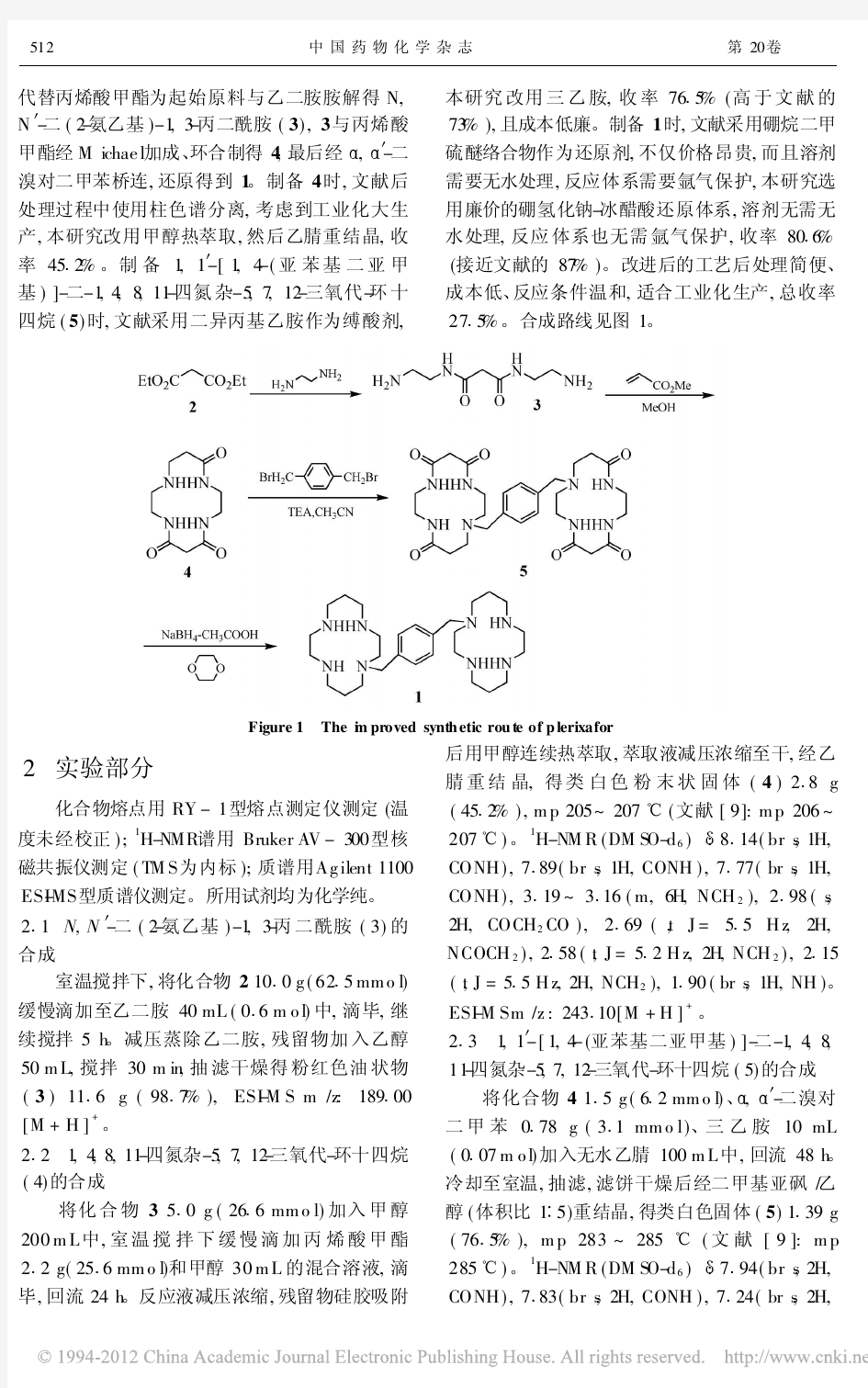
普乐沙福的合成工艺改进

瑞替加滨的合成工艺改进
收稿日期:2013-04-28 作者简介:朱磊(1987-),男(汉族),江苏泰州人,硕士研究生, E-mail :qpalzm0523@https://www.360docs.net/doc/cc13680284.html, ;*通讯作者:王浦海(1956-),男(汉族),江苏南京人,研究员,硕士生导师,主要从事药物化学教学与研究,Tel :(025)58139412,E-mail :wangpuhai@hotmail.com 。 文章编号:1005-0108(2014)01-0031-03 瑞替加滨的合成工艺改进 朱磊1,王佳乐1,王浦海 2* (1.南京工业大学药学院,江苏南京211816;2.南京工业大学江苏省药物研究所,江苏南京211816)摘要:目的改进抗癫痫药瑞替加滨的合成工艺。方法以对硝基苯胺(2)为起始原料,首先与氯甲酸乙酯反 应得到N -(4-硝基苯基)氨基甲酸乙酯(3),3经还原、氨基保护、硝化、脱保护制得N -(2-硝基-4-氨基苯基)氨基甲酸乙酯(6),6与对氟苯甲醛反应生成N -[2-硝基-4-(4-氟苯基亚甲基氨基)苯基]氨基甲酸乙酯(7), 7不经分离直接以NaBH 4还原制得N -[2-硝基-4-(4-氟苯基甲基氨基)苯基]氨基甲酸乙酯(8),最后8经三氯化 铁/水合肼还原制得抗癫痫药物瑞替加滨。结果与结论目标化合物的结构经IR、1H-NMR、13 C-NMR和HRMS (ESI )谱确证。改进后的工艺操作简单,反应选择性高,成本低,利于工业化生产,总收率为62%(以对 硝基苯胺计)。 关键词:瑞替加滨;抗癫痫药;工艺改进中图分类号:O626;R914.5文献标志码:A 瑞替加滨(retigabine ,1)化学名为N -[2-氨基-4-(4-氟苯基甲基氨基)苯基]氨基甲酸乙酯, 是由GlaxoSmithKline 和Valeant 制药公司研发的神经元钾离子通道开启剂,是一种全新作用机制的抗癫痫药。该药于2011年3月在欧盟获准上市,2011年6月在美国获准上市,用于成人部分性癫痫发作的辅助治疗。该药对耐药性部分癫痫的发作尤其有效, 可明显降低发作频率,为临床抗癫痫治疗提供了新方法[1-2] 。本文作者对瑞替加滨的合成工艺进行改进。 1合成路线 文献报道的瑞替加滨的合成方法主要有以下 4种:1)以2-硝基-1,4-苯二胺为原料,与对氟苯甲醛反应后经过两次还原,再与氯甲酸乙酯反应制 得瑞替加滨(二盐酸盐)[3-4] 。2)以2-硝基-5-氟 苯胺为原料, 与对氟苄胺反应后经还原反应,再与氯甲酸乙酯反应制得瑞替加滨(二盐酸盐)[3] 。3)以4-氟-1,2-二硝基苯为起始原料,与对氟苄胺反应制得4-(4-氟苯基甲基氨基)-1,2-二硝基苯,经还原、与焦碳酸二乙酯进行酰化制得瑞替加 滨[5-6] 。4)以N -(4-氨基苯基)氨基甲酸乙酯为原料,经氨基保护、硝化、脱保护,与对氟苯甲醛反 应制得N -[2-硝基-4-(4-氟苯基亚甲基氨基)苯基] 氨基甲酸乙酯,再经过两次还原反应制得瑞替加滨(二盐酸盐,总收率为44%)[3] 。 本文作者参考相关文献[3,7-8] ,在文献[3]报 道的方法基础上,以廉价易得的对硝基苯胺(2) 为起始原料,经取代、还原、氨基保护、硝化、脱保 护、加成消去、还原反应制得瑞替加滨(1),总收率约为62%(以对硝基苯胺计),合成路线见图1 。 Figure 1The improved synthetic route to retigabine 第24卷第1期2014年2月总117期 中国药物化学杂志Chinese Journal of Medicinal Chemistry Vol.24No.1p.31Feb.2014 Sum 117
抗高血压药缬沙坦的新合成方法
化学试剂,2009,31(4),303~304 抗高血压药缬沙坦的新合成方法 邹江,杨琰,鲁峰,王文峰3 (北京赛科药业有限责任公司,北京 101111) 摘要:以2N 2三苯甲基252(4′2溴甲基联苯222基)四氮唑为原料,与L 2缬氨酸甲酯盐酸盐反应制得N 2[[2′2(2N 2三苯甲基2四氮唑252基)2(1,1′2二苯基)242基]2甲基]2L 2缬氨酸甲酯,然后经过脱三苯甲基保护、酰化、水解得到标题化合物,总收率4918%。 关键词:缬沙坦;血管紧张素Ⅱ受体拮抗剂;合成 中图分类号:O626.2 文献标识码:A 文章编号:025823283(2009)0420303202 收稿日期:2008206217作者简介:邹江(19792),男,山东人,硕士,研究方向为原料药及医药中间体。 缬沙坦(Valsartan ,1),化学名:N 2(12氧戊 基)2N 2[42[22(1H 2四唑252基)苯基]苄基]2L 2缬氨酸,是一种血管紧张素Ⅱ的1型(AT 1)受体拮抗剂,具有全新的降压机制,降压平稳、疗效强、作用时间长、患者耐受性好;作用部位确切,降压起效温和,对心率和细胞组织影响极小,长期用药对心肾功能有较好的保护作用。 