双酶切反应时通用缓冲液的使用表

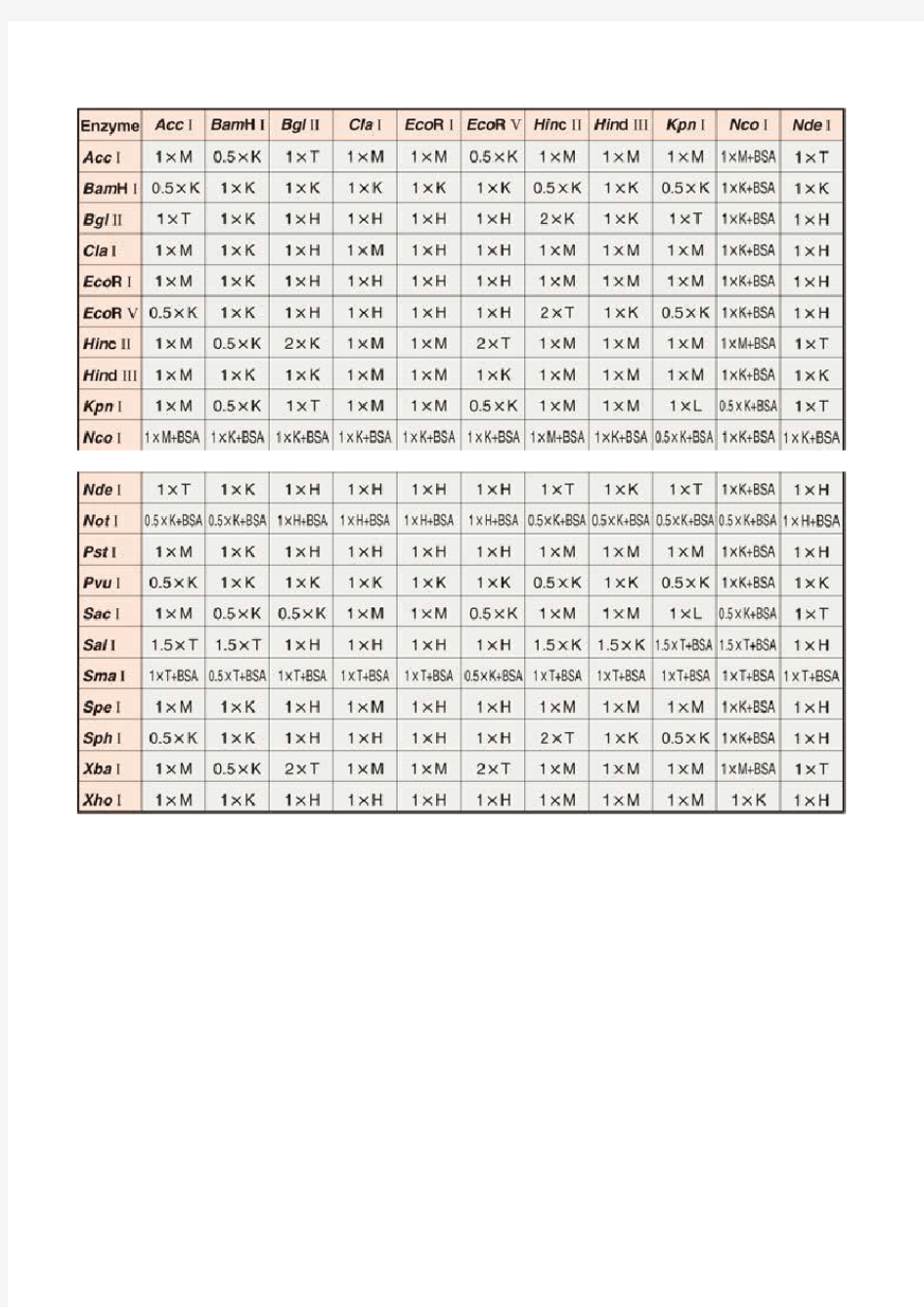
Double Digestion(双酶切反应)时Universal Buffer(通用缓冲液)的使用表
■ 说明
使用二种酶同时进行DNA切断反应(Double Digestion) 时,为了节省反应时间,通常希望在同一反应体系内进行。TaKaRa采用Universal Buffer表示系统,并对每种酶表示了在各Universal Buffer中的相对活性。尽管如此,在进行Double Digestion时,有时还会难以找到合适的Universal Buffer。
本表以在pUC系列载体的多克隆位点处的各限制酶为核心,显示了在Double Digestion可使用的最佳Universal Buffer条件。在本表中,各Universal Buffer之前表示的[数字×] 是指各Universal Buffer的反应体系中的最终浓度。TaKaRa销售产品中添附的Universal Buffer全为10倍浓度的缓冲液。终浓度为0.5×时反应体系中的缓冲液则稀释至20倍,1×时稀释至10倍,2×时稀释至5倍进行使用。
■ 注意
◇1 μg DNA中添加10 U的限制酶,在50 μl的反应体系中,37℃下反应1小时可以完全降解DNA。
◇为防止Star活性的产生,请将反应体系中的甘油含量,尽量控制在10%以下。
◇根据DNA的种类,各DNA的立体结构的差别,或当限制酶识别位点邻接时,有时会发生Double Digestion不能顺利进行的可能。
酶切注意事项
酶切 DNA酶切一般分为质粒直接酶切和PCR产物酶切。 DNA酶切及凝胶电泳 一.DNA的限制性内切酶酶切分析 限制性内切酶能特异地结合于一段被称为限制性酶识别序列的DNA序列之内或其附近的特异位点上,并切割双链DNA。它可分为三类:Ⅰ类和Ⅲ类酶在同一蛋白质分子中兼有切割和修饰(甲基化)作用且依赖于ATP的存在。Ⅰ类酶结合于识别位点并随机的切割识别位点不远处的DNA,而Ⅲ类酶在识别位点上切割DNA分子,然后从底物上解离。Ⅱ类由两种酶组成: 一种为限制性内切核酸酶(限制酶),它切割某一特异的核苷酸序列; 另一种为独立的甲基化酶,它修饰同一识别序列。Ⅱ类中的限制性内切酶在分子克隆中得到了广泛应用,它们是重组DNA的基础。绝大多数Ⅱ类限制酶识别长度为4至6个核苷酸的回文对称特异核苷酸序列(如EcoRⅠ识别六个核苷酸序列:5'- G↓AATTC-3'),有少数酶识别更长的序列或简并序列。Ⅱ类酶切割位点在识别序列中,有的在对称轴处切割,产生平末端的DNA片段(如SmaⅠ:5'-CCC ↓GGG-3');有的切割位点在对称轴一侧,产生带有单链突出末端的DNA片段称粘性未端, 如EcoRⅠ切割识别序列后产生两个互补的粘性末端。 5'…G↓AATTC…3' →5'… G AATTC…3' 3'…CTTAA↑G …5' →3'… CTTAA G…5'
DNA纯度、缓冲液、温度条件及限制性内切酶本身都会影响限制性内切酶的活性。大部分限制性内切酶不受RNA或单链DNA 的影响。当微量的污染物进入限制性内切酶贮存液中时,会影响其进一步使用,因此在吸取限制性内切酶时,每次都要用新的吸管头。如果采用两种限制性内切酶,必须要注意分别提供各自的最适盐浓度。若两者可用同一缓冲液,则可同时水解。若需要不同的盐浓度,则低盐浓度的限制性内切酶必须首先使用,随后调节盐浓度,再用高盐浓度的限制性内切酶水解。