素蛋白质序列的FASTA格式序列贴入输入栏;
(3)点击Scan;
(4)查看分析结果(注意Prosite Profile中的motif information);
5、人脂联素蛋白质序列的二级结构预测:
(1)进入下列蛋白结构预测服务器网址http://www.embl-heidelberg.de/predictprotein//predictprotein.html
(The PredictProtein Server);
(2)在You can栏点击default;
(3)填写email地址和序列名称;
(4)将人脂联素蛋白质序列的FASTA格式序列贴入输入栏点击Submit;
(5)从email信箱查看分析结果;
6、人脂联素蛋白质序列的三维结构预测:
(1)进入https://www.360docs.net/doc/9a4099134.html,/swissmod/SWISS-MODEL.html (SwissModel First Approach Mode)网页;
(2)填写email地址、姓名和序列名称;
(3)将人脂联素蛋白质序列的FASTA格式序列贴入输入栏;
(4)点击Send Request;
(5)从email信箱查看分析结果(注:需下载软件入rasmol查看三维图象)。【作业】
1、提交使用上述软件对人脂联素蛋白质序列进行基本性质分析、同源性分析、motif结构分析以及二级结构和三维结构预测的结果;
2、相互对比结果,说明产生不同结果的原因,总结进行上述分析所需注意的关键事项。
蛋白质序列分析
pfam https://www.360docs.net/doc/9a4099134.html,/蛋白质结构域预测
smart http://smart.embl-heidelberg.de/ 蛋白质结构域预测
BLOCKS https://www.360docs.net/doc/9a4099134.html,/ 蛋白质家族的序列保守区域
CluSTr https://www.360docs.net/doc/9a4099134.html,/clustr/ 将SWISS-PROT+TrEMBL蛋白自动归类InterPro https://www.360docs.net/doc/9a4099134.html,/interpro/ 蛋白质家族、结构域和位点的集成信息资源PIR-ALN https://www.360docs.net/doc/9a4099134.html,/pirwww/dbinfo/piraln.html 蛋白质序列联配
PRINTS https://www.360docs.net/doc/9a4099134.html,/dbbrowser/PRINTS/ 分层的基因家族指纹PROSITE http://www.expasy.ch/prosite/ 具有生物学意义的蛋白模式和轮廓ProtoMap https://www.360docs.net/doc/9a4099134.html,/ 对SWISS-PROT蛋白质进行自动分类
SBASE http://hydra.icgeb.trieste.it/~kristian/SBASE/ 注释了的蛋白质结构域序列SYSTERS http://systers.molgen.mpg.de/ 基于多种资源对蛋白质序列进行分类Emotif https://www.360docs.net/doc/9a4099134.html,/emotif 蛋白质序列模体查询
Iproclass https://www.360docs.net/doc/9a4099134.html,/iproclass/ 注释了的蛋白质分类数据库
PFSCAN www.isrec.isb-sib.ch/software/PFSCAN_form.html 蛋白质序列的(profile)分析
Compute pI/Mw- http://www.expasy.ch/ch2d/pi_tool.html蛋白质序列的理论等电点、分子量的计算
CATH- https://www.360docs.net/doc/9a4099134.html,/bsm/cath 通过自动、手动方式进行蛋白质结
构相关性的系统分类,
CATH是以下四个单词的第一个字母的组合
Class(类) -源于二级结构的最高级别的分类
Architecture(构造) -独立于拓扑结构的二级结构排列描述。
Topology(拓扑结构)-参照已知的折叠及观测的结构进行拓扑分析
Homology(同源)-结构间相同的拓扑布局、不同的二级结构(与功能相似性相关)描述
CRASP http://wwwmgs.bionet.nsc.ru/Programs/CRASP/default.htm 蛋白质序列的相关分析
FPAT https://www.360docs.net/doc/9a4099134.html,/fpat/-an 检索分子序列数据库模式的简易方法Hydropathy Plot (GIF image) https://www.360docs.net/doc/9a4099134.html,/nixon/webtools/hydro/default.htm绘制亲疏水图
MOTIF http://www.genome.ad.jp/SIT/MOTIF.html- 蛋白质序列模式检索
RandSeq http://www.expasy.ch/sprot/randseq.html- 随机的蛋白质序列生长子REPRO- http://www.embl-heidelberg.de/repro/repro_info.htmlA 蛋白质重复片断识别服务器
paralign https://www.360docs.net/doc/9a4099134.html,/ 蛋白质、核酸序列相似性检索
SignalP http://www.cbs.dtu.dk/services/SignalP/ 在不同的物种中预测信号肽及酶切位点
SIM- http://expasy.hcuge.ch/sprot/sim-prot.html 由用户定义的最佳两序列对比TMpred http://ulrec3.unil.ch/software/TMPRED_form.html - 蛋白质跨模区预测、定位,该方法基于统计学结果,通过权重矩阵打分进行预测分析
Coils http://ulrec3.unil.ch/software/COILS_form.html 对比分析具有双股卷曲结构的序列同源性打分,进而计算适于采用该结构的序列的几率