文献[123]报道的关于缬沙坦的合成方法主要有:1)以4′2甲基222氰基联苯为原料,经过溴化、水解、氧化合成4′2甲酰基222氰基联苯,与L 2缬氨酸苄酯对甲苯磺酸盐缩合后还原得到N 2[42(22氰基苯基)苄基]2L 2缬氨酸苄酯,戊酰化后得到N 2[42(22氰基苯基)苄基]2N 2戊酰基2L 2缬氨酸苄酯,然后与三丁基叠氮化锡反应成四氮唑环,最后由催化氢解得到缬沙坦;2)4′2溴甲基222氰基联苯与L 2缬氨酸苄酯对甲苯磺酸盐反应得到N 2[42(22氰基苯基)苄基]2L 2缬氨酸苄酯,戊酰化、氢解、环合后制得缬沙坦;3)4′2溴甲基222氰基联苯L 2缬氨酸甲酯盐酸盐缩合得到N 2[(2′2氰基联苯242基)甲基]2(L )2缬氨酸甲酯,戊酰化得到N 2[(2′2氰基联苯242基)甲基]2N 2戊酰基2(L )2缬氨 酸甲酯,与三丁基叠氮化锡反应得到四氮唑物,经水解后得到缬沙坦。以上3种方法都是应用氰基与叠氮化物高温反应制备四氮唑环,污染大,危险性高。 本文根据最近专利文献[4]并在其基础上进行了工艺优化。以2N 2三苯甲基252(4′2溴甲基联苯222基)四氮唑为原料,与L 2缬氨酸甲酯盐酸盐反应制得N 2[[2′2(2N 2三苯甲基2四氮唑252基)2(1,1′2二苯基)242基]2甲基]2L 2缬氨酸甲酯(2);原文献中需要制备化合物2的氢溴酸盐,我们在反应过程中加入缚酸剂制备化合物2的游离碱,不 经纯化直接投入下一步反应中。由化合物2制备 N 2(12氧戊基)2N 2[[2′2(1H 2四氮唑252基)2(1,1′2二苯基)242基]2甲基]2L 2缬氨酸甲酯(4),原文献是先酰化后脱三苯甲基保护;我们在实验中发现,这样的酰化过程经常会伴随脱三苯甲基保护的副反应,不利于反应控制;我们调整了反应的次序,先脱三苯甲基保护得到N 2[[2′2(1H 2四氮唑252基)2(1,1′2二苯基)242基]2甲基]2L 2缬氨酸甲酯(3),然后再酰化得到化合物4。在水解的操作中,原文献采用三甲基硅醇钠的方法,成本高,反应条件苛刻;我们采用稀氢氧化钾(浓度<6%)低温(25~ 30℃ )反应的方式来进行水解,取得了不错的结果,产品缬沙坦中手性异构体含量<012%。整个工艺过程,中间体不经纯化直接投入下一步反应。合成路线如下所示。 3 03第31卷第4期邹江等:抗高血压药缬沙坦的新合成方法o
缬沙坦粗品合成第一工序 甲酯
缬沙坦粗品合成第一工序甲酯 甲酯设备: (1)、2个反应釜1500升,(2)、2台水冲泵,(3)、1个1500升高位槽(抽氯化亚砜),(4)、1个甲醇罐2000升,(5)、1个蒸出甲醇罐,(6)、2个片冷冷却系统, 甲酯投料:缬氨酸150kg,甲醇(回收甲醇)860升+180升,氯化亚砜200kg,饮用水200升。 酯化操作工艺: 1、先将200kg的氯化亚砜抽入高位槽滴加待用。 2、打入酯化釜甲酯860升,再投入缬氨酸150kg盖好釜盖,开启冷冻系统,将釜内降温到-5—5度。 3、打开酸性废气,开始滴加氯化亚砜,滴加时控制釜内温度-10—15度滴加时控制时间2±0.5小时,滴加结束后升温30±10度,搅拌反应3小时。反应结束继续升温,将釜内加热到有回流状态,即温度在55—60度时反应回流5小时。 析晶操作工艺: 上述反应保温结束,将酯化釜内的料液转至结晶釜内开始升温,打开蒸馏阀门及配套的真空阀门、控制表压≤-0.06Mpa。釜内温度20—55度,减压蒸馏,当蒸出680±50升甲醇时停止蒸馏,打开甲醇管道阀门,向釜内加入180升新甲醇,再打开蒸汽阀门继续升温到
55—60度回流反应60±30分钟。 4、回流反应结束后,用冷却水将釜内料液温度降至40—50度。 5、打开真空系统,控制表压≤-0.06Mpa釜内温度35—75度,继续减压蒸馏,直至无液体流出(足够干)。 6、蒸馏结束打开氮气阀门,破釜内压力为0时关闭氮气,开废气阀门,将釜内降温至35±10度, 7、冷却完毕后,静止60分钟,再向釜内加入700升饮用水,搅拌至溶清,送入缩合釜内待用。 缬沙坦粗品合成第二工序缩合 缩合:设备2个5000升反应釜,一个盐酸高位槽1500升,一个螺冷,一台水冲泵抽料用,一个饮用水储罐2000升。 投料配比: 饮用水:700升、300升、300升、600;升饱和盐水300升、300升,碳酸钠275kg精致盐酸125升,联苯溴化物270kg、乙酸乙酯2700升、200升缬氨酸甲酯盐酸盐溶液700—800升 操作工艺 1、向釜内打入饮用水700升,然后抽缬氨酸甲酯盐酸盐溶液搅拌溶清。 2、向釜内投入碳酸钠275kg,搅拌到溶清。 3、向釜内加入2700升乙酸乙酯搅拌。
沙坦联苯的应用及合成工艺
沙坦联苯的应用及合成工艺 商品名:沙坦联苯; 英文名:2-Cyano-4'-methylbiphenyl; 简称: OTBN; 化学名: 2-氰基-4-甲基联苯; 化学分子式: C H N; 1411 分子量: 193.24 分子结构: ; 外观:白色或类白色粉末结晶; 熔程: 48°C~52°C; 含量≥99%;有关杂质含量不大于0.5%。 用途:用于合成新型沙坦类高血压药(洛沙坦、替迷沙坦、缬沙坦、伊普沙坦、伊贝沙坦等) 。 特性:沙坦联苯不溶于水,溶于甲醇、乙醇、THF(四氢呋喃)、苯、甲苯庚烷等有机溶剂。 目前治疗高血压、心脏病、中风、肾炎等循环系统疾病疗效较好的