也可在第一个酶切反应完成后,用等体积酚/氯仿抽提,加0.1倍体积3mol/L NaAc 和2倍体积无水乙醇,混匀后置-70℃低温冰箱30分钟,离心、干燥并重新溶于缓冲液后进行第二个酶切反应。 DNA限制性内切酶酶切图谱又称DNA的物理图谱,它由一系列位置确定的多种限制性内切酶酶切位点组成,以直线或环状图式表示。在DNA序列分析、基因组的功能图谱绘制、DNA的无性繁殖、基因文库的构建等工作中,建立限制性内切酶图谱都是不可缺少的环节,近年来发展起来的RFLP(限制性片段长度多态性)技术更是建立在它的基础上。 构建DNA限制性内切酶图谱有许多方法。通常结合使用多种限制性内切酶,通过综合分析多种酶单切及不同组合的多种酶同时切所得到的限制性片段大小来确定各种酶的酶切位点及其相对位置。酶切图谱的使用价值依赖于它的准确性和精确程度。 在酶切图谱制作过程中,为了获得条带清晰的电泳图谱,一
REMI介导红曲霉遗传转化条件的优化
REMI介导红曲霉遗传转化条件的优化 摘要:为了构建限制性内切酶介导的整合(remi)技术介导的红曲霉(monascus anka)遗传转化系统,以潮霉素b 作为抗性筛选标记,pbc-hygro、pcb1003、pan7-1作为转化质粒,采用remi技术转化红曲霉。结果表明,用remi技术介导红曲霉转化时,质粒pbc-hygro和pcb1003适合于红曲霉的转化。环状质粒与线性质粒的转化率无显著差别;在用remi技术介导转化时添加酶切缓冲液不利于转化;原生质体浓度为1×107~1×108个/ml时,每微克质粒可获得的潮霉素b抗性转化子为2800~3200个;hindⅲ介导转化时的最佳酶用量为105~120u;在质粒用量为8μg/100μl时转化率最高。 关键词:红曲霉(monascus anka);遗传转化;限制性内切酶介导的整合(remi) abstract:toestablishagenetictransformationsystemofmo nascus anka byrestrictionenzym e-mediatedintegration(remi),usingthehphgeneasselectablemarkerandpbc-hygro,pcb1003andpan7-1asvector,thefungus
m.ankawastransformedtobehygromycinb-resistantbyremi.additionofrestrictionenzymestotransformationmixturesresultedinincreaseoftransformationratewhenusingpbc-hygroandpcb1003asvectors.thetransformationratehadnosignificantdifferencenomatterifvectorsweredigestedbyrestrictionenzymeornot.additionofenzymedigestionbufferresultedinreducingoftransformation.protoplastsat the concentrationof1×107~1×108mlwerefitfortransformingm.anka,transformationnumberwas2800~3200ind./μgvectordna.theoptimumhindiiiandvectordosewas105~120uand8μg/100μl,respectively.