GuessProt http://www.expasy.ch/www/guess-prot.html 选择SwissProt蛋白的等电点、分子量
MassSearch http://cbrg.inf.ethz.ch/subsection3_1_3.html- 蛋白质消化后的聚集检索
Phospepsort http://genome.eerie.fr/gcg/phospepsort-query.html- 磷酸化位点分类SAPS http://ulrec3.unil.ch/software/SAPS_form.html- 蛋白质序列统计分析
Sleuth https://www.360docs.net/doc/9a4099134.html,/molebio/pro... du/pub/sleuth/data- 氨基酸构象及可及性分析
PEDANT-http://pedant.mips.biochem.mpg.de/frishman/pedant.html 蛋白质提炼PROVE http://www.ucmb.ulb.ac.be/%7Ejoan/survol/form.html 蛋白质原子体积计算
Naccess (ASA for proteins)https://www.360docs.net/doc/9a4099134.html,/research/themes/bioinformatics/ 利用Lee & Richards方法描述分子表面性质的计算程序,能够计算蛋白质的表面
准确地将已知的蛋白质序列转化成DNA序列
根据物种在swiss-prot里面翻转
https://www.360docs.net/doc/9a4099134.html,/index.php?id=tools/backtranslation&lang=eng
https://www.360docs.net/doc/9a4099134.html,/sms2/re_trans.html
https://www.360docs.net/doc/9a4099134.html,/sms2/re_trans.html
进去之后,点击Codon Usage Database,然后将你要的物种的拉丁文名输入QUERY Box for search with Latin name of organism ,点击submit,然后就可以看到各个物种的一个密码子列表。然后替换那个大肠杆菌就可以了
蛋白质结构分析原理及工具-文献综述
蛋白质结构分析原理及工具 (南京农业大学生命科学学院生命基地111班) 摘要:本文主要从相似性检测、一级结构、二级结构、三维结构、跨膜域等方面从原理到方法再到工具,系统地介绍了蛋白质结构分析的常用方法。文章侧重于工具的列举,并没有对原理和方法做详细的介绍。文章还列举了蛋白质分析中常用的数据库。 关键词:蛋白质;结构预测;跨膜域;保守结构域 1 蛋白质相似性检测 蛋白质数据库。由一个物种分化而来的不同序列倾向于有相似的结构和功能。物种分化后形成的同源序列称直系同源,它们通常具有相似的功能;由基因复制而来的序列称为旁系同源,它们通常有不同的功能[1]。因此,推测全新蛋白质功能的第一步是将它的序列与进化上相关的已知结构和功能的蛋白质序列比较。表一列出了常用的蛋白质序列数据库和它们的特点。 表一常用蛋白质数据库 网址可能有更新 氨基酸替代模型。进化过程中,一种氨基酸残基会有向另一种氨基酸残基变化的倾向。氨基酸替代模型可用来估计氨基酸替换的速率。目前常用的替代模型有Point Accepted Mutation (PAM)矩阵、BLOck SUbstitution Matrix (BLOSUM)矩阵[2]、JTT模型[3]。 序列相似性搜索工具。序列相似性搜索又分为成对序列相似性搜索和多序列相似性搜索。成对序列相似性搜索通过搜索序列数据库从而找到与查询序列相似的序列。分为局部联配和全局联配。常用的局部联配工具有BLAST和SSEARCH,它们使用了Smith-Waterman 算法。全局联配工具有FASTA和GGSEARCH,基于Needleman-Wunsch算法。多序列相似性搜索常用于构建系统发育树,这里不阐述。表二列举了常用的成对序列相似性比对搜索工具
蛋白质数据库
生物芯片北京国家工程研究中心 湖南中药现代化药物筛选分中心 暨湖南涵春生物有限公司 常用数据库名录 1、蛋白质数据库 PPI - JCB 蛋白质与蛋白质相互作用网络 ?Swiss-Prot - 蛋白质序列注释数据库 ?Kabat - 免疫蛋白质序列数据库 ?PMD - 蛋白质突变数据库 ?InterPro - 蛋白质结构域和功能位点 ?PROSITE - 蛋白质位点和模型 ?BLOCKS - 生物序列分析数据库 ?Pfam - 蛋白质家族数据库 [镜像: St. Louis (USA), Sanger Institute, UK, Karolinska Institutet (Sweden)] ?PRINTS - 蛋白质 Motif 数据库 ?ProDom - 蛋白质结构域数据库 (自动产生) ?PROTOMAP - Swiss-Prot蛋白质自动分类系统 ?SBASE - SBASE 结构域预测数据库 ?SMART - 模式结构研究工具 ?STRING - 相互作用的蛋白质和基因的研究工具
?TIGRFAMs - TIGR 蛋白质家族数据库 ?BIND - 生物分子相互作用数据库 ?DIP - 蛋白质相互作用数据库 ?MINT - 分子相互作用数据库 ?HPRD - 人类蛋白质查询数据库 ?IntAct - EBI 蛋白质相互作用数据库 ?GRID - 相互作用综合数据库 ?PPI - JCB 蛋白质与蛋白质相互作用网络 2、蛋白质三级结构数据库 ?PDB - 蛋白质数据银行 ?BioMagResBank - 蛋白质、氨基酸和核苷酸的核磁共振数据库?SWISS-MODEL Repository - 自动产生蛋白质模型的数据库 ?ModBase - 蛋白质结构模型数据库 ?CATH - 蛋白质结构分类数据库 ?SCOP - 蛋白质结构分类 [镜像: USA | Israel | Singapore | Australia] ?Molecules To Go - PDB数据库查询 ?BMM Domain Server - 生物分子模型数据库 ?ReLiBase - 受体/配体复合物数据库 [镜像: USA] ?TOPS - 蛋白质拓扑图 ?CCDC - 剑桥晶体数据中心 (剑桥结构数据库 (CSD))
蛋白质组学蛋白质组学相关技术及发展文献综述
