动力学方程拟合模型(DOC)

动力学方程拟合模型
动力学方程拟合模型主要分为幕函数型模型和双曲线型模型。
在幕函数型动力学方程中,温度和浓度被认为是独立地影响反应速率的,可
以表示为:
在双曲线型动力方程中强调模型方程中的吸附常数不能靠单独测定吸附性质来确定,而必须和反应速率常数一起由反应动力学实验确定。这说明模型方程中的吸附平衡常数并不是真正的吸附平衡常数,模型假设的反应机理和实际反应机理也会有相当的距离。双曲线型动力学方程的一般表达形式为
(吸附项严
上述两类动力学模型都具有很强的拟合实验数据的能力,都既可用于均相反应体系,也可用于非均相反应体系。对气固相催化反应过程,幕函数型动力学方程可由捷姆金的非均匀表面吸附理论导出,但更常见的是将它作为一种纯经验的关联方式去拟合反应动力学的实验数据。虽然,在这种情况中幕函数型动力学方程不能提供关于反应机理的任何信息,但因为这种方程形式简单、参数数目少,通常也能足够精确地拟合实验数据,所以在非均相反应过程开发和工业反应器设计中还是得到了广泛的应用。
1. 幕函数拟合
刘晓青⑴等人研究了HN03介质中TiAP萃取Th (W)的动力学模式和萃取动力学反应速率方程。
对于本萃取体系,由反应速率方程的一般形式可知:
R = - dCrJdt = klThtNOa )4? [TiAP? LHNOJ"
可用孤立变量法求得各反应物的分反应级数a、b与c,从而确立萃取动力
学方程。
第一步:分级数的求算
1.求a
固定反应物中TiAP和HNO3的浓度,
当TiAP的浓度远远大于体系中Th的初始浓度时,可以认为体系中TiAP浓度在整个萃取过程中没有变化而为一定値,则速率方程可以简化为
dCrh/ dt = ki[|h(NQ)4
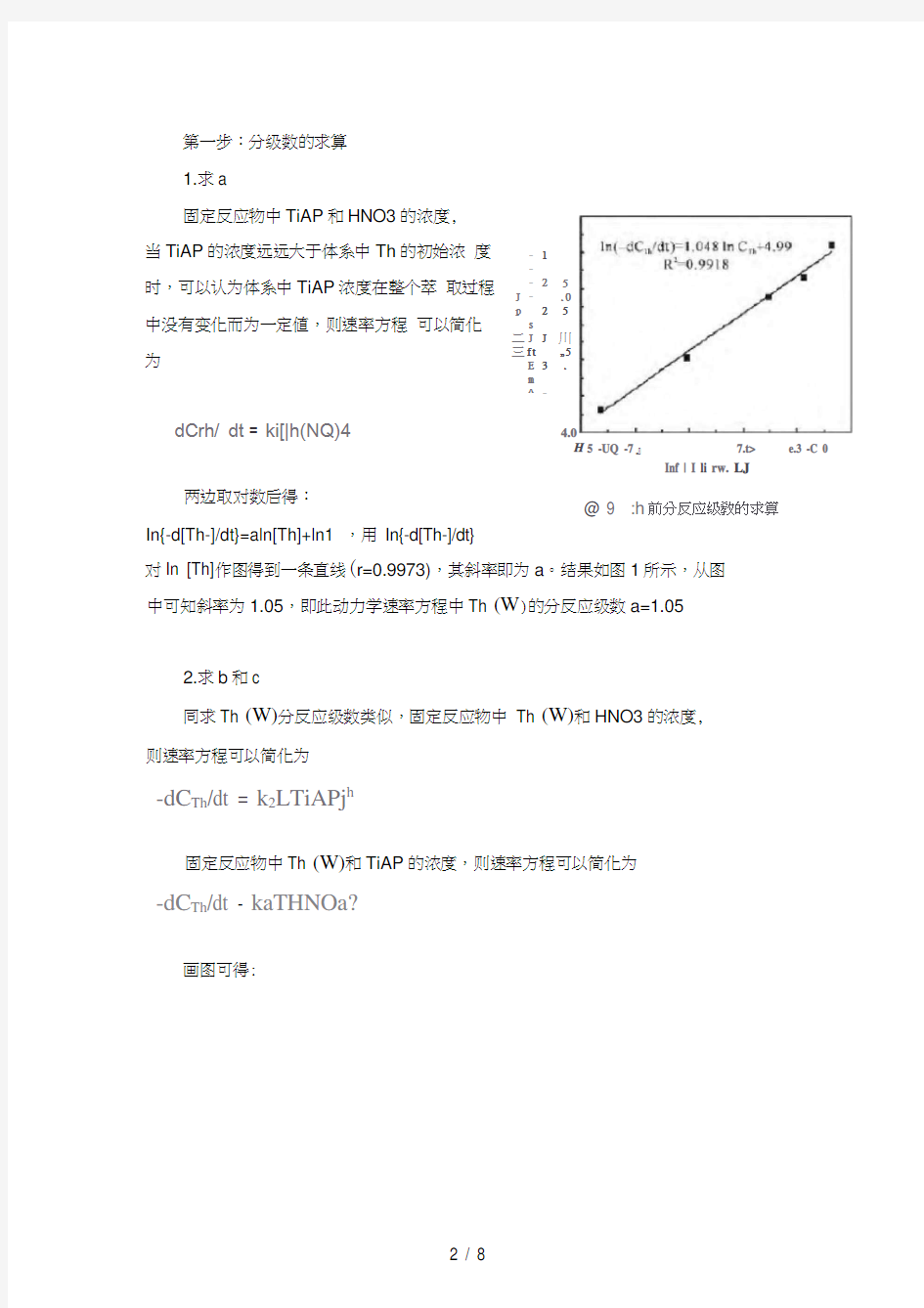
两边取对数后得:
In{-d[Th-]/dt}=aln[Th]+ln1 ,用In{-d[Th-]/dt}
4.0
H5 -UQ -7』7.t> e.3 -C 0
Inf | I li rw. LJ
5
.0
5
川
…5
.