常用限制性内切酶酶切位点汇总
Acc65I识别位点AccI识别位点AciI识别位点AclI识别位点AcuI识别位点 AfeI识别位点AflII识别位点AflIII识别位点AgeI识别位点AhdI识别位点AleI识别位点AluI识别位点AlwI识别位点AlwNI识别位点ApaI识别位点ApaLI识别位点ApeKI识别位点ApoI识别位点AscI识别位点AseI识别位点AsiSI识别位点AvaI识别位点AvaII识别位点AvrII识别位点BaeI识别位点BamHI识别位点BanI识别位点BanII识别位点
BbvCI识别位点BbvI识别位点 BccI识别位点BceAI识别位点BcgI识别位点 BciVI识别位点 BclI识别位点 BfaI识别位点 BfuAI识别位点 BglI识别位点 BglII识别位点 BlpI识别位点 Bme1580I识别位点BmgBI识别位点BmrI识别位点BmtI识别位点BpmI识别位点Bpu10I识别位点BpuEI识别位点BsaAI识别位点BsaBI识别位点BsaHI识别位点BsaI识别位点BsaJI识别位点BsaWI识别位点BsaXI识别位点BseRI识别位点BseYI识别位点
BsiEI 识别位点BsiHKAI 识别位点BsiWI识别位点BslI 识别位点BsmAI识别位点 BsmBI识别位点BsmFI识别位点BsmI识别位点BsoBI识别位点Bsp1286I识别位点BspCNI识别位点BspDI识别位点BspEI识别位点BspHI识别位点BspMI识别位点BspQI识别位点BsrBI识别位点BsrDI识别位点BsrFI识别位点BsrGI识别位点BsrI识别位点BssHII识别位点BssKI识别位点BssSI识别位点BstAPI识别位点BstBI识别位点BstEII识别位点BstNI识别位点
双酶切连接反应常见问题分析
前一阵子一直在做双酶切质粒重组,失败了很多次,不过很快改善了实验方法,用2周重组了14个质粒。现就自己的体会,谈一下质粒重组的一些个人经验。 1. 回收PCR产物: 在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶。选好酶切位点后,在各个酶的两边加上保护碱基。 双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。 2. 纯化问题: 纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。 3. 酶量的问题: 对1单位酶的定义如下:在50μl 反应液中,30℃温度下反应1小时,将1μg 的λDNA完全分解的酶量定义为1个活性单位(U)。而该酶浓度约为15单位/微升,在除外酶降解的因素外,该酶可分解15μg的DNA,而一般从1-4ml 菌液提出的DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的质量好,酶切完全切得动。 4. 酶切、回收后的PCR产物与载体的连接: 摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则非常有必要。回收的载体片段:回收的PC R产物片段=1:10,一般取前者0.03pmol,后者取0.3pmol。
pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000(注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套。1pmol 1000bp DNA=0.66μg,如载体是5380b p,则0.03pmol为0.03×5.38×0.66=0.106524μg。 5. 测DNA浓度: 测DNA浓度可以在专用机子上测,注意OD值,一般约1.8-2.0.另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的MARKER每个条带约50ng。 6. 连接反应: TAKARA的连接酶上的说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λD NA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNa段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。 7.转化: ①全量(10 μl)加入至100μl JM109感受态细胞中,冰中放置30分钟。 ②42℃加热45秒钟后,再在冰中放置1分钟。 ③加入890 μl AMP阴性培养基,37℃振荡培养60分钟。 取100μl铺板。也可离心后余100μl 几个非常重要的问题: 1 做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解。为保险起见,一般连接3小时,16度。
限制性内切酶酶切位点_方便搜索
GACGTC CTGCAG Acc65I 识别位点 GTMKAC CAKMTG AccI 识别位点 GTMKAC CAKMTG AciI 识别位点 CCGC GGCG AclI 识别位点 AACGTT TTGCAA AcuI 识别位点 CTGAAG GACTTC AfeI 识别位点 AGCGCT TCGCGA CTTAAG GAATTC AflIII 识别位点 ACRYGT TGYRCA AgeI 识别位点 ACCGGT TGGCCA AhdI 识别位点 GACNNNNNGTC CTGNNNNNCAG AleI 识别位点 CACNNNNGTG GTGNNNNCAC AluI 识别位点 AGCT TCGA AlwI 识别位点 GGATC CCTAG
CAGNNNCTG GTCNNNGAC ApaI 识别位点 GGGCCC CCCGGG ApaLI 识别位点 GTGCAC CACGTG ApeKI 识别位点 GCWGC CGWCG ApoI 识别位点 RAATTY YTTAAR AscI 识别位点 GGCGCGCC CCGCGCGG AseI 识别位点 ATTAAT TAATTA GCGATCGC CGCTAGCG AvaI 识别位点 CYCGRG GRGCYC AvaII 识别位点 GGWCC CCWGG AvrII 识别位点 CCTAGG GGATCC BaeI 识别位点 NACNGTAYCN BamHI 识别位点 GGATCC CCTAGG BanI 识别位点 GGYRCC CCRYGG
限制性内切酶酶切反应的标准操作规程
限制性内切酶酶切反应的标准操作规程(编号:007) 1、目的及适用范围 利用限制性内切酶在特异性的识别位点上或附近切割双链DNA分子,用于特定基因的克隆等分子生物学研究。 2、主要试剂及仪器 微量移液器、恒温水浴锅、限制性内切酶 EcoR I, BamH I 等、通用缓冲液10× Buffer 3、操作步骤 按顺序加入下列反应物,放入37℃水浴锅内反应2h。 反应物体积(μL) 灭菌水3 DNA40 10× Buffer K 5 EcoR I1 BamH I1 总体积50 4、问题向导 4.1 建立一个标准的酶切反应:目前大多数研究者遵循一条规则,即10个单位的内切酶可以切割1μg不同来源和纯度的DNA。通常,一个50μL的反应体系中,1μL的酶在1X NEBuffer终浓度及相应温度条件下反应1h即可降解1μg已纯化好的DNA。如果加入更多的酶,则可相应缩短反应时间;如果减少酶的用量,对许多酶来说,相应延长反应时间(不超过16h)也可完全反应。4.2 选择正确的酶:选择的酶在底物DNA上必须至少有一个相应的识别位点。识别碱基数目少的酶比碱基数目多的酶更频繁地切割底物。假设一个GC含量50%的DNA链,一个识别4个碱基的酶将平均在每44(256)个碱基中切割一次;而一个识别6个碱基的酶将平均在每46(4096)碱基切割一次。内切酶的产物可以是粘端的(3\'或5\'突出端),也可以是平端的片段。粘端产物可以与相容的其它内切酶产物连接,而所有的平端产物都可以互相连接。 4.3 内切酶:内切酶一旦拿出冰箱后应当立即置于冰上。酶应当是最后一个被加入到反应体系中(在加入酶之前所有的其它反应物都应当已经加好并已预混合)。酶的用量视在底物上的切割频率而定。例如,超螺旋和包埋法切割的DNA通常需要超过1U/μg的酶才能被完全切割。 21
双酶切连接反应之全攻略
双酶切连接反应之全攻略 1、回收PCR产物:在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基,其原则可参照: 双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。 纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒 酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。 而该酶浓度约为15单位/ 微升,在除外酶降解的 因素外,该酶可分解15μg的DNA,而一般从1-4ml菌液提出的 DNA约 为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的 质量好,酶切完全切得动。 2、酶切、回收后的PCR产物与载体的连接 摩尔比的计算: 很多人凭经验也可以。但对于初学者从头认真计算则 非常有必要。回收的载体片段:回收的PCR产物片段=1:10 ,一般取前者0.03pmol,后者取0.3pmol。 pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为 0.03×5.38×0.66=0.106524μg。 测DNA浓度: 可以在专用机子上测,注意OD值,一般约1.8-2.0.另外,如果嫌麻烦,也可用MARKER 进行估测,如MARKER2000,5微升的 MARKER每个条带约50ng。 连接反应:TAKARA的 连接酶上的 说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段 被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连 接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。 3、转化: a、全量(10 μl)加入至100 μl JM109感受态细胞中,冰中放置30分钟。 b、42℃加热45秒钟后,再在冰中放置1分钟。 c、加入890 μl AMP阴性培养基,37℃振荡培养60分钟。 取100μl铺板。也可离心后余100μl 几个非常重要的问题 1 做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解.为保险起见,一般 连接3小时,16度.