蛋白质组学蛋白质组学相关技术及发展文献综述 蛋白质组学相关技术及发展文献综述张粒植物学211070161概念及相关内容1994年澳大利亚Macquaie大学的Wilkins和Williams等在意大利的一次科学会议上首次提出了蛋白质组proteome这个概念该英文词汇由蛋白质的“prote”和基因组的“ome”拼接而成并且最初定义为“一个基因组所表达的蛋白质”1。然而这个定义并没有考虑到蛋白质组是动态的而且产生蛋白的细胞、组织或生物体容易受它们所处环境的影响。目前认为蛋白质组是一个已知的细胞在某一特定时刻的包括所有亚型和修饰的全部蛋白质2。蛋白质组学就是从整体角度分析细胞内动态变化的蛋白质组成、表达水平与修饰状态了解蛋白质之间的相互作用与联系提示蛋白质的功能与细胞的活动规律。2蛋白质组学的分类蛋白质组学从其研究目标方面可分为表达蛋白质组学和结构蛋白质组学。前者主要研究细胞或组织在不同条件或状态下蛋白质的表达和功能这将有助于识别各种特异蛋白3目前蛋白质组学的研究在这方面开展的最为广泛其运用技术主要是双相凝胶电泳Two-dimensional gel electrophoresis2DE技术以及图像分析系统当对感兴趣的蛋白质进行分析时可能用到质谱。由于蛋白质发生修饰后其电泳特性将发生改变这些技术可以直接测定蛋白质的含量并有助于发现蛋白质翻译后的修饰如糖基化和磷酸化等4。结构蛋白质组学的目标是识别蛋白质的结构并研究蛋白质间的相互作用。近年来酵母双杂交系统是研究蛋白质相互作用时常用的方法同时研究者也将此方法不断改进5。有研究者最近发现在研究蛋白质相互作用时通过纯化蛋白复合物并用质谱进行识别是很有价值的4。3蛋白质组学相关技术目前蛋白质组学研究在表达蛋白质组学方面的研究最为广泛其分析通常有三个步骤第一步运用蛋白质分离技术分离样品中的蛋白质第二步应用质谱技术或N末端测序鉴定分离到的蛋白质第三步应用生物信息学技术存储、处理、比较获得的数据。3.1蛋白质分离技术这类技术主要是电泳其中应用最多的是双向电泳技术其他还有SDS-PAGE、毛细吸管电泳等。除了电泳外还有液相色谱通常使用高效液相色谱HPLC和二维液相色谱2D-LC。另外还有用于蛋白纯化、除杂的层析技术、超离技术等。 3.1.1双相凝胶电泳双相凝胶电泳two-dimensional gel elec—trophoresis2DE这是最经典、最成熟的蛋白质组分离技术产生于20世纪70年代中叶但主要的技术进步如实验的重复性、可操作性蛋白质的溶解性、特异性等是在近lO年取得的。它根据蛋白质不同的特点分两相分离蛋白质。第一相是等电聚焦IEF电泳根据蛋白质等电点的不同进行分离。蛋白质是两性分子根据其周围环境pH可以带正电荷、负电荷或静电荷为零。等电点pI是蛋白质所带静电荷为零时的pH周围pH小于其pI时蛋白质带正电荷大于其pI时蛋白质带负电荷。IEF时蛋白质处于一个pH梯度中在电场的作用下蛋白质将移向其静电荷为零的点静电荷为正的蛋白将移向负极静电荷为负的将移向正极直到到达其等电点如果蛋白质在其等电点附近扩散那么它将带上电荷重新移回等电点。这就是IEF的聚焦效应它可以在等电点附近浓集蛋白从而分离电荷差别极微的蛋白。pH梯度的形成最初是在一个细的包含两性电解质的聚丙烯酰胺凝胶管中进行。在电流的作用下两性电解质可形成一个pH梯度。但由于两性电解质形成的pH梯度不稳定、易漂移、重复性差80年代以后研究人员研制了固定pH梯度的胶条IPG。此种胶条的形成需要一些能与丙烯酰胺单体结合的分子每个含有一种酸性或碱性缓冲基团。制作时将一种含有不同酸性基团的此分子溶液和一种含有不同碱性基团的此分子溶液混合两种溶液中均含有丙烯酰胺单体和催化剂不同分子的浓度决定pH的范围。聚合时丙烯酰胺成分与双丙烯酰胺聚合形成聚丙烯酰胺凝胶。第二相是SDS聚丙烯酰胺凝胶电泳SDS-PAGE根据蛋白质的分子量不同进行分离。此相是在包含SDS的聚丙烯酰胺凝胶中进行。SDS是一种阴离子去污剂它能缠绕在多肽骨架上使蛋白质带负电所带电荷与蛋白质的分子量成正比在SDS聚丙烯酰胺凝胶中蛋白质分子量的对数与它在胶中移动的距离基本成线性关系。SDS-PAGE装置有水平和垂直两种形式垂直装置可同时跑多块胶如Amersham pharmacia Biotech的Ettan DALT II系统可同时跑12块胶提高了操作的平行性。经过2DE
蛋白质组学技术在各领域的解决方案
蛋白质组学技术在农业生物科研领域、疾病机理机制研究、药物研究、海洋环境、植物胁迫机制研究等方面具有广泛应用。蛋白组学的研究通常遵循以下思路: 蛋白质组学研究思路 图 1 蛋白质组学研究思路 一、蛋白质组学在农业生物科研领域的应用 蛋白质组学技术在农业生物科研领域的应用为作物生长发育、病虫害防治、遗传育种、畜牧兽医学疾病诊断和治疗等方面发挥重要的作用,为现代农业发展开辟新途径。 1 .蛋白质组学在农作物研究中的应用 农业是我国人口赖以生存的基础,而提高粮食产量和品质则是农业发展的关键。蛋白质组学关键技术在作物遗传育种、品系鉴定、品质改良、逆境胁迫应答等关键环节的应用,为农业作物的进一步开发利用提供巨大的参考价值。蛋白质组学可系统研究农作物在特定环境或某个发育阶段的组织和器官中蛋白质的表达变化,有助于作物发育过程机制的理解。 Jia等人利用SWATH等技术对四种玉米组织中的蛋白质进行定量分析:包括未成熟雌穗,未成熟雄穗,授粉后20天的幼胚和14日龄幼苗的根。在玉米的4种组织中总共鉴定到4551个蛋白质,其中在雌穗,雄穗,幼胚和幼根中分别鉴定到3916、3707、3702和2871种蛋白质。利用生物信息学技术将蛋白质组和转录组进行关联分析,并且进一步分析组织特异性高表达的基因和蛋白,以了解玉米组织结构和器官发生的调节机制,为研究玉米发育生物学研究提供了新的线索。相关成果2017年发表在Journal of Proteome Research上。
