氢谱中影响化学位移的因素

氢谱中影响化学位移的因素
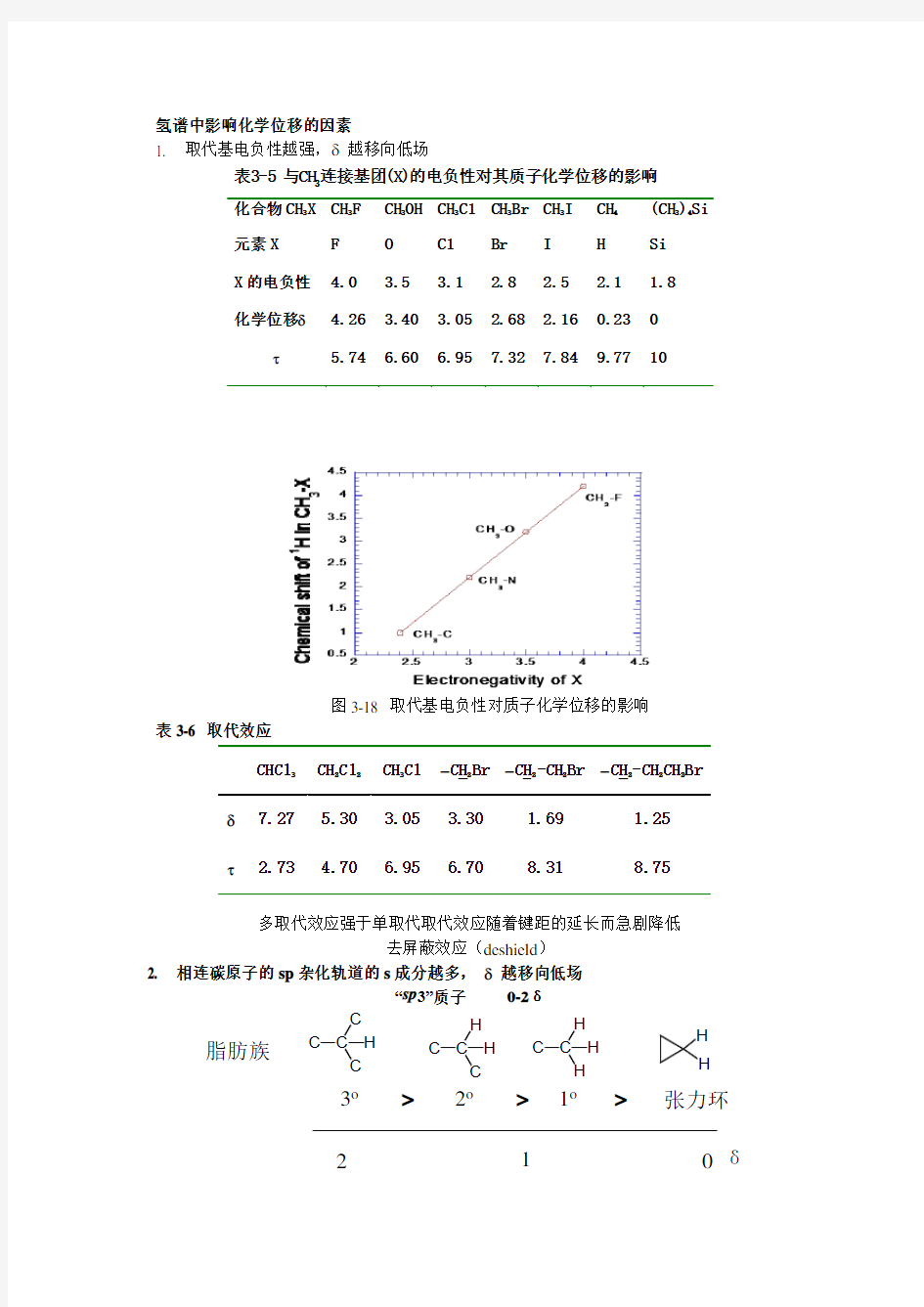
1. 取代基电负性越强,δ越移向低场
表3-5 与CH
3
连接基团(X)的电负性对其质子化学位移的影响
化合物CH
3
X CH
3
F CH
3
OH CH
3
Cl CH
3
Br CH
3
I CH
4
(CH
3
)
4
Si 元素X F O Cl Br I H Si
X的电负性 4.0 3.5 3.1 2.8 2.5 2.1 1.8
化学位移δ 4.26 3.40 3.05 2.68 2.16 0.23 0
τ 5.74 6.60 6.95 7.32 7.84 9.77 10
图3-18 取代基电负性对质子化学位移的影响
表3-6 取代效应
CHCl
3
CH
2
Cl
2
CH
3
Cl -CH
2
Br -CH
2
-CH
2
Br -CH
2
-CH
2
CH
2
Br δ7.27 5.30 3.05 3.30 1.69 1.25
τ 2.73 4.70 6.95 6.70 8.31 8.75
多取代效应强于单取代取代效应随着键距的延长而急剧降低
去屏蔽效应(deshield)
2. 相连碳原子的sp杂化轨道的s成分越多,δ越移向低场
“sp3”质子0-2δ
321
C C
C
C
H C C
H
C
H C C
H
H
H
H
H
0 δ
1
2
脂肪族
o o o张力环
>>>
“sp 2”质子 4.5-7δ
“sp ”质子 2-3δ
3. 酸性质子 10-12δ
R C O
O H
R
C O
O H
作用和O 电负性拉电子效应的双重影响
氢键的影响和可交换质子 能够形成氢键的质子(-OH ,-NH2)化学位移可变
表3-7 可变化学位移的质子
Acids RCOOH 10.5-12.0δ Phenols ArOH 4.0-7.0 Alcohols ROH 0.5-5.0 Amines RNH 2 0.5-5.0 Amides RCONH 2 5.0-8.0 Enols
CH=CH-OH
≥15
形成氢键的数目越多,质子的去屏蔽效应越强 氢键数目通常是浓度和温度的函数
R O
O
H
O R O H δ
δδ
游离羟基(稀溶液)0.5-1.0δ 形成氢键(浓溶液)4-5δ
可交换质子(活泼氢)
H H R O H a
+
R'
O H b R O H b
+
R'
O H a
R O +R O H
+
H SOLV
SOLV
R O +SOLV SOLV
R O
+
3. 共轭效应
sp 2-1s C-H 键,C 原子具有更多的s 特征,s 轨道的吸电子能力大于p 轨道
C H 4.03C H H OCH 33.88
6.27C C H
H
C H
O CH 35.90
OH H
6.73
C H
O H
7.81
p-π共轭,邻位H 的电子密度增加(正屏蔽),δ值减少
π-π共轭,邻位H 的电子密度减少(去屏蔽),δ值增加
CH 2
H 2C
H 2C H 2C H 2C
H 2C
CH 2CH 2CH 2
H 2C
2.63
1.551.080.70
0.51
5. 相邻基团电偶极矩和范德华力的影响 当分子内有强极性基团时,它在分子内产生电场,影响分子内其余部分的电子云密
度,从而影响其它核的屏蔽常数。 当氢核和邻近原子间距离小于范德华半径之和时,电子云相互排斥,导致原子周围
的电子密度降低,屏蔽减少,共振移向低场。
H c
H b H a
OH
H c
H b HO
H a
H a 4.68H b 2.40H c
1.10
H a 3.92H b 3.55H c
0.88
6. 介质的影响(溶剂效应和磁化率效应)
不同溶剂分子对溶质分子有不同的作用,因此介质影响δ值。氘代氯仿(CDCl3)是NMR中使用最普遍的溶剂。
若采用内参考,样品与标准物质处于同样介质中,不必作磁化率校正;若采用外参考法,标准物质与样品的介质不同,磁化率也不同,须作磁化率校正。
3.6.3 自旋-自旋耦合(Spin-Spin Coupling)
核自旋通过成键电子与附近相邻磁性核自旋间的相互作用所引起的NMR谱线分裂现象。
3.6.3.1 自旋-自旋耦合裂分
Fork Rule(n+1 rule)
相邻原子上的质子数以n表示,则简单氢谱(一级谱)NMR谱线的裂分数为n+1。
Cl C
H
Cl
CH2Cl
Integral ratio=2:1
3.6.3.2 自旋耦合图解
HA与HB为化学非等价核,化学位移δA与δB不相同。
图3-27 溶液中HA和HB不同自旋状态的两种分子
HA所受磁场影响: H0+HB和H0-HB
HA所受磁场影响: H0+HB和H0-HB
3.6.3.3 耦合常数J(coupling constant) 谱线裂分产生的裂距,单位Hz
J J J J J
3J2J
Chemical Shift Difference
δAδB
图3-30 乙基裂分的耦合常数定义
不同类型的质子耦合常数J 不同(0-18Hz):相邻sp3杂化C相连质子,J=7.5Hz 双键C相连质子,J cis=10Hz,J trans=17Hz
一级谱(first order spectra)
5.77 δ
3.95 δ
化学位移取
中心值
δ?:互相作用的两组核的化学位移之差δ?/J ≥ 3时,谱线裂分规律符合n+1规则
一级谱:1) 谱线数目2nI+1(与n个等价核相耦合)I=1/2时,n+1规则
2) 相邻两条谱线间距离都相等(J)
3) 谱线强度分布符合(a+b)n展开式的各项系数
n J,反映了核磁矩间相互作用能的大小,和两核之间相隔化学键的数目密切相关,与仪器的工作频率无关。n-化学键的个数
1J,13C与质子之间的耦合,13C-1H,125?250Hz;
2J,同碳上的氢之间的耦合;
磁等价,有耦合,无裂分;磁不等价,有耦合裂分;