常用限制性内切酶酶切位点
AatII 识别位点 Acc65I 识别位点 AccI 识别位点 AciI 识别位点 AclI 识别位点 AcuI 识别位点 AfeI 识别位点 AflII 识别位点 AflIII 识别位点 AgeI 识别位点 AhdI 识别位点 AleI 识别位点 AluI 识别位点 AlwI 识别位点 AlwNI 识别位点 ApaI 识别位点 ApaLI 识别位点 ApeKI 识别位点 ApoI 识别位点 AscI 识别位点 AseI 识别位点 AsiSI 识别位点
AvaI识别位点 AvaII识别位点 AvrII识别位点 BaeI识别位点 BamHI 识别位点 BanI识别位点 BanII识别位点 BbsI识别位点 BbvCI识别位点 BbvI识别位点 BccI识别位点 BceAI识别位点BcgI识别位点BciVI识别位点BclI识别位点 BfaI识别位点BfuAI识别位点BglI识别位点BglII识别位点BlpI识别位点Bme1580I识别位点BmgBI识别位点BmrI识别位点
BmtI 识别位点 BpmI 识别位点 Bpu10I 识别位点 BpuEI 识别位点 BsaAI 识别位点 BsaBI 识别位点 BsaHI 识别位点 BsaI 识别位点 BsaJI 识别位点 BsaWI 识别位点 BsaXI 识别位点 BseRI 识别位点 BseYI 识别位点 BsgI 识别位点 BsiEI 识别位点 BsiHKAI 识别位点 BsiWI 识别位点 BslI 识别位点 BsmAI 识别位点 BsmBI 识别位点 BsmFI 识别位点 BsmI 识别位点
酶切反应条件的优化
当建立内切酶酶切反应体系时有几个关键因素需要考虑。比如如何在正确的反应体系中,加入适量的DNA、内切酶和缓冲液,就可以获得最佳酶切效果。根据定义,在50μl体系中,1单位的限制性内切酶可以在60分钟内完全切割1μg的底物DNA。上述酶、DNA与总反应体积的比值可以做为建立反应体系的参考数据。但是,目前大多数科研人员会遵循下表中所列的标准反应条件,使用5-10倍的过量酶切割DNA,这样有利于克服由于DNA来源不同、质量和纯度不同而造成的实验失败。 “标准”反应体系 内切酶 ?从冰箱取出后请一直置于冰上。 ?酶最后加入到反应体系中。 ?加入酶之前将反应混合物混匀,可以用移液枪上下吹打或轻弹管壁,然后在离心机中快速离心。切忌振荡混匀! ?当切割超螺旋质粒和琼脂糖包埋DNA时,通常需要超过1unit/μg的酶量以达到完全酶切。DNA ?避免酚、氯仿、酒精、EDTA、变性剂或过多盐离子的污染。 ?甲基化的DNA会抑制某些酶的切割效率。 缓冲液 ?使用终浓度为1X的缓冲液。 ?根据实验需要加入终浓度为100μg/ml的BSA(1:100稀释)。 ?在不需要BSA即可达到最佳活性的酶切反应中如果加入BSA也不会影响酶切效果。 反应总体积 ?建议在50μl反应体系中消化1μg底物DNA。 ?为避免星号活性,甘油浓度应<5%。 ?加入内切酶(贮存于50%甘油中)的量应不超过总体积的10%。 ?使用以下技术,内切酶的反应条件可能未达到最佳反应条件:克隆、基因分型、突变检测、基因定位、探针制备、测序和甲基化检测等。 ?内切酶贮存液中的添加物(如:甘油和盐)和底物溶液中尚存的残余物(如:盐、EDTA 或乙醇)会导致小体积反应体系出现问题。NEB提供了一系列高保真内切酶(方便建立反应体系。下述为小体积反应体系反应指南。 酶切反应体系的选择
常见分子实验限制性内切酶酶切位点大全
Acc65I识别位点 AccI识别位点 AciI识别位点 AclI识别位点 AcuI识别位点 AfeI识别位点 AflII识别位点 AflIII识别位点 AgeI识别位点 AhdI识别位点 AleI识别位点 AluI识别位点 AlwI识别位点 AlwNI识别位点 ApaI识别位点 ApaLI识别位点 ApeKI识别位点 ApoI识别位点 AscI识别位点 AseI识别位点 AsiSI识别位点 AvaI识别位点 AvaII识别位点 AvrII识别位点 BaeI识别位点 BamHI识别位点 BanI识别位点 BanII识别位点
BbvCI识别位点 BbvI识别位点 BccI识别位点 BceAI识别位点 BcgI识别位点 BciVI识别位点 BclI识别位点 BfaI识别位点 BfuAI识别位点 BglI识别位点 BglII识别位点 BlpI识别位点 Bme1580I识别位点 BmgBI识别位点 BmrI识别位点 BmtI识别位点 BpmI识别位点 Bpu10I识别位点 BpuEI识别位点 BsaAI识别位点 BsaBI识别位点 BsaHI识别位点 BsaI识别位点 BsaJI识别位点 BsaWI识别位点 BsaXI识别位点 BseRI识别位点 BseYI识别位点
BsiEI识别位点 BsiHKAI识别位点 BsiWI识别位点 BslI识别位点 BsmAI识别位点 BsmBI识别位点 BsmFI识别位点 BsmI识别位点 BsoBI识别位点 Bsp1286I识别位点 BspCNI识别位点BspDI识别位点 BspEI识别位点 BspHI识别位点 BspMI识别位点 BspQI识别位点 BsrBI识别位点 BsrDI识别位点 BsrFI识别位点 BsrGI识别位点 BsrI识别位点 BssHII识别位点 BssKI识别位点 BssSI识别位点 BstAPI识别位点 BstBI识别位点 BstEII识别位点 BstNI识别位点