图 2 实验流程图 文献来源:Jia HT, Sun W, Li MF, et al. An integrated analysis of protein abundance, transcript level and tissue diversity to reveal developmental regulation of maize [J]. J. Proteome Res, December 18, 2017. 2.蛋白质组学在食品科学中的应用 在食品安全研究中,蛋白组学的出现为食品科学的研究指明了方向,同时也为食品科学的研究奠定了良好的发展平台。蛋白质组学在粮油食品、肉类食品、水产食品、乳品食品等方面的应用,不仅可以提高食品安全,并且在改善食品制作以及储存条件的同时,还可以提高食品的口感以及营养程度。 在热处理过程中,肉类的主要成分蛋白质会发生结构性变形,如氧化、降解、变性和聚集。蛋白质的这些变化对最终肉制品的质量、颜色、嫩度和风味有重要影响,并最终影响适口性和可接受性。Tian等人利用2-DE等技术手段研究了在加热中心温度为72℃时用不同的烹饪方法,例如水浴烹饪-WB、短时欧姆烹饪-STOH和长时间欧姆烹饪-LTOH,对牛肉的颜色、烹饪损失、剪切值和蛋白质组变化的影响。蛋白质组学分析表明,欧姆烹饪的烹饪损失、剪切值显著低于水浴烹饪(P<0.05)。利用2-DE蛋白组学技术成功鉴定到STOH和WB烹饪样品之间的17个差异蛋白质,并鉴定出LTOH和WB样品之间的13个差异蛋白质。大多数差异蛋白是肌原纤维和肌浆蛋白,可能与肉质的变化相关。WB烹饪可改变蛋白质溶解度并降低2-DE图像中的蛋白质斑点强度。应用欧姆烹饪会产生更高质量的牛肉产品,并减少烹饪时间。相关成果2016年发表在Innovative Food Science & Emerging Technologies上。
生物信息研究中常用蛋白质数据库的总结
生物信息研究中常用蛋白质数据库简述 内蒙古工业大学理学院呼和浩特孙利霞 2010.1.5 摘要:在后基因组时代生物信息学的研究当中,离不开各种生物信息学数据库。尤其在蛋白质从序列到功能的研究当中,目前各种行之有效的方法都是基于各种层次和结构的蛋白质数据库。随着计算机技术及网络技术的发展,目前的蛋白质数据库不论是所包含数据量还是功能都日新月异,新的数据库层出不穷。一个新手面对如此浩瀚的数据量往往无从下手。本文粗浅地为目前蛋白质数据库的使用勾画出一个轮廓,作为自己蛋白质研究入门的一个引导。 关键词:蛋白质;数据库 0 引言 随着科技的发展,个人的知识往往赶不上快速膨胀的信息量,人们为了解决这个问题,便创建了形形色色的数据库。蛋白质数据库是指:在蛋白质研究领域根据实际需要,对蛋白质序列、蛋白质结构以及文献等数据进行分析、整理、归纳、注释,构建出具有特殊生物学意义和专门用途的数据库。蛋白质数据库总体上可分为两大类:蛋白质序列数据库和蛋白质结构数据库,蛋白质序列数据库来自序列测定,结构数据库来自X-衍射和核磁共振结构测定(详见图1)。这些数据库是分子生物信息学的基本数据资源。上世纪90年代,我国从事蛋白质研究的学者使用的蛋白质数据库储存介质还是国外实验室发布的激光光盘[1]。信息的传播储存甚为不便。随着蛋白质研究的发展飞快,同时伴随着计算机和因特网发展,蛋白质数据库的储存传播方式也发生的巨大的变化。进入21世纪后,我们所用的各种蛋白质数据库都发展成为存储在网络服务器上,基于“服务器—客户机”的访问查询方式。伴随着计算机及物理测试技术的发展数据库的容量和功能成数量级膨胀。但是面对如此浩瀚的数据,新手往往感到无从下手,在需要时找不到自己需要的合适数据库。 本文从目前蛋白质数据库建立的的逻辑层次出发,系统地简绍了常用蛋白质数据的概况,它们的查询方法以及它们相互之间的联系。同时尽量不涉及数据库建设和维护方面的计算机和网络这些数据库底层的技术,为蛋白质研究的入门者及对蛋白质感兴趣的人员的一个引导。
质谱技术在蛋白质组学研究中的应用_甄艳
第35卷 第1期2011年1月 南京林业大学学报(自然科学版) J o u r n a l o f N a n j i n g F o r e s t r y U n i v e r s i t y (N a t u r a l S c i e n c e E d i t i o n ) V o l .35,N o .1 J a n .,2011 h t t p ://w w w .n l d x b .c o m [d o i :10.3969/j .i s s n .1000-2006.2011.01.024] 收稿日期:2009-12-31 修回日期:2010-10-26 基金项目:国家自然科学基金项目(31000287);江苏省高校自然科学基础研究项目(10K J B 220002) 作者简介:甄艳(1976—),副教授,博士。*施季森(通信作者),教授。E -m a i l :j s h i @n j f u .e d u .c n 。 引文格式:甄艳,施季森.质谱技术在蛋白质组学研究中的应用[J ].南京林业大学学报:自然科学版,2011,35(1):103-108. 质谱技术在蛋白质组学研究中的应用 甄 艳,施季森 * (南京林业大学,林木遗传与生物技术省部共建教育部重点实验室,江苏 南京 210037) 摘要:随着蛋白质组学研究的迅速发展,质谱技术已成为应用于蛋白质组学研究中的强有力工具和核心技术。质谱技术的先进性在于为蛋白质组学研究提供的通量和分子信息。笔者重点概述了基于质谱路线的蛋白质组学研究,介绍了基于质谱的定量蛋白质组学﹑翻译后修饰蛋白质组学、定向蛋白质组学、功能蛋白质组学以及基于串联质谱技术的蛋白质组学数据解析的研究 进展。 关键词:质谱;蛋白质组学;定量蛋白质组学;翻译后修饰;定向蛋白质组学;功能蛋白质组学中图分类号:Q 81 文献标志码:A 文章编号:1000-2006(2011)01-0103-06 A p p l i c a t i o n o f m a s s s p e c t r o m e t r y i n p r o t e o m i c s s t u d i e s Z H E NY a n ,S H I J i s e n * (K e y L a b o r a t o r y o f F o r e s t G e n e t i c s a n d B i o t e c h n o l o g y M i n i s t r y o f E d u c a t i o n , N a n j i n g F o r e s t r y U n i v e r s i t y ,N a n j i n g 210037,C h i n a ) A b s t r a c t :W i t ht h e r a p i d d e v e l o p m e n t o f p r o t e o m i c s ,m a s s s p e c t r o m e t r y i s m a t u r i n g t o b e a p o w e r f u l t o o l a n dc o r e t e c h -n o l o g y f o r p r o t e o m i c s s t u d i e s d u r i