药物是血管紧张素Ⅱ[简称A ( Ⅱ) ]拮抗体药品。 血管紧张素Ⅱ受体拮抗剂(ATⅡ)是作用于肾素--血管紧张素系统的一类药物,近年来广泛用于一线抗高血压临床用药,这类药物目前上市的有:络沙坦、缬沙坦、厄贝沙坦、替米沙坦、坎地沙坦、奥美沙坦、依普罗沙坦品种,到1999 年底在国外上市的该类药物已达9个, 这无疑对高血压疾病的治疗是一大进步。沙坦类药物具有高效、长效、安全、可以口服、耐受性好、靶器官保护等特点,并避免了非选择性ACEI 类药物引起咳嗽的不良反应,优势明显,市场占有率不断提高,成为21 世纪市场上最具发展潜力的降压药物之一。 沙坦类抗高血压药物具有巨大的潜在市场, 这些药品售价昂贵,每吨高达数万美元。据统计, 2010年全球这类药物的市场已达到266亿美元。 大多数沙坦类药物都是以沙坦联苯(2-氰基-4-甲基联苯, 2-Cyano-4'-methylbiphenyl)作为其关键的中间体,但由于其生产技术难度大、设备繁杂、可操作性差、工业生产投入高、专利保护等原因,只有少数外国公司拥有此项产品的生产技术,国内尚处于开发阶段。因此这种中间体的开发研究和生产,备受国内各化工、制药企业的重视。 沙坦联苯是沙坦类药品的基础中间体,目前沙坦类药物的市场扩大速度越来越快,发展规模越来越大。
化学化工浅谈缬沙坦的生产方法剖析
浅谈缬沙坦的生产方法 目录 目录..................................................... I 浅谈缬沙坦的生产方法...................................... II 摘要.................................................... II Abstract.................................................. III 1引言 .. (1) 1.1高血压疾病的概况 (1) 1.2关于高血压药物研制和开发的意义 (1) 1.3高血压药物研制现状 (2) 1.3.1血管紧张素II受体拮抗剂 (2) 1.3.2 AT1受体拮抗剂 (2) 1.3.3 AT2受体拮抗剂 (3) 1.3.4 ATI/AT2双重受体拮抗剂 (4) 1.3.5 AⅡ受体拮抗剂的前景介绍 (4) 2抗高血压药物缬沙坦的合成方法 (4) 2.1抗高血压药物缬沙坦 (4) 2.1.1缬沙坦的简介 (4) 2.1.2 缬沙坦的药理分析 (5) 2.2缬沙坦的合成方法 (5) 2.2.1先d后a路线合成法 (6) 2.2.2先a后d路线合成法 (8) 2.2.3先c后ad路线合成法 (10) 2.3缬沙坦的生产 (11) 2.3.1原料的检验 (11) 2.3.2各物质的合成 (15) 2.3.3缬沙坦的合成 (16) 2.3.4 结论 (16) 参考文献 (18) 致谢 (19) I
浅谈缬沙坦的生产方法 浅谈缬沙坦的生产方法 摘要 本文详细介绍了缬沙坦的各种合成方法,综合比较了各种合成方法的利弊,并分析了缬沙坦合成的国内外研究现状和发展趋势。 关键词:缬沙坦;合成;工艺流程 II
异烟肼说明书
异烟肼说明书 1 2020年4月19日
篇一:异烟肼片说明书 异烟肼片说明书 药品名称】通用名:异烟肼片曾用名:商品名:汉语拼音: yiyanjing pian 英文名:isoniazid tablets 化学名称:4-吡啶甲酰肼化学 结构式: 分子式:c6h7n3o 分 子量:137.14 【 性状】本品为白色片。 【 药理毒理】 本 品是一种具有杀菌作用的合成抗菌药,本品只对分枝杆菌,主要是 生长繁殖期的细菌有效。其作用机制尚未阐明,可能抑制敏感细菌 分枝菌酸(mycolicacid)的合成而使细胞壁破裂。 【 药代动力学】 2 2020年4月19日
品口服后迅速自胃肠道吸收,并分布于全身组织和体液中,包括脑脊液、胸水、腹水、皮肤、肌肉、乳汁和干酪样组织。并可穿过胎盘屏障。蛋白结合率仅0~10%。口服1~2小时血药浓度可达峰值,但4~6小时后血药浓度根据患者的乙酰化快慢而不一,快乙酰化者,t1/2为0.5~1.6小时,慢乙酰化者为2~5小时,肝、肾功能损害者可能延长。代谢主要在肝脏中乙酰化而成无活性代谢产物,其中有的具有肝毒性。乙酰化的速率由遗传所决定。慢乙酰化者常有肝脏n-乙酰转移酶缺乏,未乙酰化的异烟肼可被部分结合。 本品主要经肾排泄(约70%),在24小时内排出,大部分为无活性代谢物。快乙酰化者中93%以乙酰化型在尿液中排出,慢乙酰化者为63%。快乙酰化者尿液中7%的异烟肼呈游离或结合型,而慢乙酰化者则为37%。本品易经过血脑屏障,亦可从乳汁排出,少量可自唾液、痰液和粪便中排出。相当量的异烟肼可经血液透析与腹膜透析清除。 【适应症】 3 2020年4月19日
指示剂甲基橙的合成方法以及应用
目录 摘要: (2) 关键词: (2) 1前言 (2) 1.1偶氮染料的研究现状 (2) 1.2甲基橙的研究 (4) 1.2.1甲基橙的名称 (4) 1.2.2甲基橙的性质 (4) 1.3甲基橙的应用 (5) 1.3.1在酸碱滴淀中的应用。 (5) 1.3.2在氧化还原中的应用。 (5) 1.3.3在氧化还原光度分析法中的应用 (6) 1.3.4在配合物水相光度分析中的应用 (6) 三:实验部分: (6) 1、实验原理 (6) 2、实验所使用到的仪器及药品 (7) 3、实验步骤 (7) 一、低温下合成甲基橙 (7) 二、常温下合成甲基橙 (8) 四、操作重点及注意事项 (8) 五、结果与讨论 (9) 六、讨论 (10) 七、参考文献 (10) 八、致谢词 (11)