固定环上的的CH2及与手性碳相连的CH2,两个氢的δ值常不相等,耦合常数J 约为几到十几Hz。>C=CH2(端烯)也常能反映出2J引起的耦合裂分,约2Hz。
3J,邻位C原子上的两个氢核直接的耦合,1H-12C-12C-1H
4J,相隔4个化学键,耦合作用很弱。
3.7 耦合机制和耦合常数n J
两个氢核相距奇数根键时,自旋相反,J > 0
两个氢核相距偶数根键时,自旋相同,J < 0
耦合作用随键的数目的增加而迅速下降
耦合常数J,反映了核磁矩间相互作用能的大小,与外加磁场无关,而与成键的数目密切相关。
原子核之间的自旋耦合的相互干扰作用,是通过成键电子传递的;
饱和烃化合物中,自旋耦合作用一般只能传递三个单键;
耦合作用又与立体结构有关,有时在相隔四个键以上的质子间仍存在耦合,称远程耦合;
双键或三键较单键更易传递耦合;
质子之间的耦合产生的J值,一般不超过20Hz。
3.7.2 核的等价性质
化学等价(Chemical equivalent):分子中同种类的核或由于分子的对称而具有相同化学位移的核
分子中的两基团(或质子)通过旋转操作可互换,则为化学等价,是等频的。
C C H F
F
C
H
H
F
F C
H
H
F
磁等价(Magnetic equivalent):
两个核(或基团)磁等价必须同时满足下列两条件:
1)它们是化学等价的。
2)它们对任意另一核的耦合常数J相同(数值及符号)。
既是化学等价,也是磁等价核,有耦合,无裂分
自旋体系内,自旋耦合始终存在,只有当相互耦合的核的化学位移值不等时才引起峰的裂分。偕质子之间的耦合
Ha和Hb化学位移相等,对其它核的耦合常数相等,因此是磁等价的质子,可视为一体;
氘的I=1,2nI+1规则,两个等价的质子裂分为三个强度相等的三重峰。
J HH和J HD
同碳上质子与质子之间和质子与其它原子核之间两个耦合常数之比等于质子与该原子核的
核磁常见杂质化学位移表
proton mult CDC13 (CD 3)2CO (CD 3)2SO C 6D 6 CD 3CN CD 3OD D 2O solvent residual peak 7.26 2.05 2.50 7.16 1.94 3.31 4.79 H 2O s 1.56 2.84 3.33 0.40 2.13 4.87 acetic acid CH 3 s 2.10 1.96 1.91 1.55 1.96 1.99 2.08 acetone CH 3 s 2.17 2.09 2.09 1.55 2.08 2.15 2.22 acetonitrile CH 3 s 2.10 2.05 2.07 1.55 1.96 2.03 2.06 benzene CH s 7.36 7.36 7.37 7.15 7.37 7.33 tert -butyl alcohol CH 3 s 1.28 1.18 1.11 1.05 1.16 1.40 1.24 OH s 4.19 1.55 2.18 chloroform CH s 7.26 8.02 8.32 6.15 7.58 7.90 cyclohexane CH 2 s 1.43 1.43 1.40 1.40 1.44 1.45 1,2-dichlorbethane CH 2 s 3.73 3.87 3.90 2.90 3.81 3.78 dichloromethane CH 2 s 5.30 5.63 5.76 4.27 5.44 5.49 diethyl ether CH 3 t, 7 1.21 1.11 1.09 1.11 1.12 1.18 1.17 CH 2 q, 7 3.48 3.41 3.38 3.26 3.42 3.49 3.56 1,2-dimethoxyethane CH 3 s 3.40 3.28 3.24 3.12 3.28 3.35 3.37 CH 2 s 3.55 3.46 3.43 3.33 3.45 3.52 3.60 dimethylformamide CH s 8.02 7.96 7.95 7.63 7.92 7.97 7.92 CH 3 d 2.96 2.94 2.89 2.36 2.89 2.99 3.01 CH 3 s 2.88 2.78 2.73 1.86 2.77 2.86 2.85 dimethyl sulfoxide CH 3 s 2.62 2.52 2.54 1.68 2.50 2.65 2.71 dioxane CH 2 s 3.71 3.59 3.57 3.35 3.60 3.66 3.75 ethanol CH 3 t, 7 1.25 1.12 1.06 0.96 1.12 1.19 1.17 CH 2 q, 7 3.72 3.57 3.44 3.34 3.54 3.60 3.65 OH s 1.32 3.39 4.63 2.47 ethyl acetate CH 3CO s 2.05 1.97 1.99 1.65 1.97 2.01 2.07 CH 2CH 3 q, 7 4.12 4.05 4.03 3.89 4.06 4.09 4.14 CH 2CH 3 t, 7 1.26 1.20 1.17 0.92 1.20 1.24 1.24 ethylene glycol CH s 3.76 3.28 3.34 3.41 3.51 3.59 3.65 “grease” CH 3 m 0.86 0.87 0.92 0.86 0.88 CH 2 br, s 1.26 1.29 1.36 1.27 1.29 n -hexane CH 3 t 0.88 0.88 0.86 0.89 0.89 0.90 CH 2 m 1.26 1.28 1.25 1.24 1.28 1.29 HMPA CH 3 d, 9.5 2.65 2.59 2.53 2.40 