n g t h e r e c e n t y e a r s .T h e s u p e r i o r i t y o f m a s s s p e c t r o m e t r y l i e s i n p r o v i d i n g t h e t h r o u g h -p u t a n d t h e m o l e c u l a r i n f o r m a t i o n ,w h i c hn o o t h e r t e c h n o l o g y c a n b e m a t c h e di np r o t e o m i c s .I nt h i s r e v i e w ,w e m a d e a g l a n c e o n t h e o u t l i n e o f m a s s s p e c t r o m e t r y -b a s e d p r o t e o m i c s .A n dt h e nw e a d d r e s s e d o n t h e a d v a n c e s o f d a t a a n a l y s i s o f m a s s s p e c t r o m e t r y -b a s e dp r o t e o m i c s ,q u a n t i t a t i v em a s ss p e c t r o m e t r y -b a s e dp r o t e o m i c s ,p o s t -t r a n s l a t i o n a l m o d i f i c a t i o n s b a s e d m a s s s p e c t r o m e t r y ,t a r g e t e d p r o t e o m i c s a n df u n c t i o n a l p r o t e o m i c s b a s e d -m a s s s p e c t r o m e t r y . K e yw o r d s :m a s ss p e c t r o m e t r y ;p r o t e o m i c s ;q u a n t i t a t i v ep r o t e o m i c s ;p o s t -t r a n s l a t i o n m o d i f i c a t i o n ;t a r g e t e d p r o -t e o m i c s ;f u n c t i o n a l p r o t e o m i c s 蛋白质组学(P r o t e o m i c s )是从整体水平上研究细胞内蛋白质的组成、活动规律及蛋白质与蛋白质的相互作用,是功能基因组学时代一门新的学科。目前蛋白质组学的研究主要有两条路线:一是基于双向电泳的蛋白质组学;二是基于质谱的蛋白质组学,其中基于双向电泳的蛋白质组学研究路线最终也离不开质谱技术的应用。自20世纪80年代末,两种质谱软电离方式即电喷雾电离(e l e c t r o s p r a y i o n i z a t i o n ,E S I )和基质辅助激光解析离子化(m a -t r i x a s s i s t e d l a s e r d e s o r p t i o n i o n i z a t i o n ,M A L D I )的发明和发展解决了极性大、热不稳定蛋白质和多肽分 析的离子化和分子质量大的测定问题[1] ,蛋白质组学研究中常用的质谱分析仪包括离子阱(i o n t r a p ,I T ),飞行时间(t i m e o f f l i g h t ,T O F ),串联飞行时间(T O F -T O F ),四级杆/飞行时间(q u a d r u p o l e /T O F h y b r i d s ),离子阱/轨道阱(I T /o r b i t r a ph y b r i d ) 和离子阱/傅里叶变换串联质谱分析仪(I T /F o u r i e r t r a n s f o r m i o n c y c l o t r o nr e s o n a n c em a s s s p e c t r o m e t e r s h y b r i d s ,I T /F T M S ),这些质谱仪具有不同的灵敏度、分辨率、质量精确度和产生不同质量的M S /M S 谱[2] 。质谱作为蛋白质组学研究的一项强有力的工具日趋成熟,并作为样品制备及数据分析的信息学工具被广泛地应用。因此,有学者指出质谱技术 已在蛋白质组学研究中处于核心地位[3] 。目前在通量及所包含的分子信息内容上,基于质谱的蛋白质组学技术在细胞生物学研究中可以鉴定和量化
蛋白质组学及其主要技术
蛋白质组学及其主要技术 朱红1 周海涛2 (综述) 何春涤1, (审校) (1.中国医科大学附属第一医院皮肤科,辽宁沈阳110001; 2.北京大学深圳医院核医学 科,广东深圳518036) 【摘要】蛋白质组是指一种细胞、组织或有机体所表达的全部蛋白质。蛋白质组学是以蛋白质组为研究对象的新兴学科,近年来发展迅速,已成为后基因组时代的研究热点。目前,蛋白质组学研究技术主要包括:样品的制备和蛋白质的分离、蛋白质检测与图像分析、蛋白质鉴定及信息查询。本文就蛋白质组学概念及主要技术进行综述。 【关键词】蛋白质组,蛋白质组学 1蛋白质组学的概念 随着人类基因组测序计划的完成,人们对生命科学的研究重点由结构基因组转向功能基因组,1994年Wilkins和Williams首先提出蛋白质组一词[1],蛋白质组是指一种细胞、组织或有机体所表达的全部蛋白质。从基因到蛋白质存在转录水平、翻译水平及翻译后水平的调控,组织中mRNA丰度与蛋白质丰度不完全符合[2]。蛋白质复杂的翻译后修饰、蛋白质的亚细胞定位或迁移、蛋白质-蛋白质相互作用等也无法从DNA/mRNA水平来判断。因此,只有将功能基因组学与蛋白质组学相结合,才能精确阐明生命的生理及病理机制。 蛋白质组学是以蛋白质组为研究对象,对组织、细胞的整体蛋白进行检测,包括蛋白质表达水平、氨基酸序列、翻译后加工和蛋白质的相互作用,在蛋白质水平上了解细胞各项功能、各种生理、生化过程及疾病的病理过程等[3,4]。蛋白质组学有两种研究策略。一种是高通量研究技术,把生物体内所有的蛋白质作为对象进行研究,并建立蛋白质数据库,从大规模、系统性的角度来看待蛋白质组学,更符合蛋白质组学的本质。