1
2
2
J
3
-
-
-
-
-
s
J
ft
E
m
^
J
p
二
三
@ 9 :h前分反应级敎的求算
对In [Th]作图得到一条直线(r=0.9973),其斜率即为a。结果如图1所示,从图中可知斜率为1.05,即此动力学速率方程中Th (W)的分反应级数a=1.05
2.求b和c
同求Th (W)分反应级数类似,固定反应物中Th (W)和HNO3的浓度, 则速率方程可以简化为
-dC Th/dt = k2LTiAPj h
固定反应物中Th (W)和TiAP的浓度,则速率方程可以简化为
-dC Th/dt - kaTHNOa?
画图可得:
国⑷TiAP 的分反应级数的求胃
闽11 HNn 的分反应毅数的求筲
TiAP 的分反应级数为1.77, HN03的分反应级数为0.38
第二步:写出反应反应速率方程
r--dcTh (N():s )4 dt-k LTh (NO, )4]105 LTiAI J J
L77
LHN()d^ 38
⑼
则293K 时,该反应的平均速率常数
k 为 1.6*10-2( mol/L )-2.2 ? s -1
2. 双曲线拟合
双曲线型反应动力学方程是由
Hinshelwood 在研究气固相催化反应动力学
时,根据Langmuir 的均匀表面吸附理论导出的,其后 Hougen 和Watson 用此模
型成功地处理了许多气固相催化反应,
使它成为一种广泛应用的方法。
因此,双
曲线型动力学方程又被称为 Langmuir-Hin-shelwood 方程或Hougen-Watson 方程。
王志良⑵等人用半连续式无梯度反应器在
130?210 C 范围内研究了苯与
乙烯在FX-02沸石催化剂上烷基化反应的本征动力学。应用改进的Gauss-Newton 法对常微分形式的动力学模型进行了参数估值,得到了双曲函数形式的本征动力 学方程。体系的反应情况如下:
EB
0.6
-0 4
-0,3 0.0 0,3 fafLTiAPL motl )
K
二二--
E 77
二
「
】■].二二-2 H
0 0
0 4 0.8 1 2 1.6
inlLHNQJ, JIIC L L }
乙烯(E)、苯(B)、乙苯(EB)、二乙苯(DEB)
根据Langmuir-Hinshelwood机理,对上述两个反应用如下的动力学模型来表示: kt\ K H K E C H C社—仏-】K EH C仙
I I + K E C E +K H「H+K FH C EH+K DEH C IJKK)
ks2 K EB 人 F C F fit E —Rj- 2 K DEB Cl>E B
{ 1+ A L:C E+应日(71$4 A A^DEB C DLU )~序贯试验设计选用最小体积判别式(MVD ):
i ■* *
niii^ 二max |S Df 旷'D
对ksl、ks- 1、ks2、ks- 2、KE、KB、KEB、KDEB 八个参数采用改进的Gauss-Newton法,直接对常微分形式的动力学模型进行参数估值,目标函数由最
小二乘估值准则确定:
.H .Xj (If .k J
¥Xn
v= i ?k
ALU
x= h= ( TB );
XVB
经过动力学的预试验和序贯试验后,下图给出了参数估值的相对置信区间大
小与试验次数的关系。可以看出,当进行5次序贯试验后,△ -1/2的值迅速下降,表
yj -
6
痕J动力学樓塑蠢豔佶值结果
动力学替进ifitr因于治活化歳/kJ nF 1 bi127,0544^ fW4
kr-i Q 019 SJ J7 9J6
氐強1 22h 74|屛1(1 020 計 2. 957
0. I .M KI *■ 37. 741
^11:.(?} i(r 3-3 工794
J^ra玉耳静nr *-39.