酶切和连接5页
双酶切: 载体大小为3000bp左右,在SfiⅠ和BssHⅡ位点之间有370bp左右的片段存在。 我想通过SfiⅠ和BssHⅡ双酶切,将370bp的片段切掉,然后装入不同的片段。 我的酶切体系如下: 质粒(载体+老片段)1ul(约100ng) (NEBlack Eye SfiⅠ1ul (NEBlack Eye BssHⅡ1ul 10×buf 2.2ul 100×BSA 0.2ul 水14.8ul 总体积20ul 50度,2小时。切出了370bp左右的片段,回收载体。 然后取回收载体的1ul自连,铺平板,但是长出了300多个克隆。证明酶切不完全,怀疑有大量载体只是单酶切。50度3小时我试过,但是质粒有降解。酶量应该说是过量的。请问有什么办法可以酶切完全? 粘性连接 (一)外源DNA和质粒载体的连接反应 外源DNA片段和线状质粒载体的连接,也就是在双链DNA5’磷酸和相邻的3'羟基之间形成的新的共价链。如质粒载体的两条链都带5'磷酸,可生成4个新的磷酸二酯链。但如果质粒DNA已去磷酸化,则吸能形成2个新的磷酸二酯链。在这种情况下产生的两个杂交体分子带有2个单链切口,当杂本导入感受态细胞后可被修复。相邻的5'磷酸和3'羟基间磷酸二酯键的形成可在体外由两种不同的DNA连接酶催化,这两种酶就是大肠杆菌DNA连接酶和T4噬菌体DNA连接酶。实际上在有克隆用途中,T4噬菌体DNA连接酶都是首选的用酶。这是因为在下沉反应条件下,它就能有效地将平端DNA片段连接起来。 DNA一端与另一端的连接可认为是双分子反应,在标准条件下,其反应速度完全由互相匹配的DNA末端的浓度决定。不论末端位于同一DNA分子(分子内连接)还是位于不同分子(分子间连接),都是如此。现考虑一种简单的情况,即连接混合物中只含有一种DNA,也就是用可产生粘端的单个限制酶切割制备的磷酸化载体DNA。在瓜作用的底物。如果反应中DNA浓度低,则配对的两个末端同一DNA分子的机会较大(因为DNA分子的一个末端找到同一分子的另一末端的概率要高于找到不同DNA分子的末端的概率)。这
限制性内切酶的一般原则和建议!
限制性内切酶的一般原则和建议! 1.如何做酶切反应? 该问题看似什么简单: DNA中加上酶,然后保温一段时间就可以了。但是在实际操作过程中,我们不断听到:切不动,装不上。问题在什么地方?能系列生产限制性内切酶的公司国际上,就那么几个,位列前 3 的是NEB, Fermentas, SibEnzyme。这些公司提供酶的品质一般都能得到保证。您可以怀疑酶的质量问题,但是更多的问题来源于模板是否合适酶切要求。下面几点对你的酶切是有帮助的。 1) 成功酶切的关键是准备好模板DNA。DNA样品中不能含有有机溶剂(会使酶变性或产生星号货性),不能含有干扰酶活性的污染物质,不能含有高浓度的EDTA (TE中的EDTA浓度较低,对Mg的浓度影响较小);同时要对DNA甲基化程度及其对酶切效率的影响要做到心中有数。 2) 选用合适的酶。根据酶切序列选用,特别注意选用甲基化对酶活性的干扰。 3) 正确使用和保存酶。酶需要保存在-20度的低温环境中,只是在需要用酶才从冰箱中取出来。运输和临时存放时需要将酶至于冰上。手拿酶管时不要接触酶管下步含酶的部分,移酶时尽可能用长TIP, 避免污染。用完后需要及时送回原处。注意:酶通常是最后加。所有4) 反应体积需要根据实验目的定,常规的酶切一般要维持在10-50ul,酶切鉴定10-20ul就可以了。 5) 模板浓度问题:浓度过高,溶液黏度过大,酶不能有效扩散,酶切效果不会好。浓度过低,也会影响酶活性。 6) 注意模板用量和反应体积的关系。对酶用量,模板用量,反应体积等要素的确定需要的是时间和经验的积累。 7) 酶切反应的各个组分加完后,需要用TIP小心混匀几次,short spin 一下就可以保温了。一般不能使用振荡器混匀。 8) 反应温度的选择。一般反应都用37度,但是 Sma I 的最适合温度是25度,37度时酶仍表现出活性,但是效率下降50%。部分从耐热菌制备的酶需要在37度以上的温度反应,如Taq I的最适温度为65度,37度保温,效率仅为前者的1/10。 9) 反应时间的选择。一般酶切鉴定30分钟就可以了。要完全酶切可以采用少量的酶长时间反应,或较高的酶量短时间处理都可以达到。在使用高酶量的时候需要注意甘油的最终浓度不要超过5%,也就是说10ul的体系,酶的用量不要超过1ul。 10) 是否和如何终止反应?酶切鉴定之类的实验不需要特殊处理。灭活的手段:加入高浓度的EDTA;65度或80度热处理20-30分钟;部分从高温菌纯化出来的内切酶由于最适的反应温度比较高,热处理灭活不一定完全,需要用苯酚/氯仿/乙醇方法纯化;电泳回收也是实验室常用除酶的手段。 2.如果遇到酶切不动或切不完全,该怎么办? 要回答这么问题常常需要了解酶活性单位是如何确定,我们多次接到这样的问题:1个单位的酶能在60分钟内切1ug的DNA,为什么我们的DNA那么少切那么长时间也不能切开或切完全?从下面几个因素去考虑: 1) 酶是否有活性:酶的活性单位通常是在60分钟酶切1ug lambda DNA或特定线状DNA所需要的酶量。鉴定酶的活性高低不是用您待切的DNA模板,也不是别的公司的酶来判定。因为不同公司酶可能是从不同系统中纯化的,虽然识别位点相同,但是酶的特性可能是有差异的。鉴定酶必须使用使用说明书上认定的酶活确定的方式,通常需要用lambda DAN做模板来判定。同时如果酶对甲基化敏感,还需要用Dcm-, Dam-的DNA.不排除由于运输或分装不当导致酶活性下降,这种情况是很少发生。我们公