但是,由于剪切变异和翻译后修饰,蛋白质数量极其庞大,且表达随空间和时间不断变化,所以分析生物体内所有的蛋白质是一个耗时费力,难以实现的理想目标。另一种策略是研究不同状态或不同时期细胞或组织蛋白质组成的变化,主要目标是研究有差异蛋白质及其功能,如正常组织与肿瘤组织间的差异蛋白质,寻找肿瘤等疾病标记物并为其诊断治疗提供依据。 2蛋白质组学的常用技术 2.1样品的制备和蛋白质的分离技术 2.1.1样品的制备样品制备包括细胞裂解与蛋白质溶解,以及去除核酸等非蛋白质成分。 激光捕获显微切割(Laser-captured microdissection, LCM)[5]技术可大量获得足够用于蛋白质组学研究的单一细胞成分,避免其他蛋白成分对电泳结果的干扰。尤其是肿瘤的蛋白质组学研究常用LCM技术来获取单一的肿瘤细胞。 2.1.2蛋白质的分离技术 ①双向凝胶电泳(Two-dimensional electrophoresis, 2-DE):双向电泳方法于 l975年由O'Farrell[6]首先提出,根据蛋白质等电点和分子量的差异,连续进行成垂直方向的两次电泳将其分离。 第一向为等电聚焦(Isoelectric focusing,IEF)电泳,其基本原理是利用蛋白质分子的等电点不同进行蛋白质的分离。较早出现的IEF是载体两性电解质pH梯度,即在电场中通过两性缓冲离子建立pH梯度;20世纪80年代初建立起来的固相pH梯度(Immobilized pH gradients,IPG)IEF,是利用一系列具有弱酸或弱碱性质的丙烯酰胺衍生物形成pH梯度并参与丙烯酰胺的共价聚合,形成固定的、不随环境电场条件变化的pH梯度。IPG胶实验的重复
蛋白常用数据库
搞蛋白质的童鞋们,甭要只查NCBI了~蛋白质相关数据库启蒙~ ★ 小木虫(金币+1):奖励一下,谢谢提供资源 qinhy:恭喜,您的帖子被版主审核为资源贴了,别人回复您的帖子对资源进行评价后,您就可以获得金币了理由:资源贴2011-11-26 16:56 本来是带图的,可是弄过来就变成米图了,附件里面一个是PDF版、一个是WORD版均是带图的,童鞋们看带图的可能比较方便点哦~ 基于蛋白质序列的蛋白质相互作用位点预测(闲谈版) 这个不是论文不是论文啊~~这个是应某某的要求帮他找的,所以都是用现成的免费的网站数据库做的预测分析。无论文为依托,无原理为根据,纯粹就是流连各大网站作个的闲谈。 1、用这些网站先查查你要研究的蛋白质的底细。 这些网站的数据库大多数是实验或者一些相关文献报道的数据的组成。 ★String http://string.embl.de/ 输入你要搜寻的蛋白,它就把这个蛋白相关的数据反映给你,分confidence、evidence的数据可信度参考,同时还具有actions选项,反应它们之间可能是激活/抑制的关系。按按+、-号可以扩大缩小关联蛋白的数量范围。 往下拉一点点就是数据,哈哈,我们都要看数据吃饭啊~~ 分析的数据源自Neighborhood、Fusion、Occurrence、Coexpression、Experiments Database、Textminin及Homology,表示点得证明有数据,根据各项数据给出综合评分。评分越高相互存在关系可能性越高。点击下方各项图标等详细看到各项数据内容。 设条件确定筛选范围。 ★DIP https://www.360docs.net/doc/9a4099134.html,/dip/Main.cgi 跟上面的大同小异的功能,装上它附带的软件可能操作性会好一点,不过我米有试过哦。倒是跟它有链接的几个数据库都很强大,大家可以点击看看。 ★BIND http://www.bind.ca 文献有介绍的网站,不过我不能理解为什么我注册就注不了……. 2、继续查,用这些网站将要研究的蛋白质的家庭背景,月收入也大起底。 这里的网站可能跟相互作用方面的关系不大,但是如果知道这些,可以对研究的蛋白有更深的了解。 ★PDB https://www.360docs.net/doc/9a4099134.html,/pdb/home/home.do 要查3D结构就往这里查~通常说的PDB号为文献号末4位。 ★PIR https://www.360docs.net/doc/9a4099134.html,/pirwww/index.shtml 在蛋白质方面如NCBI般强大的网站,去上面晃荡下吧,会有收获滴。 ★KEGG http://www.genome.jp/kegg/ 粉强大的一个网站,我只说说它的KEGG PA THW AY子项,能迅速掌握一个蛋白质的功能通路,对于小白的偶们来说,很有用,有木有。 3、正题正题,做完上面那些后,接着就是纯预测的成分。也因为如此,要找着这些网站是很悲催的一件事。就算你找着了,你不懂语言,不懂算法,到底结果的可靠性怎样,见人见智。 需要PDB号作分析: promate http://bioinfo.weizmann.ac.il/promate/
SWISS-MODEL_蛋白质结构预测教程
SWISS-MODEL 蛋白质结构预测 SWISS-MODEL是一项预测蛋白质三级结构的服务,它利用同源建模的方法实现对一段未知序列的三级结构的预测。该服务创建于1993年,开创了自动建模的先河,并且它是讫今为止应用最广泛的免费服务之一。 同源建模法预测蛋白质三级结构一般由四步完成: 1. 从待测蛋白质序列出发,搜索蛋白质结构数据库(如PDB,SWISS-PROT等),得到许多相似序列 (同源序列),选定其中一个(或几个)作为待测蛋白质序列的模板; 2. 待测蛋白质序列与选定的模板进行再次比对,插入各种可能的空位使两者的保守位置尽量对齐; 3. 建模:调整待测蛋白序列中主链各个原子的位置,产生与模板相同或相似的空间结构——待测蛋白 质空间结构模型; 4. 利用能量最小化原理,使待测蛋白质侧链基团处于能量最小的位置。 最后提供给用户的是经过如上四步(或重复其中某几步)后得到的蛋白质三级结构。 SWISS-MODEL工作模式 SWISS-MODEL服务器是以用户输入信息的最小化为目的设计的,即在最简单的情况下,用户仅提供一条目标蛋白的氨基酸序列。由于比较建模程序可以具有不同的复杂性,用户输入一些额外信息对建模程序的运行有时是有必要的,比如,选择不同的模板或者调整目标模板序列比对。该服务主要有以下三种方式: ?First Approach mode(简捷模式):这种模式提供一个简捷的用户介面:用户只需要输入一条氨基酸序列,服务器就会自动选择合适的模板。