2.57 2.64 2.61 methanol CH 3 s 3.49 3.31 3.16 3.07 3.28 3.34 3.34 OH s 1.09 3.12 4.01 2.16 nitromethane CH 3 s 4.33 4.43 4.42 2.94 4.31 4.34 4.40 n -pentane CH 3 t, 7 0.88 0.88 0.86 0.87 0.89 0.90 CH 2 m 1.27 1.27 1.27 1.23 1.29 1.29 2-propanol CH 3 d, 6 1.22 1.10 1.04 0.95 1.09 1.50 1.17 CH sep, 6 4.04 3.90 3.78 3.67 3.87 3.92 4.02 pyridine CH(2) m 8.62 8.58 8.58 8.53 8.57 8.53 8.52 CH(3) m 7.29 7.35 7.39 6.66 7.33 7.44 7.45 CH(4) m 7.68 7.76 7.79 6.98 7.73 7.85 7.87 silicone grease CH 3 s 0.07 0.13 0.29 0.08 0.10 tetrahydrofuran CH 2 m 1.85 1.79 1.76 1.40 1.80 1.87 1.88 CH 2O m 3.76 3.63 3.60 3.57 3.64 3.71 3.74 toluene CH 3 s 2.36 2.32 2.30 2.11 2.33 2.32 CH(o /p ) m 7.17 7.1-7.2 7.18 7.02 7.1-7.3 7.16 CH(m ) m 7.25 7.1-7.2 7.25 7.13 7.1-7.3 7.16 triethylamine CH 3 t,7 1.03 0.96 0.93 0.96 0.96 1.05 0.99 CH 2 q, 7 2.53 2.45 2.43 2.40 2.45 2.58 2.57 丙酮三乙胺
核磁共振氢谱(1H-NMR)
第二章核磁共振氢谱(1H-NMR) §1 概述 基本情况 1H 天然丰度:99.9844%, I=1/2, γ=26.752(107radT-1S-1) 共振频率:42.577 MHz/T δ: 0~20ppm §2 化学位移 1.影响δ值的因素 A.电子效应 (1)诱导效应 a电负性 电负性强的取代基使氢核外电子云密度降低,其共振吸收向低场位移,δ值增大 b.多取代有加和性 c.诱导效应通过成键电子传递,随着与电负性取代基距离的增大,诱导效应的影响逐渐减弱,通常相隔3个以上碳的影响可以忽略不计 (2).共轭效应 氮、氧等杂原子可与双键、苯环共轭。 苯环上的氢被推电子基取代,由于p-π共轭,使苯环电子云密度增大, δ值向高场移动苯环上的氢被吸电子基取代,由于p-π共轭或π-π共轭,使苯环电子云密度降低, δ值向低场移动 (3). 场效应 在某些刚性结构中,一些带杂原子的官能团可通过其电场对邻近氢核施加影响,使其化学
位移发生变化.这些通过电场发挥的作用称为场效应 (4). 范德华(Van der Waals)效应 在某些刚性结构中,当两个氢核在空间上非常接近,其外层电子云互相排斥使核外电子云不能很好地包围氢核,相当于核外电子云密度降低,δ值向低场移动 B.邻近基团的磁各向异性 某些化学键和基团可对空间不同空间位置上的质子施加不同的影响,即它们的屏蔽作用是有方向性的。磁各向异性产生的屏蔽作用通过空间传递,是远程的。 (1)芳环 在苯环的外周区域感应磁场的方向与外加磁场的方向相同(顺磁屏蔽),苯环质子处于此去屏蔽区,其所受磁场强度为外加磁场和感应磁场之和,δ值向低场移动。 (2)双键 >C=O, >C=C<的屏蔽作用与苯环类似。在其平面的上、下方各有一个锥形屏蔽区 (“+”),其它区域为去屏蔽区。 (3)三键 互相垂直的两个π键轨道电子绕σ键产生环电流,在外加磁场作用下产生与三键平行但方向与外加磁场相反的感应磁场。三键的两端位于屏蔽区(“+”),上、下方为去锥形屏蔽区(“-”)δ值比烯氢小。 (4)单键和环己烷 单键各向异性方向与双键相似,直立键质子的化学位移一般比平伏键小0.05-0.8 C.氢键 氢键的缔合作用减少了质子周围的电子云密度, δ值向低场移动。 氢键质子的δ值变化范围大,与缔合程度密切相关。 分子内氢键,质子的δ值与浓度无关 分子间氢键,质子的δ值与浓度有关,浓度大,缔合程度密切。 D.非结构因素 1.介质因素 2.浓度 3.温度 2.各类质子的化学位移 (1).sp3杂化(饱和烷烃) a.化学位移的范围 δ<-CH3 < CH2 < CH, 0-2ppm 与同碳上有强电子基团(O,N,CL,Br)相连, 或邻位有各项异性基团(=,=O,Ph),δ值上升,<5ppm b.化学位移的计算 1)-CH2- δ(CH2R1R2) =1.25+Σσ δ(CHR1R2R3) =1.50+Σσ
溶剂的化学位移(氢谱)