双酶切连接反应
【原创】双酶切连接反应之全攻略(原创) 双酶切连接反应之全攻略 前一阵子一直在做双酶切质粒重组,失败了很多次,不过很快改善了实验方法,用2周重组了14个质粒。现就自己的体会,结合战友的宝贵经验,谈一下质粒重组的一些个人经验。 1、回收PCR产物:在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基,其原则可参照: 双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。 纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒 酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。而该酶浓度约为15单位/微升,在除外酶降解的因素外,该酶可分解15μg的DNA,而一般从1-4ml菌液提出的DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的质量好,酶切完全切得动。 2、酶切、回收后的PCR产物与载体的连接 摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则非常有必要。回收的载体片段:回收的PCR产物片段=1:10,一般取前者,后者取。pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套.1pmol 1000bp DNA=μg,如载体是5380bp,则为 ××=μg。 测DNA浓度可以在专用机子上测,注意OD值,一般约另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的MARKER每个条带约50ng。连接反应:TAKARA的连接酶上的说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为个小时足已。3,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间350 U/μl 3、转化: a、全量(10 μl)加入至100 μl JM109感受态细胞中,冰中放置30分钟。 b、42℃加热45秒钟后,再在冰中放置1分钟。 c、加入890 μl AMP阴性培养基,37℃振荡培养60分钟。 取100μl铺板。也可离心后余100μl 几个非常重要的问题 1 做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解.为保险起见,一般连接3小时,16度. 2 对含有AMP-RESISTENCE的质粒铺板时,注意加AMP时的温度,温度过高,会使克隆株无法筛选出来.我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为 55-60度,然后做的时候拿出来,这样好掌握温度。铺板前后注意用吹风机吹干 3对照的设立: 为验证双酶切是否成功,可做如下对照: A 酶切反应时加各单酶分别切,两管,用同一种BUFFER,跑胶,看单切的两管是否成线性.如两管均成线性可初步判断双酶切成功. 做转化时,也要进行对照. 设4个: A.即拿双酶切的质粒产物也进行连接反应,这个对照可进一步看双酶切是否成功,如果长出克隆,说明很有可能只进行了单酶切,如没长出克隆,则证明双酶切成功, 当然要保证感受态,培基,连接酶都'正常'的情况下. B.酶切过的未进行连接反应的双酶切产物,进行转化,这一步可以证明是否有残留的未被任何酶切的原始质粒 C.设原始质粒为对照,意为检测整个操作过程中是否有误. 阴性板上用同一批感受态细胞铺板20微升足够,检测感受态状况. 4.所有的试剂切记低温保存.一步一个脚印.不要偷懒,图省事最后却更费事.注意设立对照。
酶切反应注意事项
1、DNA纯度 一般而言,纯度高的DNA(即混入的蛋白质、RNA或多糖类物质较少)容易被限制性内切酶消化,基因重组是严重洪的所有酶促反应都有类似共性,因此应尽量提高DNA纯度。若DNA不能被限制性内切核酸酶切割,应对DNA样品进行酚抽提、乙醇沉淀等操作,以提高DNA纯度。下列因素影响DNA纯度: (1)DNA样品中常常杂有RNA,虽然它的存在不影响酶的反应速度,但RNA可和酶蛋白 发生非特异性结合而减少酶的有效浓度,使酶解不彻底。另外,RNA在凝胶电泳中 呈现的区带会掩盖该区带范围内的DNA片段的呈现,干扰DNA片段的观察。 (2)一般来说,少量蛋白质的污染对DNA酶解的影响不大,但如果杂有核酸酶等蛋白质, 就会干扰酶切反应,并影响酶解产物。有一类蛋白质叫结合蛋白,他能与DNA发生 非特异结合,不仅封闭DNA上的识别序列而影响酶切反应,而且DNA与蛋白质结 合物在凝胶电泳上迁移甚慢,从而改变DNA片段在电泳图谱中的位置,影响DNA 片段的定性分析。 (3)样品中杂有其他DNA,如制备的质粒DNA中含有染色体DNA片段等,它们不影响 酶解作用,但干扰酶解产物电泳图谱并影响以后的重组连接反应。 (4)DNA样品中的其他杂志,如Hg2+、酚、氯仿、乙醇、EDTA、SDS、NaCl等,这些杂 质常常是制备过程中不慎带进的,它们影响酶切速度,甚至改变识别特异性,出现 酶的第二活性。 2、DNA甲基化 限制性内切核酸酶的识别序列若被修饰酶产生了修饰反应(如甲基化酶的甲基化反应),则该DNA不能被限制酶再切割。甲基化酶是大多数大肠杆菌菌株中都存在 的酶系之一,有dam甲基化酶和dcm甲基化酶两大类。Dam甲基化酶在5’GATC3’ 的A上甲基化,dcm甲基化酶在5’CCAGG3’或5’CCTGG3’的C上甲基化。若出现甲基 化影响,则应使用甲基化酶缺陷菌株(dcm或dam),或使用不受甲基化酶影响的限 制性内切核酸酶,如MboI和Sau3A是同裂酶,因MboI受甲基化影响程度大,则宜 使用几乎不受甲基化酶影响的Sau3A。 3、星号活力 又称第二活力,是指该改变了酶切反应条件后特异序列识别特性降低的一种现象。 由于识别特异性降低,可能对原识别序列相似的序列也产生切割反应。如EcoRI的典型识别序列为GAATTC,条件改变后,其识别序列由原来的六核苷酸降为四核苷酸AATT。 高浓度甘油、高PH、低离子强度、β-巯基乙醇、DMSO、Mn2+等的存在可能导致酶产生星号活力。厂家提供的酶一般存放在50%甘油溶液中,若酶量站反应体系的1/10以上,将可能导致星号活力。小的反应体系若长时间在恒温水浴锅中进行酶切反应,因Eppendorf管里的内外温差将导致水分蒸发,蒸气凝结在盖子上而使反应体积缩小非常明显,将改变反应体系的组成,而容易产生星号活力,因此长时间进行酶切反应,宜使用空气恒温箱为保温设备,可避免水分蒸发和凝结。 4、终止限制性内切核酸酶反应的方法 (1)加EDT A以Mg2+,使酶失去辅助因子而终止酶切反应; (2)65℃下保温5-10min而使酶失活。但是有些酶在此温度下仍有活性,这种酶就不能 用高温失活方法来终止酶切反应; (3)加SDS至终浓度0.1%或加尿素至0.5mol/L,使酶蛋白解聚变形; (4)用等体积酚抽提酶解产物,这种方法使酶活性丧志最彻底,灭火后的样品用乙醇沉 淀法回收DNA;
酶切
DNA的酶切实验 采用粘末端连接必须对目的DNA分子和载体分子进行酶切以获得相应的粘末端进行连接。酶切可以是单酶切也可以是双酶切。单酶切操作比较简单,但双酶切如果两种酶所用缓冲液成分不同(主要是盐离子浓度不同)或反应温度不同,这时可以采用如下措施解决:1)先用一种酶切,然后乙醇沉淀回收DNA分子后再用另外一种酶切;2)先进行低盐要求的酶酶切,然后添加盐离子浓度到高盐的酶反应要求,加入第二种酶进行酶切;3)使用通用缓冲液进行双酶切。具体要根据酶的反应要求进行,尽量避免星号活力。一材料、试剂和仪器: 1 材料:质粒DNA 2 试剂:限制性内切酶、ddH2O 3 仪器:微量移液枪,离心机,水浴锅,电泳仪,紫外透射观测仪 实验程序: I. .单酶切: II. 双酶切: 注:酶切的选择原则一般是尽量扩大酶切体系,这样抑制因素得以稀释;基因组DNA或质粒DNA酶的用量较一般DNA大,一般为1μg/10U;所加酶的体积不能超过酶切总体积的1/10,否则甘油浓度会超过5%,会产生星号活力;对难切的质粒或基因组DNA应延长反应时间4—5hr, 甚至过夜。灭火限制性内切酶活性可以采用加热灭活,乙醇沉淀,酚/氯仿抽提,添加EDTA或SDS等方法,具体每一种酶可能有些方法不能完全灭活,这一点需要注意。 二. 结果与分析: 假若一种酶在环状质粒DNA中只有一个酶切位点, 且酶切彻底,紫外灯下检测电泳结果, 则单酶切应为一条带, 而双酶切则为两条带。如果条带数目多于理论值,那么有可能是酶切不完全。如果酶切结果与酶切前的质粒条带一样(超螺旋、线性和开环三条带),则说明质粒完全没有被切开。 图4 重组质粒HindIII XbaI双酶切琼脂糖凝胶电泳分析
酶切体系中不可忽视的13个实验要素
摘要: 本文展示了建立一个标准的酶切体系需要注意的13个要素,例如怎么去选择正确的酶,DNA甲基化问题,缓冲液,反应体积,反应时间和温度,终止反应,对照反应,稳定性,贮存条件等等。一、建立一个标准的酶切反应目前大多数研究者遵循一条规则,即10个单位的内切酶可以切割1g不同来源和纯度的DNA。通常,一个50l的反应体系中,1l 的酶在1X NEBuffer终浓度及相应温度条件下反应。 一、建立一个标准的酶切反应 目前大多数研究者遵循一条规则,即10个单位的内切酶可以切割1μg不同来源和纯度的DNA。通常,一个50μl的反应体系中,1μl的酶在1X NEBuffer终浓度及相应温度条件下反应1小时即可降解1μg已纯化好的DNA。如果加入更多的酶,则可相应缩短反应时间;如果减少酶的用量,对许多酶来说,相应延长反应时间(不超过16小时)也可完全反应。 二、选择正确的酶 不言而喻,选择的酶在底物DNA上必须至少有一个相应的识别位点。识别碱基数目少的酶比碱基数目多的酶更频繁地切割底物。假设一个GC含量50%的DNA链,一个识别4个碱基的酶将平均在每44(256)个碱基中切割一次;而一个识别6个碱基的酶将平均在每46(4096)碱基切割一次。内切酶的产物可以是粘端的(3''或5''突出端),也可以是平端的片段。粘端产物可以与相容的其它内切酶产物连接,而所有的平端产物都可以互相连接。相关信息参见目录的Compatible Cohesive ends And Recleavable Blunt Ends一文。 三、酶 内切酶一旦拿出冰箱后应当立即置于冰上。酶应当是最后一个被加入到反应体系中(在加入酶之前所有的其它反应物都应当已经加好并已预混合)。酶的用量视在底物上的切割频率而定。例如,超螺旋和包埋法切割的DNA通常需要超过1U/μg的酶才能被完全切割。参见目录第244和264之"切割质粒dna"和"包埋法切割DNA"。 四、DNA 待切割的DNA应当已去除酚、氯仿、乙醇、EDTA、去污剂或过多盐离子的污染,以免干扰酶的活性。DNA的甲基化也应该是酶切要考虑到的因素。 五、缓冲液 对于每一种酶NEB都提供相应的最佳缓冲液,可保证几乎100%的酶活性。使用时的缓冲液浓度应为1X。有的酶要求100μg/ml的BSA以实现最佳活性。在这种情况下,我们也相应提供100X的BSA(10mg/ml)。不需要BSA的酶如果加了BSA也不会受太大影响。关于缓冲液更详细的信息参见第234页。 六、反应体积 内切酶活力单位的定义是:1小时内,50μl反应体积中,降解1μg的底物DNA所需的酶
史上最全限制性内切酶酶切位点汇总
A系列 AatII识别位点 Acc65I识别位点 AccI识别位点 AciI识别位点 AclI识别位点 AcuI识别位点 AfeI识别位点 AflII识别位点 AflIII识别位点 AgeI识别位点 AhdI识别位点 AleI识别位点 AluI识别位点 AlwI识别位点 AlwNI识别位点 ApaI识别位点 ApaLI识别位点 ApeKI识别位点 ApoI识别位点 AscI识别位点 AseI识别位点 AsiSI识别位点 AvaI识别位点 AvaII识别位点 AvrII识别位点 BaeI识别位点 BamHI识别位点 BanI识别位点 BanII识别位点
BbvCI识别位点 BbvI识别位点 BccI识别位点 BceAI识别位点 BcgI识别位点 BciVI识别位点 BclI识别位点 BfaI识别位点 BfuAI识别位点 BglI识别位点 BglII识别位点 BlpI识别位点 Bme1580I识别位点 BmgBI识别位点 BmrI识别位点 BmtI识别位点 BpmI识别位点 Bpu10I识别位点 BpuEI识别位点 BsaAI识别位点 BsaBI识别位点 BsaHI识别位点 BsaI识别位点 BsaJI识别位点 BsaWI识别位点 BsaXI识别位点 BseRI识别位点 BseYI识别位点
BsiEI识别位点 BsiHKAI识别位点 BsiWI识别位点 BslI识别位点 BsmAI识别位点 BsmBI识别位点 BsmFI识别位点 BsmI识别位点 BsoBI识别位点 Bsp1286I识别位点 BspCNI识别位点BspDI识别位点 BspEI识别位点 BspHI识别位点 BspMI识别位点 BspQI识别位点 BsrBI识别位点 BsrDI识别位点 BsrFI识别位点 BsrGI识别位点 BsrI识别位点 BssHII识别位点 BssKI识别位点 BssSI识别位点 BstAPI识别位点 BstBI识别位点 BstEII识别位点 BstNI识别位点
原核表达条件优化
【专题讨论】原核表达条件优化! 之所以首先介绍Novagen公司的产品是因为用过它的pET系列载 体,感觉很好用。Novagen的母公司是德国默克(Merck)公司,它是国际著名的化学及制药公司总部位于德国的Darmstadt,已有300多年的历史。已在全世界55个主要国家设立了分公司,其中在28个国家建有62个生产基地。 Novagen公司出品的pET系列载体是目前应用最为广泛的原核表达系统,已经成功地在大肠杆菌中表达了各种各样的异源蛋白。pET系列载体是利用大肠杆菌T7噬菌体转录系统进行表达的载体,其表达原理见下图。 T7噬菌体具有一套专一性非常强的转录体系,利用这一体系中的元件为基础构建的表达系统称为T7表达系统。T7噬菌体基因编码的T7RNA聚合酶选择性的激活T7噬菌体启动子的转录。它是一种高活性的RNA 聚合酶,其合成mRNA的速度比大肠杆菌RNA聚合酶快5倍左右。并可以转录某些不能被大肠杆菌RNA聚合酶有效转录的序列。在细胞中存在T7 RNA聚合酶和T7噬菌体启动子的情形下,大肠杆菌宿主本身基因的转录竞争不过T7噬菌体转录体系,最终受T7噬菌体启动子控制的基因的转录能达到很高的水平。 T7噬菌体启动子的转录完全依赖于T7 RNA聚合酶,因此T7 RNA聚合酶的转录调控模式就决定了表达系统的调控方式。噬菌体DE3是λ噬菌体的衍生株,一段含有lacⅠ,lacUV5启动子和T7 RNA聚合酶基因的 DNA片段倍插入其int基因中,用噬菌体DE3的溶源菌,如BL21(DE3)、 HMS174(DE3)等作为表达载体的宿主菌,调控方式为化学信号诱导型,类似于Lac表达系统。 从开始涉及表达的时候可以根据是否要用基因本身的起始密码子进行选择,Novagen公司仅提供三个载体:pET-21(+),pET-24(+)和 pET-23(+)。如果你打算利用载体的起始密码子,那么就有许多选择。 根据是否要可溶性表达,选择加有不同标记的载体。一般说来在大肠杆菌中不加标记外源蛋白都会以不溶的包涵体形式表达。为了让外源蛋白融合表达一般说来有三个策略: 1. 与一个高度可溶的多肽联合一起表达,比如:谷胱甘肽S转移酶 (glutathione S transferase, GST)、硫氧还蛋白 (thioredoxin, Trx)和N利用质A(N utilization substance