甲基橙的合成 焦婷 摘要:以对氨基苯磺酸结晶为先导化合物,利用重氮盐的制备、偶合反应来合成目的物—甲基橙。用两种方法来制备甲基橙:一,甲基橙在低温下的合成。二,常温下合成甲基橙。实验结果表明在常温上合成甲基橙的产率最高。 利用甲基橙在不同的PH值条件下所显现的颜色不同来证明所得的产品是甲基橙。 关键词:甲基橙,对氨基苯磺酸,重氮盐,偶合反应,常温 1前言 1.1偶氮染料的研究现状 偶氮染料迄今为止为止子仍然是普遍使用的最重要的染料之一。它是指偶氮基(—N=N—)连接两个芳环形成的一类有机化合物。偶氮染料是合成染料中品种最多的一类,广泛用于多种天然和合成纤维的染色和印花,也用于油漆、塑料、橡胶等的着色。 由于部分偶氮染料与人体接触过程中可释放出有致癌危险的芳香胺化合物。这种化合物致癌机理是被人体吸收后,经过一系列活化作用,使人体的DNA发生结构和功能的变化,成为人体病变的诱因。偶氮染料也因为环保问题受到了禁用,受禁品种已达100种以上,但因偶氮染料有色谱范围广,色种齐全,牢固度高等优点,仍广泛用于纺织品,皮革制品等染色及印花工艺,有机光信息记录,临床医疗诊断等生命科学领域。 随着偶氮染料禁用政策的出台,对我国这样一个纺织品和服装出口大国的影响已显现出来,在目前的国际贸易中的“绿色”已经成为一个话题,而且将一直持续下去,面对咄咄逼人的“绿色壁垒”国内染料行业也应加大力度,加紧开发替代产品。
缬沙坦工艺流程
缬沙坦半成品生产工艺 投料前检查确保合成系统、管道系统、放空系统、尾气吸收系统运行正常。生产过程中的尾气经放空总管去尾气吸收塔吸收处理。 反应原理及工艺流程简述 (1)甲酯缩合工序 L-缬氨酸与氯化亚砜反应生成L-缬氨酸甲酯盐酸盐,L-缬氨酸甲酯盐酸盐与溴代沙坦联苯反应生成N-【(2,-氰基联苯-4-基)甲基】-L-缬氨酸甲酯盐酸盐。 从人孔向甲酯反应釜R401中投入L-缬氨酸,真空抽入一定量的甲醇,夹套通入冰盐水降温至20℃以下,从高位槽经流量计向釜内滴加入氯化亚砜。约2小时滴完,滴加结束,夹套通入蒸汽升温至80℃回流反应5小时生成L-缬氨酸甲酯盐酸盐。反应结束,打开真空阀,压力保持在-0.09MPa减压蒸馏,蒸馏出来的甲醇冷凝后进入甲醇接收罐回收再利用。向釜内加入饮用水制成水溶液备用。 预先向缩合反应釜R402中加入饮用水,从人孔投入碳酸钠、溴代沙坦联苯,真空抽入一定量的醋酸乙酯,然后真空抽入上述甲酯反应釜中的备用水溶液,夹套通入蒸汽加热升温至40℃反应4小时。反应结束,静置半小时分层,从高位槽经流量计向釜内滴加盐酸,边滴加边搅拌2小时成盐,然后将物料通离心机离心甩料,滤饼烘干得缩合物N-【(2,-氰基联苯-4-基)甲基】-L-缬氨酸甲酯盐酸盐。 (2)粗品工序 N-【(2,-氰基联苯-4-基)甲基】-L-缬氨酸甲酯盐酸盐与特戊酰
氯反应生成N-(2,-氰基联苯-4-亚甲基)-N-戊酰基-L-缬氨酸甲酯,N-(2,-氰基联苯-4-亚甲基)-N-戊酰基-L-缬氨酸甲酯再与叠氮钠和三乙胺反应生成N-(1-戊酰基)-N-【4-【2-(1H-四氮唑-5-基)苯基】苄基】-L-缬氨酸(缬氨酸) 向戊酰化反应釜R403中真空抽入一定量甲苯、加入经计量的饮用水,从人孔投入缩合物N-【(2,-氰基联苯-4-基)甲基】-L-缬氨酸甲酯盐酸盐和碳酸钠,搅拌溶解,然后从高位槽向釜内缓慢滴加特戊酰氯,保持温度在60℃以下,1小时滴完后继续反应2小时。反应结束,静置分层。下层水层去污水处理,夹套通入蒸汽,开启真空阀,将釜内的有机层在90℃和-0.09MPa条件下减压蒸馏甲苯,蒸馏出的甲苯冷凝后进入甲苯接收罐回收再利用。釜内剩余物料真空抽料到四氮唑反应釜R404。从人孔向四氮唑反应釜中加入三乙胺盐酸盐、叠氮钠,夹套通入蒸汽加热至100℃,回流反应15小时。反应结束,停止通入蒸汽,加入经计量的饮用水,从人孔投入亚硝酸钠,通入冰盐水降温至20℃以下,真空抽入一定量的盐酸,静置分层,下层水层真空抽入三乙胺回收釜;上层甲苯层在四氮唑反应釜中,加入经计量的饮用水,真空抽入一定量的液碱,控制温度50℃反应5小时。反应结束,真空进料到甲苯回收釜R406中静置分层,上层甲苯回收再利用,下层水层真空抽入一定量的醋酸乙酯和盐酸,静置分层,下层水层去污水处理。夹套通入冰盐水将上层有机层降温到0-5℃结晶,析晶半小时,物料进入离心机离心甩料,所得滤饼烘干得缬沙坦粗品,离心母液进入醋酸乙酯母液接收槽。从高位槽向三乙胺回收釜R405中的
第十二章_嘌呤代谢最终版本_王忠超、孙晓娟汇总
第十二章嘌呤代谢系统 第一节概述 嘌呤代谢是指核酸碱基腺嘌呤及鸟嘌呤等的嘌呤衍生物的活体合成及分解。动物,其嘌呤化合物几乎全部氧化为尿酸,分别以不同形式而排出。人体尿酸主要由细胞代谢分解的核酸和其他嘌呤类化合物以及食物中的嘌呤,经酶的作用分解而来。为了了解尿酸的生成机制,首先要了解嘌呤代谢及其调节机制。 一、嘌呤代谢调节 嘌呤代谢速度受1-焦磷酸-5-磷酸核糖(PRPP)和谷氨酰胺的量以及鸟嘌呤核苷酸、腺嘌呤核苷酸和次黄嘌呤核苷酸对酶的负反馈控制来调节。次黄嘌呤-鸟嘌呤磷酸核糖转移酶和黄嘌呤氧化酶,为嘌呤磷酸核糖焦磷酸酰胺移换酶,是嘌呤代谢过程中的关键酶,它们的作用点见下图12-1。 注:E1:磷酸核糖焦磷酸酰胺移换酶;E2:次黄嘌呤脱氢酶;E3腺苷酸代琥珀酸合成酶;E4次黄嘌呤-鸟嘌呤磷酸核糖转移酶;E5黄嘌呤氧化酶;→表示负反馈控制。 由核酸分解代谢为尿酸是一个十分复杂的过程,主要有以下三种生成途径:
(1)核酸→鸟嘌呤核苷酸→鸟嘌呤→黄嘌呤→尿酸。 (2)核酸→腺嘌呤核苷酸→腺嘌呤→黄嘌呤→尿酸。 (3)5-磷酸核糖+ATP→次黄嘌呤核苷酸→次黄嘌呤→黄嘌呤→尿酸。 此乃尿酸生成的一个总轮廓,中间有许多环节已被省略,在尿酸生成的过程中,有多种酶的参与和调节。但从上述尿酸生成的简要过程中可以看出,嘌呤是尿酸生成的主要来源。因此,嘌呤合成代谢增高及(或)尿酸排泄减少均可造成血清尿酸值增高。 生物化学研究表明,人体体内约有8种酶参与了尿酸的生成过程,其中有7种酶均促进尿酸生成,它们包括:①磷酸核糖焦磷酸酰胺转移酶;②磷酸核糖焦磷酸合成酶;③腺嘌呤磷酸糖核糖苷转移酶;④腺苷去胺基酶;⑤嘌呤核苷酸磷酸酶;⑥5-核苷酸酶;⑦黄嘌呤氧化酶。这些酶的活性增加时,尿酸生成即增加;在这些酶中,以黄嘌呤氧化酶最为重要。另一种次黄嘌呤-鸟嘌呤磷酸核糖转移酶,其作用和上述7种酶正好相反,当其活性增强时可抑制尿酸生成,活性减弱时则尿酸生成增加。酶缺陷包括某种酶的数量增多或活性增强和某种酶的完全性缺乏或部分缺乏,皆可导致嘌呤合成加速和尿酸生成增多。酶缺陷在痛风发病中占有十分重要的地位,但大多数很难得到证实,仅少数病人可以鉴定出酶缺陷。嘌呤排出物的多样性,可能与在进化过程中发生的酶缺失现象(eezymaphresis)有关[1、2]。对导致过量嘌呤生物合成的机制,有嘌呤代谢酶的数量增多或活性过高,或酶活性降低或缺乏。 二、尿酸代谢的平衡 血清中尿酸浓度,取决于尿酸生成和排泄速度之间的平衡。尿酸是嘌呤代谢的终末产物,体内尿酸的积聚,可见于如下的5种情况:①外源性吸收增多,即摄食富含嘌呤的食物增多; ②内源性生物合成增加,包括酶缺陷,如核酸分解加速和嘌呤基氧化产生尿酸增多;③排泄减少,即由肾脏经尿排出减少和由胆汁、胃肠分泌后,肠道细菌分解减少;④体内代谢减少,即尿酸内源性破坏减少;⑤上述综合因素或不同因素的组合。 拥有尿酸(氧化)酶的物种,能将尿酸转化为溶解性较高、更易排出的尿囊素(allantoin),故血清尿酸水平低而无痛风存在,人和几种类人动物是在进化过程中发生尿酸氧化酶基因突变性灭活的,从这点来说,人类的高尿酸血症是由尿酸分解代谢的先天性缺陷造成[3]。高尿酸血症血清中尿酸浓度取决于尿酸生成和排泄速度之间的平衡,人体内尿酸有两个来源,一是从富含核蛋白的食物中核苷酸分解而来的,属外源性,约占体内尿酸的20%;二是从体内氨基酸、磷酸核糖及其他小分子化合物合成和核酸分解代谢而来的,属内源性,约占体内总尿酸的80%。对高尿酸血症的发生,显然内源性代谢紊乱较外源性因素更为重要。核素示踪研究,正常人体内尿酸池的尿酸平均为1200mg,每天产生约750mg,排出500~1000mg,约2/3经尿排泄,另1/3由肠道排出,或在肠道内被细菌尿酸氧化酶分解。
沙坦联苯的应用及合成工艺模板
沙坦联苯的应用及合成工艺 商品名: 沙坦联苯; 英文名: 2-Cyano-4'-methylbiphenyl; 简称: OTBN; 化学名: 2-氰基-4-甲基联苯; 化学分子式: C H N; 1411 分子量: 193.24 分子结构: ; 外观:白色或类白色粉末结晶; 熔程: 48°C~52°C; 含量≥99%;有关杂质含量不大于0.5%。 用途:用于合成新型沙坦类高血压药(洛沙坦、替迷沙坦、缬沙坦、伊普沙坦、伊贝沙坦等) 。 特性: 沙坦联苯不溶于水,溶于甲醇、乙醇、THF( 四氢呋喃) 、苯、
甲苯庚烷等有机溶剂。 当前治疗高血压、心脏病、中风、肾炎等循环系统疾病疗效较好的药物是血管紧张素Ⅱ[简称A ( Ⅱ) ]拮抗体药品。 血管紧张素Ⅱ受体拮抗剂( ATⅡ) 是作用于肾素--血管紧张素系统的一类药物, 近年来广泛用于一线抗高血压临床用药, 这类药物当前上市的有: 络沙坦、缬沙坦、厄贝沙坦、替米沙坦、坎地沙坦、奥美沙坦、依普罗沙坦品种, 到1999 年底在国外上市的该类药物已达9个, 这无疑对高血压疾病的治疗是一大进步。沙坦类药物具有高效、长效、安全、能够口服、耐受性好、靶器官保护等特点,并避免了非选择性ACEI 类药物引起咳嗽的不良反应, 优势明显, 市场占有率不断提高, 成为21 世纪市场上最具发展潜 力的降压药物之一。 沙坦类抗高血压药物具有巨大的潜在市场, 这些药品售价昂贵, 每吨高达数万美元。据统计, 全球这类药物的市场已达到266亿美元。 大多数沙坦类药物都是以沙坦联苯(2-氰基-4-甲基联苯, 2-Cyano-4'-methylbiphenyl)作为其关键的中间体,但由于其生产技 术难度大、设备繁杂、可操作性差、工业生产投入高、专利保护等原因,只有少数外国公司拥有此项产品的生产技术,国内尚处于开发阶段。因此这种中间体的开发研究和生产,备受国内各化工、制药企业的重视。 沙坦联苯是沙坦类药品的基础中间体, 当前沙坦类药物的市 场扩大速度越来越快, 发展规模越来越大。
抗结核病新药研发进展
龙源期刊网 https://www.360docs.net/doc/cc13680284.html, 抗结核病新药研发进展 作者: 来源:《上海医药》2008年第11期 肺结核仍是全球最常见和最严重的感染性疾病之一。世界卫生组织估计,全球每年新诊出肺结核病例数约达880万人,每年有近160万人死于该病。肺结核的发生率逐年上升,而多药耐药(特别是泛耐药)肺结核的发生率也在全球许多地区持续升高,由此成为全球一大严重公共卫生问题。相比之下,新药的开发却滞后于肺结核的治疗和预防需求。自40多年前利福平(rifampin)上市后,全球范围内仅批准了利福布丁(rifabutin)和利福喷丁(rifapentine)两 个新药治疗肺结核。为控制肺结核的流行趋势,临床迫切需要有新的抗结核病药物,尤其是能用于潜伏性结核杆菌感染患者化学预防及对难治性肺结核即多药耐药和泛耐药肺结核治疗有效的抗结核病新药。 开发抗结核病新药的主要目标包括寻找快速作用药物,以期能够缩短现行肺结核长治疗期,及开发对耐药结核杆菌有活性药物、开发对持续性和休眠性结核杆菌均具活性药物等。尽管目前抗结核病药物开发管线有限,但还是有一些新型结构类别化合物如硝基咪唑类、二芳基喹啉类、噁唑烷酮类、乙二胺类和氟喹诺酮类等化合物正在进行临床研究。 1 硝基咪唑类化合物 许多硝基咪唑类化合物(特别是咪唑并噁唑和咪唑并噁嗪衍生物)都已经体内、外试验证实具有强力的抗分枝杆菌活性,它们对结核杆菌的最低抑菌浓度(MIC)为0.015~1.95 μg/mL。不过,这些物质的致癌性和致突变性水平相对较高,因而阻碍了它们进入临床研究。值得注意的是,常用于治疗厌氧菌感染的抗菌药甲硝唑(metronidazole)已显示其在MIC时对处于厌氧条件下的休眠性结核杆菌存活有显著活性,同时对多药耐药肺结核株也具活性。近期一项研究还指出,甲硝唑联合利福平治疗休眠性结核杆菌患者有效。甲硝唑目前正在进行用作抗多药耐药肺结核药物的Ⅱ期临床研究,但此类化合物中最有希望获准治疗肺结核的可能还是两个在研药物——PA-824和OPC-67683。 1.1PA- PA-824为硝基咪唑并吡喃衍生物,对多药耐药肺结核株呈高度活性且对休眠性结核杆菌具杀灭作用。PA-824在结核杆菌中的靶的尚不明了,推测与分枝菌酸和核酸的生物合成有关。PA-824属硝基咪唑类化合物的前体药物,须在其结构中的硝基经低氧化还原电位的氢化物转移辅酶的作用还原性地激活才会呈现抗结核杆菌效应,而此过程是由Rv3547基因编码的依赖性硝基还原酶催化的。PA-824不仅对多药耐药肺结核株的MIC值低至
沙坦联苯的合成工艺研究
沙坦联苯的合成工艺 摘要:综合分析沙坦联苯的各种合成工艺的利弊之后 ,确定以对溴甲苯和邻氯苯甲腈为原料一步催化合成。并对该工艺的反应配比、催化剂配比、反应温度、滴加速度进行了深入细致的研究 ,确定摩尔比邻氯苯甲腈:对溴甲苯: NiCl 2·6H2O:PPh 3 :Zn =0.3:0.25:0.125:0.5:0.275可得到收率85%、 含量 99.0%以上产品。 关键词:对溴甲苯;邻氯苯甲腈;沙坦联苯;合成 Study on Syntheti c Techn ique of Sartanbiphenyl LU J in2liang (Chemistry Institute of J iangxi, J iangxiNanchang 330029) Abstract: The writer of this essay tries t o catalyze and synthesize the raw material of p2 br omot oluene and o2 chl orobenzonitrile int o sartabi phenyl according t o analysis on the advantages and disadvantages in different kinds of synthesis technics . And the reacti on rati o, reacti on temperature and titrati on rate of this technics are further studied in the paper . When the rati o of o2 chl orobenzonitrile t o p2 br omot ol2 uene to NiCl 2·6H2O:PPh 3 :Zn =0.3:0.25:0.125:0.5:0.275 (mol) . The yield and content of the p roduct can reach 85% and more than 99 . 0 % res pectively . Key words: p2 B r omot oluene, o2 Chl orobenzonitrile, Sartanbi pheny, Synthesis
发酵法生产鸟苷的工艺优化
发酵法生产鸟苷的工艺优化 摘要以菌株Bacillus Subtilis-BB518为生产菌株,对该菌株进行摇瓶发酵试验优化培养基组成。然后,利用该培养基配方进行50L全自动发酵罐发酵试验选择适当的溶解氧条件。最终,该菌种在优化的培养条件下鸟嘌呤核苷发酵水平达到17.8g/L,比出发菌株发酵产苷提高了18.7%。 关键词鸟嘌呤核苷优化溶解氧 1960年,Kuninaka[1]发现5′鸟苷酸具有较强烈的增鲜功能,在谷氨酸钠中加入1.5~2%的鸟苷酸钠可使其鲜味增加10~25倍,而鸟嘌呤核苷(简称:鸟苷)是生产鸟苷酸的原料;同时鸟苷又是抗病毒药物三氮唑核苷和无环鸟苷的前体。因此,大批量生产鸟苷前景广阔。 目前,核苷酸的工业化生产方法主要有三种:酶水解RNA法、菌体自溶法和发酵法。菌体自溶法由于产量低,提取困难而基本上不用;而两步发酵法生产鸟苷酸由于其产率高、周期短、控制易、产量大等优点,以及鸟苷可以直接透过细菌细胞膜,溶解度较低,基本上不会对嘌呤合成造成反馈抑制,通过化学或生物磷酸化形成鸟苷酸的工艺又相当成熟,因而目前已经成为工业生产鸟苷酸的主要方法。 虽然我国自80年代初已经开展了鸟苷类物质发酵的研究[2~4],但是一直进展不大,因而鸟苷生产成本居高不下,成为限制鸟苷酸工业发展的重要环节。本课题通过摇瓶正交试验以及50L全自动发酵罐溶解氧控制试验对过程的工艺进行了优化,从而使鸟苷的发酵产率提高了18.7%,发酵产苷达到了17.8g/L。 1 材料与方法 1.1 菌种:枯草芽孢杆菌(B.Subtilis)BB518 1.2 培养基 1.2.1 斜面及茄子瓶培养基配方 葡萄糖10 g/L,酵母膏10 g/L,蛋白胨10 g/L,氯化钠5 g/L,琼脂20 g/L,pH值7.0,0.1Mpa灭菌20min。 1.2.2 种子培养基配方 葡萄糖20 g/L,酵母膏10 g/L,硫酸铵5 g/L,氯化钠5 g/L,玉米浆10 g/L,pH值7.0,0.1Mpa灭菌20min。 1.2.3 发酵培养基配方 葡萄糖120 g/L,酵母粉20 g/L,玉米浆20 g/L,硫酸铵18 g/L,磷酸二氢钾4g/L,硫酸镁8g/L ,碳酸钙20g/L, pH值7.0,0.1Mpa灭菌15min。 1.3 仪器设备 FUS-50L(A)全自动发酵罐,往复式摇床。 1.4 分析方法 pH测定:Mettler Toledo在线pH检测系统,pH计。 DO测定:Mettler Toledo在线DO检测系统。 鸟苷测定:纸层析法,展开剂为异丙醇:氨:水=7:2:1(v/v)。 还原糖测定:改良斐林法。 细菌生长比浊度测定: 752分光光度计在590nm下测定OD值。 1.5 培养方法
结核分枝杆菌异烟肼耐药性研究进展
=综述>结核分枝杆菌异烟肼耐药性研究进展 朱艳伶1,2,万康林2,沈国顺1 近20余年来,由于耐药结核杆菌尤其是耐多药结核杆菌(MDR-TB)的肆意横行,各国的结核病疫情均呈回升趋势,全球发病率和死亡率不断升高112。我国的耐药情况尤为严重。据统计目前初始耐药率为1816%,获得性耐药率为4615%,全国现有耐药的涂阳肺结核病人约42万;在全球对53个国家的耐药调查中,河南省的原发MDR-TB耐药率位居第2位。因此在WHO最近公布的全球38个国家和地区的结核病耐药监测资料中,中国被列为/特别引起警示的国家和地区0之一122。近年来,各国学者采用分子生物学技术对结核杆菌耐药机制进行深入研究,定位了结核分枝杆菌包括异烟肼在内的耐药基因的位置和基因突变位点,从而促进了第一代快速鉴定耐药突变菌株方法的建立和新型抗结核药物的开发。 异烟肼作为最主要的抗结核药物之一,是多种药物联合化疗治疗结核病的基本组成部分,但也是最易产生耐药性的抗结核药物。已发现结核分枝杆菌对异烟肼耐药涉及多个基因变化:katG,inhA,kasA,ndh,及oxyR-ah pC基因连接区,突变形式多样化,并且突变位点又不确定更加剧了结核病防治的困难。因此,对耐药菌的耐药机制进行深入全面的了解,建立方便快捷经济的检测方法,从而开发新的抗结核药物和开展更有效的化疗是目前亟待解决的问题。 1结核杆菌异烟肼耐药机制 异烟肼(Isoniazid,INH)是1912年由两个捷克生化学家首次合成的,但它强有力的抗生素活性直到1951年才被发现。作为使用了40余年的抗结核一线化疗药物,其作用主要是通过抑制结核杆菌分枝菌酸的生物合成,造成细胞壁破损而杀菌。但是在单药化疗和不适当化疗期间容易产生耐药性。INH 的耐药性与一个或多个基因的各种突变有关,这些基因主要包括编码触酶-过氧化物酶的katG基因,编码烯酰基载体蛋白还原酶的inh A基因,编码烷基-氢过氧化物还原酶的ahpC 基因,编码B-酮酰基载体蛋白合成酶的Kas A基因及调节分枝杆菌氧化-应激的oxyR基因。 111katG 研究表明INH实际上是一个药物前体,需要经结核分枝杆菌触酶-过氧化物酶活化后才发挥抗结核作用,而触酶-过氧化物酶则由katG基因所编码。最新研究认为katG基因的变异(包括突变,缺失,插入等)导致的结核分枝杆菌触酶-过氧化物酶活性降低或缺失可以解释90%以上的INH耐药。20世纪90年代早期,张132等将ka tG基因克隆导入耐INH的耻垢分枝杆菌可恢复INH敏感性。 作者单位:11沈阳农业大学畜牧兽医学院,辽宁省沈阳市东陵区120号110161;21中国疾病预防控制中心传染病预防控制所,北京昌平流字五号102206 通讯作者:万康林 近年来研究发现INH耐药性结核分枝杆菌中katG基因突变最主要的是点突变。不同地区菌株点突变的位置变化较大,但约40%的变异出现在S315T位置。此位置变异通过对katG 活性位点甲基化阻碍了INH与katG的结合,导致酶失去活化INH的能力。Coll等142对来源于巴塞罗纳的61株异烟肼耐药株进行分析,其中55%分离株被检测到katG基因有所改变,最普遍的变异是发生在第315位密码子(占32%),这些菌株显示了高水平的耐药,但大多数仍保留了相当的触酶-过氧化物酶的活性。ka tG基因其他位置上的变异如D63E,H108Q, T262R,A350S等对于酶的稳定性和催化能力也很重要,可以导致细菌对INH产生不同水平的耐药性和保留不同的触酶-过氧化物酶活性152。KatG第315位点突变率的特点还体现在地区的差异上。如国内的陈曦162等对临床分离的101株耐药菌株进行分析,结果第315位点的突变率占3816%(39/101)。这一结果明显低于南非(64%,80/124)172、俄罗斯西北部地区(93%,191/204)182,而又明显高于芬兰(7%,4/54)、新加坡(26%,41/160)192等地区的报道。 