show their degree of variability.Occasionally,in order to distinguish between peaks whose assignment was ambiguous,a further 1-2μL of a specific substrate were added and the spectra run again. Table 1. 1H NMR Data 2acetic acid CH 3s 2.10 1.96 1.91 1.55 1.96 1.99 2.08acetone CH 3s 2.17 2.09 2.09 1.55 2.08 2.15 2.22acetonitrile CH 3s 2.10 2.05 2.07 1.55 1.96 2.03 2.06benzene CH s 7.367.367.377.157.377.33tert -butyl alcohol CH 3s 1.28 1.18 1.11 1.05 1.16 1.40 1.24OH c s 4.19 1.55 2.18tert -butyl methyl ether CCH 3s 1.19 1.13 1.11 1.07 1.14 1.15 1.21OCH 3s 3.22 3.13 3.08 3.04 3.13 3.20 3.22 BHT b ArH s 6.98 6.96 6.877.05 6.97 6.92OH c s 5.01 6.65 4.79 5.20ArCH 3 s 2.27 2.22 2.18 2.24 2.22 2.21ArC(CH 3)3s 1.43 1.41 1.36 1.38 1.39 1.40chloroform CH s 7.268.028.32 6.157.587.90cyclohexane CH 2s 1.43 1.43 1.40 1.40 1.44 1.451,2-dichloroethane CH 2s 3.73 3.87 3.90 2.90 3.81 3.78dichloromethane CH 2s 5.30 5.63 5.76 4.27 5.44 5.49diethyl ether CH 3t,7 1.21 1.11 1.09 1.11 1.12 1.18 1.17CH 2q,7 3.48 3.41 3.38 3.26 3.42 3.49 3.56diglyme CH 2m 3.65 3.56 3.51 3.46 3.53 3.61 3.67CH 2m 3.57 3.47 3.38 3.34 3.45 3.58 3.61OCH 3s 3.39 3.28 3.24 3.11 3.29 3.35 3.371,2-dimethoxyethane CH 3s 3.40 3.28 3.24 3.12 3.28 3.35 3.37CH 2 s 3.55 3.46 3.43 3.33 3.45 3.52 3.60dimethylacetamide CH 3CO s 2.09 1.97 1.96 1.60 1.97 2.07 2.08NCH 3s 3.02 3.00 2.94 2.57 2.96 3.31 3.06NCH 3s 2.94 2.83 2.78 2.05 2.83 2.92 2.90dimethylformamide CH s 8.027.967.957.637.927.977.92CH 3s 2.96 2.94 2.89 2.36 2.89 2.99 3.01CH 3s 2.88 2.78 2.73 1.86 2.77 2.86 2.85dimethyl sulfoxide CH 3s 2.62 2.52 2.54 1.68 2.50 2.65 2.71dioxane CH 2s 3.71 3.59 3.57 3.35 3.60 3.66 3.75ethanol CH 3t,7 1.25 1.12 1.060.96 1.12 1.19 1.17CH 2q,7d 3.72 3.57 3.44 3.34 3.54 3.60 3.65OH s c,d 1.32 3.39 4.63 2.47ethyl acetate CH 3CO s 2.05 1.97 1.99 1.65 1.97 2.01 2.07C H 2CH 3q,7 4.12 4.05 4.03 3.89 4.06 4.09 4.14CH 2C H 3t,7 1.26 1.20 1.170.92 1.20 1.24 1.24ethyl methyl ketone CH 3CO s 2.14 2.07 2.07 1.58 2.06 2.12 2.19C H 2CH 3q,7 2.46 2.45 2.43 1.81 2.43 2.50 3.18CH 2C H 3t,7 1.060.960.910.850.96 1.01 1.26ethylene glycol CH s e 3.76 3.28 3.34 3.41 3.51 3.59 3.65 “grease”f CH 3m 0.860.870.920.860.88CH 2br s 1.26 1.29 1.36 1.27 1.29n -hexane CH 3t 0.880.880.860.890.890.90CH 2m 1.26 1.28 1.25 1.24 1.28 1.29HMPA g CH 3d,9.5 2.65 2.59 2.53 2.40 2.57 2.64 2.61methanol CH 3s h 3.49 3.31 3.16 3.07 3.28 3.34 3.34OH s c,h 1.09 3.12 4.01 2.16nitromethane CH 3s 4.33 4.43 4.42 2.94 4.31 4.34 4.40n -pentane CH 3t,70.880.880.860.870.890.90CH 2m 1.27 1.27 1.27 1.23 1.29 1.292-propanol CH 3d,6 1.22 1.10 1.040.95 1.09 1.50 1.17CH sep,6 4.04 3.90 3.78 3.67 3.87 3.92 4.02pyridine CH(2)m 8.628.588.588.538.578.538.52CH(3)m 7.297.357.39 6.667.337.447.45CH(4)m 7.687.767.79 6.987.737.857.87silicone grease i CH 3s 0.070.130.290.080.10tetrahydrofuran CH 2m 1.85 1.79 1.76 1.40 1.80 