或者,用户也可以自己指定模板(最多5条),这些模板可以来自ExPDB 模板数据库(也可以是用户选择的含坐标参数的模板文件)。如果一条模板与提交的目标序列相似度大于25%,建模程序就会自动开始运行。但是,模板的可靠性会随着模板与目标序列之间的相似度的降低而降低,如果相似度不到50%往往就需要用手工来调整序列比对。这种模式只能进行大于25个残基的单链蛋白三维结构预测。 ?Alignment Interface(比对界面):这种模式要求用户提供两条已经比对好的序列,并指定哪一条是目标序列,哪一条是模板序列(模板序列应该对应于ExPDB模板数据库中一条已经知道其空间结构的蛋白序列)。服务器会依据用户提供的信息进行建模预测。 ?Project mode(工程模式):手工操作建模过程:该模式需要用户首先构建一个DeepView工程文件,这个工程文件包括模板的结构信息和目标序列与模板序列间的比对信息。这种模式让用户可以控制许多参数,例如:模板的选择,比对中的缺口位置等。此外,这个模式也可以用于“first approach mode简捷模式”输出结果的进一步加工完善。 此外,SWISS-MODEL还具有其他两种内容上的模式: ?Oligomer modeling(寡聚蛋白建模):对于具有四级结构的目标蛋白,SWISS-MODEL提供多聚模板的模式,用于多单体的蛋白质建模。这一模式弥补了简捷模式中只能提交单个目标序列,不能同时预测两条及以上目标序列的蛋白三维结构的不足。 ?GPCR mode(G蛋白偶联受体模式):是专门对7次跨膜G蛋白偶联受体的结构预测。
简述定量蛋白质组学技术
定量蛋白质组(quantitative proteomics)是把一个基因组所表达的全部蛋白或者是一个复杂体系所有的全部蛋白进行鉴定和定量的方法。蛋白质组丰度的动态变化对各种生命过程都有重 要影响。例如在许多疾病的发生和发展进程中,常常伴随着某些蛋白质的表达异常。 发展至今,传统的基于双向电泳的2D和2D-DIGE技术正在逐渐被基于NanoLC-MS/MS的液质联用技术取代;后者需要的样品量更少(25ug蛋白),灵敏度更高(ng级),通量也更高(一次分析可以鉴定和定量超过5000种蛋白)。定量蛋白质组学常见技术如iTRAQ/TMT、Label Free、三类定量方法,百泰派克均可为您提供服务。在这里我们给大家简要介绍一下这 三种定量蛋白质组学方法: iTRAQ(Isobaric Tag for Relative Absolute Quantitation)和TMT(Tandem Mass Tags)技术分别由 美国AB Sciex公司和Thermo Fisher公司研发的多肽体外标记定量技术。该技术采用多个 (2-10)稳定同位素标签,特异性标记多肽的氨基基团进行串联质谱分析,能够同时比较多 达10种不同样本中蛋白质的相对含量,可用于研究不同病理条件下或者不同发育阶段的组织样品中蛋白质表达水平的差异。 分析原理 iTRAQ/TMT标签包括三部分,如下图: 1. 报告基团(reporter group):指示蛋白样品丰度水平。 2. 平衡基团(balance group):平衡报告基团的质量差,使等重标签重量一致,保证标记的 同一肽段m/z相同。 3. 肽反应基团(amine-specific reactive group):能与肽段N端及赖氨酸侧链氨基发生共价连接,从而标记上肽段。
蛋白质相互作用数据库和分析方法
蛋白质相互作用数据库和分析方法 1. 蛋白质相互作用的数据库 蛋白质相互作用数据库见下表所示: 数据库名 说明 网址 BIND 生物分子相互作用数据库 http://bind.ca/ DIP 蛋白质相互作用数据库 https://www.360docs.net/doc/9a4099134.html,/ IntAct 蛋白质相互作用数据库 https://www.360docs.net/doc/9a4099134.html,/intact/index.html InterDom 结构域相互作用数据库 https://www.360docs.net/doc/9a4099134.html,.sg/ MINT 生物分子相互作用数据库 http://mint.bio.uniroma2.it/mint/ STRING 蛋白质相互作用网络数据库 http://string.embl.de/ HPRD 人类蛋白质参考数据库 https://www.360docs.net/doc/9a4099134.html,/ HPID 人类蛋白质相互作用数据库 http://wilab.inha.ac.kr/hpid/ MPPI 脯乳动物相互作用数据库 http://fantom21.gsc.riken.go.jp/PPI/ biogrid 蛋白和遗传相互作用数据,主要来自于酵母、线虫、果蝇和人 https://www.360docs.net/doc/9a4099134.html,/ PDZbase 包含PDZ 结构域的蛋白质相互作用数据库 https://www.360docs.net/doc/9a4099134.html,/services/pdz/start Reactome 生物学通路的辅助知识库 https://www.360docs.net/doc/9a4099134.html,/ 2. 蛋白质相互作用的预测方法 蛋白质相互作用的预测方法很非常多,以下作了简单的介绍 1) 系统发生谱 这个方法基于如下假定:功能相关的(functionally related)基因,在一组完全测序的基因组中预期同时存在或不存在,这种存在或不存在的模式(pattern)被称作系统发育谱;如果两个基因,它们的序列没有同源性,但它们的系统发育谱一致或相似.可以推断它们在功能上是相关的。
蛋白质结构预测在线软件
蛋白质预测分析网址集锦? 物理性质预测:? Compute PI/MW?? ?? SAPS?? 基于组成的蛋白质识别预测? AACompIdent???PROPSEARCH?? 二级结构和折叠类预测? nnpredict?? Predictprotein??? SSPRED?? 特殊结构或结构预测? COILS?? MacStripe?? 与核酸序列一样,蛋白质序列的检索往往是进行相关分析的第一步,由于数据库和网络技校术的发展,蛋白序列的检索是十分方便,将蛋白质序列数据库下载到本地检索和通过国际互联网进行检索均是可行的。? 由NCBI检索蛋白质序列? 可联网到:“”进行检索。? 利用SRS系统从EMBL检索蛋白质序列? 联网到:”,可利用EMBL的SRS系统进行蛋白质序列的检索。? 通过EMAIL进行序列检索?