112oxyR-ahpC Christman1102等在研究大肠杆菌和伤寒沙门菌的基础上推断出在肠菌属和其他的微生物里oxyR调节子控制着一套复杂的氧化调节系统,它在对环境刺激反应时被激活。oxyR作为一个调节蛋白它即是氧压的感应器又是基因转录的活化剂, oxyR控制编码解毒酶基因如触酶-过氧化物酶(katG编码)和烷基氢过氧化物酶(ah p C编码)的表达。研究表明在结核分枝杆菌复合体中的oxyR基因发生大量的移码突变和缺失、失活,使其成为一个假基因。Deretic1112等用这种突变体与携带具有活性的oxyR-ah p C基因的质粒组成一个重组体,结果对异烟肼耐药,表明结核分枝杆菌对异烟肼的敏感性在很大程度上由于oxyR调节子的畸变。 一般研究者发现在异烟肼耐药菌株中oxyR启动子序列的突变常伴随着katG活性的不足。因此一般将ah pC突变作为katG基因损伤的标志1122。另外Dhandayu thapani1132等通过Wes-tern免疫斑点分析得出突变的等位基因能增强转录活性,即某些katG突变的耐异烟肼分离株存在ahpC启动子的突变,以增强ah p C表达来补偿触酶-过氧化物酶的缺乏,从而抵抗宿主巨噬细胞的氧化。Dhandayuthapani又将纯化的ah pC蛋白导入耻垢分枝杆菌,结果它对INH高度敏感,而在此菌中过表达ah p C蛋白引起对INH耐药。AhpC编码基因突变极少,可能与耐异烟肼无关。 113inhA inh A基因编码的NADH依赖的enoyl-Acp还原酶催化$-不饱和脂肪酸转化为饱和脂肪酸的还原反应,分枝杆菌利用该产物合成的超长链A-脂肪酸是其细胞壁的重要成分。INH
第九章 药物合成设计原理和方法 练习题
第九章药物合成设计原理和方法练习题 一、名词解释 1、靶分子: 2、合成子: 二、完成下列反应 1、生物碱鹰爪豆碱的合成 2、喜树碱中间体的喹啉环的合成 3、β- 咔啉的合成 4、合成 ArCHCOOH CH3 5、全身麻醉药氟烷的合成。 F3C CHBrCl
三、按要求完成下列化合物全合成。 1、采用逆合成分析法完成布洛芬(Ibuprofen)的逆推过程并写出合成的反应。 i-Bu COOH 2、采用逆合成分析法完成下面化合物的逆推过程并写出合成的反应。 CHO OH 3、采用逆合成分析法完成茉莉酮的逆推过程并写出合成的反应。 O 4、采用逆合成分析法完成下面化合物的逆推过程并写出合成的反应。 MeO2C CHO COOMe
5、采用逆合成分析法完成下面化合物的逆推过程并写出合成的反应。 CO 2Me OH 6、完成非那西丁(Phenacetin)的合成反应。 NHCOCH 3 OH 7、完成吲哚美辛(Indometacin)的合成反应。 CH 2COOH CH 3 CO Cl O H 3C 8、 完成芬太尼(Fentany Citrate)的合成。 CH 3CH 2CON N CH 2CH 2
9、完成盐酸多巴(Dopamine Hydrochloride)的合成。 OH OH NH2 HCl 10、完成盐酸可乐(Clonidine Hydrochloride)的合成。 Cl Cl N H H N HCl 11、完成硫酸沙丁胺醇(Sulbutamol sulfate)的合成。 OH CH2OH CHCH2NH(CH3)3 OH 1/2 H2SO4
异烟肼说明书
篇一:异烟肼片说明书 异烟肼片说明书 药品名称】通用名:异烟肼片曾用名:商品名:汉语拼音:yiyanjing pian 英文 名:isoniazid tablets 化学名称:4-吡啶甲酰肼化学结构式: 分子式:c6h7n3o 分子 量:137.14 【性 状】本品为白色片。 【药 理毒理】 本品 是一种具有杀菌作用的合成抗菌药,本品只对分枝杆菌,主要是生长繁殖期的细菌有效。其作 用机制尚未阐明,可能抑制敏感细菌分枝菌酸(mycolicacid)的合成而使细胞壁破裂。 【药 代动力学】 本品 口服后迅速自胃肠道吸收,并分布于全身组织和体液中,包括脑脊液、胸水、腹水、皮肤、肌 肉、乳汁和干酪样组织。并可穿过胎盘屏障。蛋白结合率仅0~10%。口服1~2小时血药浓 度可达峰值,但4~6小时后血药浓度根据患者的乙酰化快慢而不一,快乙酰化者,t1/2为 0.5~1.6小时,慢乙酰化者为2~5小时,肝、肾功能损害者可能延长。代谢主要在肝脏中 乙酰化而成无活性代谢产物,其中有的具有肝毒性。乙酰化的速率由遗传所决定。慢乙酰化者 常有肝脏n-乙酰转移酶缺乏,未乙酰化的异烟肼可被部分结合。 本品 主要经肾排泄(约70%),在24小时内排出,大部分为无活性代谢物。快乙酰化者中93%以 乙酰化型在尿液中排出,慢乙酰化者为63%。快乙酰化者尿液中7%的异烟肼呈游离或结合 型,而慢乙酰化者则为37%。本品易通过血脑屏障,亦可从乳汁排出,少量可自唾液、痰液和 粪便中排出。相当量的异烟肼可经血液透析与腹膜透析清除。 【适 应症】 (1) 异烟肼与其它抗结核药联合,适用于各型结核病的治疗,包括结核性脑膜炎以及其他分枝杆菌 感染。 (2) 异烟肼单用适用于各型结核病的预防: ①新 近确诊为结核病患者的家庭成员或密切接触者; ②结 核菌素纯蛋白衍生物试验(ppd)强阳性同时胸部x射线检查符合非进行性结核病,痰菌阴 性,过去未接受过正规抗结核治疗者; ③正 在接受免疫抑制剂或长期激素治疗的患者,某些血液病或网状内皮系统疾病(如白血病、霍奇 金氏病)、糖尿病、尿毒症、矽肺或胃切除术等患者,其结核菌素纯蛋白衍生物试验呈阳性反 应者;④35岁以下结核菌素纯蛋白衍生物试验阳性的患者;