1.87 1.88CH 2O m 3.76 3.63 3.60 3.57 3.64 3.71 3.74 toluene CH 3 s 2.36 2.32 2.30 2.11 2.33 2.32CH(o/p )m 7.177.1-7.27.187.027.1-7.37.16CH(m )m 7.257.1-7.27.257.137.1-7.37.16triethylamine CH 3t,7 1.030.960.930.960.96 1.050.99CH 2 q,7 2.53 2.45 2.43 2.40 2.45 2.58 2.57 a In these solvents the intermolecular rate of exchange is slow enough that a peak due to HDO is usually also observed;it appears at 2.81and 3.30ppm in acetone and DMSO,respectively.In the former solvent,it is often seen as a 1:1:1triplet,with 2J H,D )1Hz.b 2,6-Dimethyl-4-tert -butylphenol.c The signals from exchangeable protons were not always identified.d In some cases (see note a ),the coupling interaction between the CH 2and the OH protons may be observed (J )5Hz).e In CD 3CN,the OH proton was seen as a multiplet at δ2.69,and extra coupling was also apparent on the methylene peak.f Long-chain,linear aliphatic hydrocarbons.Their solubility in DMSO was too low to give visible peaks.g Hexamethylphosphoramide.h In some cases (see notes a ,d ),the coupling interaction between the CH 3and the OH protons may be observed (J )5.5Hz).i Poly(dimethylsiloxane).Its solubility in DMSO was too low to give visible peaks. Notes https://www.360docs.net/doc/a03595803.html,.Chem.,Vol.62,No.21,19977513 乙酸丙酮乙腈叔丁醇 叔丁基醚氯仿二氯甲烷DMF 三乙胺 硅脂四氢呋喃 DMSO 乙醚甲醇
氢谱中各种基团的化学位移
1.化学位移能够提供的信息? 化学位移可以提示质子所处化学环境,氢谱中各种基团的化学位移变化很大,不易记忆,但只要牢记几个典型基团的化学位移就可以解决很多问题。如:甲基0.8~1.2ppm,连苯环的甲基2ppm附近,乙酰基上的甲基2ppm附近,甲氧基和氮甲基3~4ppm,双键5~7ppm,苯环7~8ppm,醛基8~10ppm,不连氧的亚甲基1~2ppm,连氧的亚甲基3~4ppm。 2.偶合常数可以给出哪些结构信息? 可以从偶合常数看出基团间的关系,邻位偶合常数较大,远程偶合常数较小。还可以利用Kapulus公式计算邻位氢的二面角。对于有双键的化合物,顺式的氢之间偶合常数为6~10Hz,反式的氢之间偶合常数为12~16Hz。 3.质子偏共振去偶可以用来确定碳的类型,为什么现在常用DEPT谱,而不同质子偏共振去偶谱? 质子偏共振去偶区分伯、仲、叔、季碳的方法是根据裂分成四重、三重、二重和单峰,如果峰离得近会产生重叠,不容易解析,而DEPT区分伯、仲、叔、季碳的方法是根据峰向上或向下,峰不会重叠,并且质子偏共振去偶的灵敏度比DEPT法的灵敏度低得多,所以现在常用DEPT谱区分碳的类型。 5.解析合成化合物的谱、植物中提取化合物的谱和未知化合物的谱,思路有什么不同? 合成化合物的结果是已知的,只要用谱和结构对照就可以知道化合物和预定的结构是否一致。对于植物中提取化合物的谱,首先应看是哪一类化合物,然后用已知的文献数据对照,看是否为已知物,如果文献中没有这个数据则继续测DEPT谱和二维谱,推出结构。对于一个全未知的化合物,除测核磁共振外,还要结合质谱、红外、紫外和元素分析,一步步推测结构。
溶剂的化学位移(碳谱和氢谱)
NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities Hugo E.Gottlieb,*Vadim Kotlyar,and Abraham Nudelman* Department of Chemistry,Bar-Ilan University, Ramat-Gan52900,Israel Received June27,1997 In the course of the routine use of NMR as an aid for organic chemistry,a day-to-day problem is the identifica-tion of signals deriving from common contaminants (water,solvents,stabilizers,oils)in less-than-analyti-cally-pure