当网络不是很畅通时或并不急于得到较多数量的蛋白质序列时,可采用EMAIL方式进行序列检索。? 蛋白质基本性质分析? 蛋白质序列的基本性质分析是蛋白质序列分析的基本方面,一般包括蛋白质的氨基酸组成,分子质量,等电点,亲水性,和疏水性、信号肽,跨膜区及结构功能域的分析等到。蛋白质的很多功能特征可直接由分析其序列而获得。例如,疏水性图谱可通知来预测跨膜螺旋。同时,也有很多短片段被细胞用来将目的蛋白质向特定细胞器进行转移的靶标(其中最典型的例子是在羧基端含有KDEL序列特征的蛋白质将被引向内质网。WEB中有很多此类资源用于帮助预测蛋白质的功能。? 疏水性分析? 位于ExPASy的ProtScale程序(?)可被用来计算蛋白质的疏水性图谱。该网站充许用户计算蛋白质的50余种不同属性,并为每一种氨基酸输出相应的分值。输入的数据可为蛋白质序列或SWISSPROT数据库的序列接受号。需要调整的只是计算窗口的大小(n)该参数用于估计每种氨基酸残基的平均显示尺度。? 进行蛋白质的亲/疏水性分析时,也可用一些windows下的软件如,bioedit,dnamana等。? 跨膜区分析? 有多种预测跨膜螺旋的方法,最简单的是直接,观察以20个氨基酸为单位的疏水性氨基酸残基的分布区域,但同时还有多种更加复杂的、精确的算法能够预测跨膜螺旋的具体位置和它们的膜向性。这些技术主要是基于对已知
第三讲:Uniprot蛋白数据库及其他蛋白质分析工具
第三讲 Uniprot蛋白数据库及其他蛋白质 分析工具
2013/03/19
Uniprot数据库
? Uniprot(Universal?protein?resource)是蛋白 质序列的联合数据库。
– SIB:?Swiss?Institute?of?Bioinformatics – EBI:?European?Bioinformatics?Institute – PIR:?Protein?Information?Resource – 2002年三家联合形成了Uniprot
Swiss‐Prot
? 1986年建立 ? 低冗余度 ? 功能导向 ? 由Swiss?Institute?of?Bioinformatics?和EBI共同 建立并维护
TrEMBL
? TrEMBL=Translation?from?EMBL ? EBI建立并维护 ? 是一个自动数据库 ? 冗余度高,可信度低
UniprotKB
? 部分经过专家注释的数据库 ? 具有很高的可信度 ? 包括两部分UniprotKB/Swiss‐Prot和 UniprotKB/TrEMBL ? UniprotKB/Swiss‐Prot包括539,165条序列 ? UniprotKB/TrEMBL包括29,769,971?条序列 ? 具有非冗余性
Uniparc
? 非冗余性 ? 给予序列的特异性,非同一物种的相同序 列被认为是同一个蛋白质 ? 每一条序列被給予一个特异的编号
整理(蛋白质序列数据库)
蛋白质序列数据库 我们可以根据基因组序列预测新基因,预测编码区域,并推测其产物(即蛋白质)的序列。因此,随着基因组序列的不断增长,蛋白质序列也在不断增加。 PIR 历史上,蛋白质数据库的出现先于核酸数据库。在1960年左右,Dayhoff和其同事们搜集了当时所有已知的氨基酸序列,编著了《蛋白质序列与结构图册》。从这本图册中的数据,演化为后来的蛋白质信息资源数据库PIR(Protein Information Resource)。 PIR是由美国生物医学基金会NBRF(National Biomedical Research Foundation)于1984年建立的,其目的是帮助研究者鉴别和解释蛋白质序列信息,研究分子进化、功能基因组,进行生物信息学分析。它是一个全面的、经过注释的、非冗余的蛋白质序列数据库。所有序列数据都经过整理,超过99%的序列已按蛋白质家族分类,一半以上还按蛋白质超家族进行了分类。PIR提供一个蛋白质序列数据库、相关数据库和辅助工具的集成系统,用户可以迅速查找、比较蛋白质序列,得到与蛋白质相关的众多信息。目前,PIR已经成为一个集成的生物信息数据源,支持基因组研究和蛋白质组研究。至2004年,PIR 有近30万个蛋白质的登录数据项,包括来自不同生物体的蛋白质序列。 除了蛋白质序列数据之外,PIR还包含以下信息: (1)蛋白质名称、蛋白质的分类、蛋白质的来源; (2)关于原始数据的参考文献; (3)蛋白质功能和蛋白质的一般特征,包括基因表达、翻译后处理、活化等; (4)序列中相关的位点、功能区域。 对于数据库中的每一个登录项,有与其它数据库的交叉索引,包括到GenBank、EMBL、DDBJ、GDB、MELINE等数据库的索引。PIR中一个具体的登录项如图4.4所示。
蛋白质组学主要研究技术
蛋白质组学主要研究技术 目前蛋白质组学的研究手段主要依靠分离技术、质谱技术和生物信息学的发展。分离技术要求达到高分辨率和高重复率,质谱技术主要包括MALDI-TOF、Q-TOF与MS/MS等质谱设备以及样品的预处理,生物信息学则利用算法的改进和数据库查询比对的完善提高数据结果的判断。 1. 蛋白质组学的分离技术 目前蛋白质组学研究广泛采用的是双向电泳技术。高通量性、对实验要求低、操作简便快速是双向电泳具有的最大优点,它特别适合大规模的蛋白质组学研究。尽管当前蛋白质的分离技术多种多样,但目前仍然没有一种可以彻底地取代双向电泳技术。 从1975年,O’Farrells[8]等将IEF与SDS-PAGE结合创立了2D-PAGE电泳技术以来。双向电泳技术在多个方面都得到了提高和改进:(1) IPG胶条的使用。传统的载体两性电解质等电聚焦存在上样量小、长时间电泳过程中pH梯度不稳定、阴极漂移现象及其导致的碱性蛋白损失、不同批次间重复性差等问题。IPG 胶条的使用使这些问题得到了极大的改善,这使蛋白质双向电泳数据库的建立成为现实;(2) 样品制备:蛋白质样品的质量好坏从根本上决定了电泳最终结果的好坏。双向电泳的样品制备有两个关键点,即如何使样品中蛋白质充分溶解以及尽可能减少影响等电点聚焦的杂质,特别是带电杂质。采用超声或核酸酶处理的方法可以去除核酸,超速离心可除去脂类和多糖,透析、凝胶过滤或沉淀/重悬法可以降低盐浓度。近来的研究发现磺基甘氨酸三甲内盐(ASB14-16)的裂解效果最好,而2mol/l的硫脲和4%的表面活性剂CHAPS的混合液能促使疏水蛋白从IPG到第二相胶的转换。以三丁基膦(TBP)取代β-巯基乙醇或DTT,可以完全溶解链间或链内的二硫键,增强了蛋白质的溶解度,并促进蛋白质向第二向的转移。 另外,双向电泳中对低丰度蛋白的分离识别比较困难,除了显色技术的局限外,还存在容易被高丰度蛋白掩盖的问题,这样得到的蛋白质图谱很不完整,经常会忽略那些在生命过程中发挥重要功能的微量活性分子。解决方案包括增加上样量、对样品进行分级纯化从而富集低丰度蛋白、采用更高灵敏度的显色方法,