I 7M 6-th in
明试验点的安排较合理,在有效的试验次数内使参数的估值达到了相当高的置信度。
参数估计是在序贯试验下进行的,对参数的联合置信区间检验结果证明,参数具有很高的精度。模型检验其方差分析结果表明,计算值与试验数据的相符性良好,残差分析进一步证明模型无缺陷。
3. 其他算法在动力学方程中的应用
在查找文献的时候,找到多篇与动力学有关的文献采用了不同的算法对动力学参数进行拟合估算。
3.1遗传算法
遗传算法(Genetic Algorithm )是模拟达尔文生物进化论的自然选择和遗传学机理的生物进化过程的计算模型,是一种通过模拟自然进化过程搜索|最优解的方法。黄晓峰[3]等用改进的实数编码遗传算法进行了估计反应动力学参数的研究,提出了一种优化分布线性交叉操作策略,使子代个体在搜索空间内达到均匀
分布,从而提高了搜索的效率。
作者用这种改进的实数编码遗传算法进行了正丁烷选择氧化反应动力学参数的估计。正丁烷在VPO催化剂上选择氧化制顺酐,是催化晶格氧参与催化循环、按照氧化还原(RE-DOX)机理进行的重要烃类选择氧化反应,其简化后的基元反应序列为:
(*2+ K ——X
K-,
B + X \ + H
R和X分别代表还原态和氧化态催化剂,B为正丁烷,MA为顺酐。
_ mm
f'H心+ a Mm
主要操作策略和控制参数为:适应度线性调整、带最优个体保存的期望值选
择,优化分布的线性交叉操作和连续变异操作。种群数目N =50,交叉概率Pc
=0.8 , 变异概率Pm =0.05 ,交叉系数a =2.0 。
优化问题描述为估计反应速率常数K 1、K 2或活化能E 1、E 2与指前因子k01、k 02 ,以使反应速率的估计值rBE与测量值rBM的偏差平方和RSS 极小化:
RSS= \「丄1^(1 )—曲⑴)厂
最后结果如下,表现出较高的精度。
詹晓力⑷等利用蒙特卡罗方法模拟计算了化学反应动力学参数,由基元反应
确定蒙特卡罗模拟的具体做法,将蒙特卡罗方法的模拟结果与动力学实验结果进行比较,根据比较结果自动调整和优化动力学参数,从而无需事先确定动力学方
程,即可有效地估算各种化学反应的动力学参数。用该方法模拟Mo-Bi系丙烯
氨氧化催化剂上的氨分解基元反应。无丙烯存在下的氨分解基元反应如下:
人I
Nllj + 仃庐
A> 1 - 3
N1 1^(7 + (T -- —N2(T + —I
AS
I l2rr + (Kr ----------- l l2O + 2 A(H,) H2/T < ------------ ^H2 + er 岭 \2fT' ^2 + 叫二kiK \p\/( 1+ K i p\ 4 Kipz) 下图给出了用蒙特卡罗方法对该问题进行模拟的结果, m 为催化剂质量, nAO 为初始氨物质的量,c R 为转化率,r 为速率。从图中可见,按估算的动力 学参数所计算的转化率-时间和反应速率-时间曲线与实验数据吻合得较好。 总结 化学反应的机理通常是十分复杂的。一些看起来相当简单的反应的机理至今 也没有完全搞清。因此,不论是双曲线型模型还是幕函数型模型,都只是可以用 来拟合反应动力学实验数据的一种函数形式。由于这两种方程在数学上的适应性 极强,对同一组实验数据可同时用这两种方程拟合的例子也是屡见不鲜的。 从这 个意义上讲,目前工程上应用的绝大多数动力学模型都不是机理模型, 在原实验 范围之外作大幅度的外推都是有风险的。 参考文献 [1] 刘晓青等.HN03介质中TiAP 萃取Th(W)的动力学研究[J].四川大学学报,2014, 51( 6): 1249-1254. [2] 王志良等.FX-02沸石催化剂上苯与乙烯烷基化的反应动力学 [J]?石油炼制与化工,1999,30 (2): 52-55. [3] 黄晓峰,潘立登,陈标华,等.用改进的实数编码遗传算法估计反应动力学参数 [J ].高 校化学工程学报,1999,13( 1) : 50-55 . [4] 詹晓力,罗正鸿,陈丰秋,等.基于Monte Carlo 模拟的化学反应动力学参数估算 等学校化学学报, 2003,24( 8) : 1511-1514 . 100 刖 20 :J ].高 伽C —SiijnLLlatiDn 1x23 nJ 一泯: 3讪C 10 OCD ?0 000 W QUU 皿?打飞厂nwl 1 >