samples.This data may be available in the literature,but the time involved in searching for it may be considerable.Another issue is the concentration dependence of chemical shifts(especially1H);results obtained two or three decades ago usually refer to much more concentrated samples,and run at lower magnetic fields,than today’s practice. We therefore decided to collect1H and13C chemical shifts of what are,in our experience,the most popular “extra peaks”in a variety of commonly used NMR solvents,in the hope that this will be of assistance to the practicing chemist. Experimental Section NMR spectra were taken in a Bruker DPX-300instrument (300.1and75.5MHz for1H and13C,respectively).Unless otherwise indicated,all were run at room temperature(24(1°C).For the experiments in the last section of this paper,probe temperatures were measured with a calibrated Eurotherm840/T digital thermometer,connected to a thermocouple which was introduced into an NMR tube filled with mineral oil to ap-proximately the same level as a typical sample.At each temperature,the D2O samples were left to equilibrate for at least 10min before the data were collected. In order to avoid having to obtain hundreds of spectra,we prepared seven stock solutions containing approximately equal amounts of several of our entries,chosen in such a way as to prevent intermolecular interactions and possible ambiguities in assignment.Solution1:acetone,tert-butyl methyl ether,di-methylformamide,ethanol,toluene.Solution2:benzene,di-methyl sulfoxide,ethyl acetate,methanol.Solution3:acetic acid,chloroform,diethyl ether,2-propanol,tetrahydrofuran. Solution4:acetonitrile,dichloromethane,dioxane,n-hexane, HMPA.Solution5:1,2-dichloroethane,ethyl methyl ketone, n-pentane,pyridine.Solution6:tert-butyl alcohol,BHT,cyclo-hexane,1,2-dimethoxyethane,nitromethane,silicone grease, triethylamine.Solution7:diglyme,dimethylacetamide,ethyl-ene glycol,“grease”(engine oil).For D2O.Solution1:acetone, tert-butyl methyl ether,dimethylformamide,ethanol,2-propanol. Solution2:dimethyl sulfoxide,ethyl acetate,ethylene glycol, methanol.Solution3:acetonitrile,diglyme,dioxane,HMPA, pyridine.Solution4:1,2-dimethoxyethane,dimethylacetamide, ethyl methyl ketone,triethylamine.Solution5:acetic acid,tert-butyl alcohol,diethyl ether,tetrahydrofuran.In D2O and CD3OD nitromethane was run separately,as the protons exchanged with deuterium in presence of triethylamine. Results Proton Spectra(Table1).A sample of0.6mL of the solvent,containing1μL of TMS,1was first run on its own.From this spectrum we determined the chemical shifts of the solvent residual peak2and the water peak. It should be noted that the latter is quite temperature-dependent(vide infra).Also,any potential hydrogen-bond acceptor will tend to shift the water signal down-field;this is particularly true for nonpolar solvents.In contrast,in e.g.DMSO the water is already strongly hydrogen-bonded to the solvent,and solutes have only a negligible effect on its chemical shift.This is also true for D2O;the chemical shift of the residual HDO is very temperature-dependent(vide infra)but,maybe counter-intuitively,remarkably solute(and pH)independent. We then added3μL of one of our stock solutions to the NMR tube.The chemical shifts were read and are presented in Table 1.Except where indicated,the coupling constants,and therefore the peak shapes,are essentially solvent-independent and are presented only once. For D2O as a solvent,the accepted reference peak(δ)0)is the methyl signal of the sodium salt of3-(trimeth-ylsilyl)propanesulfonic acid;one crystal of this was added to each NMR tube.This material has several disadvan-tages,however:it is not volatile,so it cannot be readily eliminated if the sample has to be recovered.In addition, unless one purchases it in the relatively expensive deuterated form,it adds three more signals to the spectrum(methylenes1,2,and3appear at2.91,1.76, and0.63ppm,respectively).We suggest that the re-sidual HDO peak be used as a secondary reference;we find that if the effects of temperature are taken into account(vide infra),this is very reproducible.For D2O, we used a different set of stock solutions,since many of the less polar substrates are not significantly water-soluble(see Table1).We also ran sodium acetate and sodium formate(chemical shifts: 1.90and8.44ppm, respectively). Carbon Spectra(Table2).To each tube,50μL of the stock solution and3μL of TMS1were added.The solvent chemical shifts3were obtained from the spectra containing the solutes,and the ranges of chemical shifts (1)For recommendations on the publication of NMR data,see: IUPAC Commission on Molecular Structure and Spectroscopy.Pure Appl.Chem.1972,29,627;1976,45,217. (2)I.e.,the signal of the proton for the isotopomer with one less deuterium than the perdeuterated material,e.g.,C H Cl3in CDCl3or C6D5H in C6D6.Except for CHCl3,the splitting due to J HD is typically observed(to a good approximation,it is1/6.5of the value of the corresponding J HH).For CHD2groups(deuterated acetone,DMSO, acetonitrile),this signal is a1:2:3:2:1quintet with a splitting of ca.2 Hz. (3)In contrast to what was said in note2,in the13C spectra the solvent signal is due to the perdeuterated isotopomer,and the one-bond couplings to deuterium are always observable(ca.20-30Hz). Figure1.Chemical shift of H DO as a function of tempera-ture. https://www.360docs.net/doc/a03595803.html,.Chem.1997,62,7512-7515 S0022-3263(97)01176-6CCC:$14.00?1997American Chemical Society
氢谱中影响化学位移的因素
氢谱中影响化学位移的因素 1. 取代基电负性越强,δ 越移向低场 aí3-5 óèCH 3á??óoùí?(X)μ?μ?·1D??????ê?óoˉ?§?oò?μ?ó??ì化合物CH 3X CH 3F CH 3OH CH 3Cl CH 3Br CH 3I CH 4 (CH 3)4Si 元素X F O Cl Br I H Si X 的电负性 4.0 3.5 3.1 2.8 2.5 2.1 1.8 化学位移δ 4.26 3.40 3.05 2.68 2.16 0.23 0 τ 5.74 6.60 6.95 7.32 7.84 9.77 10 图3-18 取代基电负性对质子化学位移的影响 表3-6 取代效应 CHCl 3 CH 2Cl 2 CH 3Cl -CH 2Br -CH 2-CH 2Br -CH 2-CH 2CH 2Br δ 7.27 5.30 3.05 3.30 1.69 1.25 τ 2.73 4.70 6.95 6.70 8.31 8.75 多取代效应强于单取代取代效应随着键距的延长而急剧降低 去屏蔽效应(deshield ) 2. 相连碳原子的sp 杂化轨道的s 成分越多, δ 越移向低场 “sp 3”质子 0-2δ 321C C C C H C C H C H C C H H H H H 0 |? 1 2 ??????o o o ??á|o? > >>
“sp 2”质子 4.5-7δ “sp ”质子 2-3δ 3. 酸性质子 10-12δ R R C O O 作用和O 电负性拉电子效应的双重影响 氢键的影响和可交换质子 能够形成氢键的质子(-OH ,-NH2)化学位移可变 aí3-7 ?éa?oˉ?§?oò?μ??ê?ó Acids RCOOH 10.5-12.0δ Phenols ArOH 4.0-7.0 Alcohols ROH 0.5-5.0 Amines RNH 2 0.5-5.0 Amides RCONH 2 5.0-8.0 Enols CH=CH-OH ≥15 形成氢键的数目越多,质子的去屏蔽效应越强 氢键数目通常是浓度和温度的函数 R O O H δ 游离羟基(稀溶液)0.5-1.0δ 形成氢键(浓溶液)4-5δ 可交换质子(活泼氢) H H R O H a + R' O H b R O H b + R' O H a R O +R O H + H SOLV SOLV R O +SOLV SOLV H R O + 3. 共轭效应
核磁共振氢谱中的几个重要参数
2.1核磁共振氢谱中的几个重要参数 1、化学位移 (1)影响化学位移的主要因素: a.诱导效应。 电负性取代基降低氢核外电子云密度,其共振吸收向低场位移,δ值增大,如 CH3F CH3OH CH3Cl CH3Br CH3I CH4TMS δ(ppm) 4.06 3.40 3.05 2.68 2.16 0.23 0 X电负性 4.0 3.5 3.0 2.8 2.5 2.1 1.6 对于X-CH<Y Z型化合物,X、Y、Z基对>CH-δ值的影响具有加合性,可用shoolery公式估算,式中0.23为CH4的δ,C i值见下表。 例如:BrCH2Cl(括号内为实测值) δ=0.23+2.33+2.53=5.09ppm(5.16ppm) 利用此公式,计算值与实测值误差通常小于0.6ppm,但有时可达1pmm。 值得注意的是,诱导效应是通过成键电子传递的,随着与电负性取代基距离的增大,诱导效应的影响逐渐减弱,通常相隔3个碳以上的影响可以忽略不计。例如:
b.磁各向异性效应。 上面所述的质子周围的电子云密度,能阐明大多数有机化合物的化学位移值。但是还存在用这一因素不能解释的事实:如纯液态下的乙炔质子与乙烯质子相比,前者在高场共振;相反苯的质子又在低场下发生共振。这些现象可用磁各向异性效应解释。 当分子中某些基团的电子云排布不是球形对称时,即磁各向异性时,它对邻近的H核就附加一个各向异性磁场,使某些位置上核受屏蔽,而另一些位置上的核受去屏蔽,这一现象称为各向异性效应。在氢谱中,这种邻近基团的磁各向异性的影响十分重要。现举例说明一下:
叁键的磁各向异性效应:如乙炔分子呈直线型,叁键轴向的周围电子云是对称分布的。乙炔质子处于屏蔽区,使质子的δ值向高场移动。 双键:π电子云分布于成键平面的上、下方,平面内为去屏蔽区。与SP2杂化碳相连的氢位于成键的平面内(处于去屏蔽区),较炔氢低场位移。乙烯:5.25ppm;醛氢:9-10ppm。 化学键的各向异性还可由下述化合物(1)至(4)看出: