临床试验监查报告模板总结模板计划模板.doc

精品文档
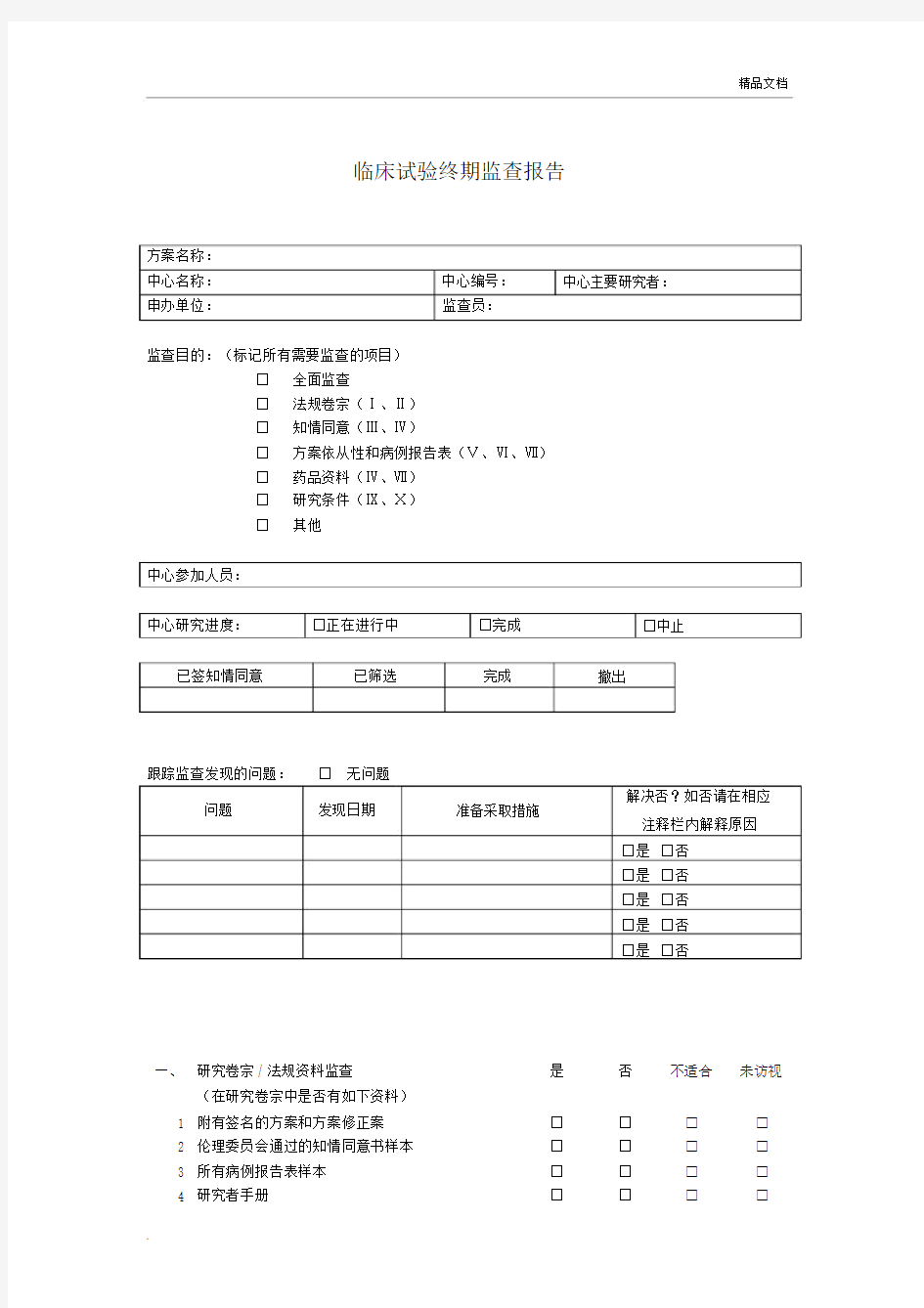
临床试验终期监查报告
方案名称:
中心名称:中心编号:中心主要研究者:
申办单位:监查员:
监查目的:(标记所有需要监查的项目)
□全面监查
□法规卷宗(Ⅰ、Ⅱ)
□知情同意(Ⅲ、Ⅳ)
□方案依从性和病例报告表(Ⅴ、Ⅵ、Ⅶ)
□药品资料(Ⅳ、Ⅶ)
□研究条件(Ⅸ、Ⅹ)
□其他
中心参加人员:
中心研究进度:□正在进行中□完成□中止已签知情同意已筛选完成撤出
跟踪监查发现的问题:□ 无问题
解决否?如否请在相应问题发现日期准备采取措施
注释栏内解释原因
□是□否
□是□否
□是□否
□是□否
□是□否
一、研究卷宗 / 法规资料监查是否不适合未访视
(在研究卷宗中是否有如下资料)
1 附有签名的方案和方案修正案□□□□
2 伦理委员会通过的知情同意书样本□□□□
3 所有病例报告表样本□□□□
5 研究人员履历 /资格说明□□□□
6 研究人员相关培训文件□□□□
7 伦理委员会通过的研究相关资料□□□□
8 实验室资格及正常值范围□□□□注释:
二、中心监察
1所有需要的文件是否都有
2对开展临床试验至关重要的设施是否都正常运转
3研究者和其工作人员保留着和开展试验相关的记录
注释:是否不适合未访视□□□□□□□□
□□□□
三、知情同意 / 入选情况是否不适合未访视
1 已签署的原始知情同意书都在受试者档案里□□□□
2 是否有筛选日志□□□□
3 是否有入选日志□□□□注释:
四、研究药物是否不适合未访视
1 试验药物是否保存在安全的地方□□□□
2 试验药物是否由专人分发□□□□
3 药物运送记录及时准确并有日期及签名□□□□
4 药物分发和数量记录是否及时准确□□□□
5 药物是否按照方案分发□□□□
6 过期的药物是否回收□□□□注释:
五、方案依从性是否不适合未访视
1 筛选过程正确□□□□
2 所有受试者都符合入选 /排除标准□□□□
3 不良事件是否正确报告,记录,评价和解决□□□□
4 受试者到诊及过程是否按照方案规定顺序完成□□□□
5 是否按照方案给予研究干预□□□□
6 是否有严重不良事件( SAEs)□□□□注释:
六、病例报告表( CRFs)和源文件是否不适合未访视
1 能否提供 CRFs 和相应的源文件查阅□□□□
2 源文件是否足以供 CRFs 核查□□□□
3 CRFs 是否完整,清晰,准确□□□□
4 CRFs 是否及时完成和递交(如数据录入)□□□□注释:
七、违背方案是否不适合未访视
1 以前没有注意到的违背方案之处,监查是否□□□□
发现以及正确记录
注释:
八、中心实验室规程是否不适合未访视
1 样本是否按照方案收集和保存□□□□
2 医务人员是否对实验室数据的临床意义作出□□□□
合适的评价和记录
注释:
九、研究设施 / 人员招募情况是否不适合未访视
1 研究中心的设施是否还合适□□□□
2 工作人员是否还合适□□□□
3 目前受试者入选率是否符合要求□□□□注释:
十、中心现状是否不适合未访视
1中心是否有可以影响开展临床试验的变化□□□□
注释:
小结
监察员签字:
日期:
临床试验总结报告的体例格式和内容要求内容
临床试验总结报告体例和容要求 1.题目封面 封面题页应包括如下容: ——试验题目 ——试验药物/研究产品的名称 ——试验用药品的适应症 ——如不能在题目中表明、则简要描述(以1~2句话)设计(平行、交叉、双盲、随机)比较(安慰剂、活性成分、剂量/反应),间隔,剂量和病例数。 ——申办者 ——原始规划与计划的确认(编码或数目,确认日期) ——试验预期进度与进程安排 ——研究开始数据(受试的第一位病人,或任何其它数据) ——后期研究终末数据(最终一个受试者的名称、或终末研究者完成的数据) ——试验研究完整的数据(最后的受试者) ——参加者或合作者或主办者责任医务官等人士的名称和职责。 ——药业公司名称/主办方签名(在公司/主办者中负责研究报告的人,公司/主办方联络者的,和传真,以备在回顾研究报告提出问题时,在此页码或应用的字母有所表明)。 ——表明该项研究是否在优良临床试验管理规(GCP)条件下进行的包括应达到的基本文件、研究设施、人员知识结构,统一培训等。 ——报告的资料(通过题目和资料,确定与该项研究相同的任何其它较早报告) 2.容概述 应提供—个临床试验的主要容提要(通常限于3页容),该提要应包括试验题目、研究人员、研究出版物名称、试验时间、试验目的、试验方法、试验研究样本、诊断及进入研究的主要标准、试验的产品、剂量、给药方式及批号、治疗持续时间、参考的治疗、剂量、给药方式及批号、评价标准(有效性、安全性)、统计方法、总结——结论(效能结论、安全性结论、结论)及报告日期。该提要应包括表明结果的数字资料,而不仅仅是文字和P值(见附件l,研究容概述)。 3.容目录 容表格应包括: ——页码或其它每一个部分的局部资料,包括归纳性表格、图和图表。 ——附录、列表和任何能提供的病例报告形式的汇总和定位。 4.名词、术语缩写和定义 应提供报告中使用的—系列缩写符、特殊的或不常用的术语,或测量单位定义,应拼写出缩写术语,并在文中首次出现时表明其全称。
会议情况汇报模板
业内人士回答:会议情况汇报主要是全面精确传达会议精神,把涉及本单位要做的事讲清楚,还可提出自己的贯彻落实意见。一般如下: 标题为“关于****会议的情况汇报” 第一段描述会议简况,大致为“**年**月**日,***召开了***会议,***主持,***出席,***等单位负责同志参加了会议。按照领导要求,我单位***同志(你自己)参加了会议。会上,****(简要描述会议议程,如:听取了***关于***的情况汇报,与会人员就***进行了讨论,对***工作进行了部署,最后***作了重要讲话,就****提出了具体要求)。 第二段重点写会议精神(实际上就是领导讲话精神,如有几个领导先后讲过话,主要写最大的领导也就是最后讲话领导的话)。大致这样写:“***(最大的领导)在讲话中要求:一是***;二是***;三是***。会上,****(其他领导)也分别作了讲话,要求***(概括几句即可)”。 此段描述后,如有会议特别要求本单位要做什么而且内容十分具体又很多的时候,可专门写一段“会议要求****(本单位名称)要抓紧做好以下具体工作:****”。一般如会议只是普遍部署工作,此段可不写(有关要求已在上面的领导讲话中了)。 最后,如你有点想法或是领导还要求你回来后把自己单位应该怎么做也在报告中一并写入,可提一段贯彻落实意见(建议)。一般这样“根据此次会议精神,建议我单位抓紧做好有关贯彻落实工作:一是认真学些传达会议精神,***;二是制定专门工作方案,**;三是***”。此段是可以不写的,但有的领导会希望这样,因为可以省去自己再开会研究提出具体贯彻落实要求,只需在你的报告上画圈批字再转给该看的部门就行了。 提醒要点:重点是把会议上领导的讲话也就是会议要求讲明白,尽量忠实领导讲话原意。你们的领导要看的其实就是上级领导讲什么,今后好对照工作。其他单位在会上发什么言等等不用写内容,只交代一下即可。
临床试验总结报告的设计与撰写
一、临床试验方案设计 临床试验方案由研究者或申办者拟订,应符合GCP要求。研究者和申办者均应在已制定的临床试验方案上签名并签署日期。 临床试验设计的基本原则:①代表性:受试者样本符合总体规律;②重复:结果经得起重复验证;③随机:受试者随机分配入组;④对照与盲法:避免条件误差与主观因素。 试验方案的格式包括:①封页:包括题目、申办者和临床试验机构的名称与地址,拟订日期;②正文:GCP要求的23项;③封底:各参与的临床试验机构与主要研究者、申办者的名称与联系方式;④主要参考文献。 临床试验方案设计主要内容(GCP第四章第十七条)有以下23条: (一)试验题目: (二)试验目的,试验背景,临床前研究中有临床意义的发现和与该试验有关得临床试验结果、已知对人体的可能危险与受益,及试验药物存在人种差异的可能; (三)申办者的名称和地址,进行试验的场所,研究者的姓名、资格和地址; (四)试验设计的类型,随机化分组方法及设盲的水平; (五)受试者的入选标准,排除标准和剔除标准,选择受试者的步骤,受试者分配的方法; (六)根据统计学原理计算要达到试验预期目的所需的病例数; (七)试验用药品的剂型、剂量、给药途径、给药方法、给药次数、疗程和有关合并用药的规定,以及对包装和标签的说明; (八)拟进行临床和实验室检查的项目、测定的次数和药代动力学分析等; (九)试验用药品的登记与使用记录、递送、分发方式及储存条件; (十)临床观察、随访和保证受试者依从性的措施; (十一)中止临床试验的标准,结束临床试验的规定; (十二)疗效评定标准,包括评定参数的方法、观察时间、记录和分析; (十三)受试者的编码、随机数字表及病例报告表的保存手续; (十四)不良事件的记录要求和严重不良事件的报告方法、处理措施、随访的方式、时间和转归; (十五)试验用药品编码的建立和保存,揭盲方法和紧急情况下破盲的规定; (十六)统计分析计划,统计分析数据集的定义和选择; (十七)数据管理和数据可溯源性的规定; (十八)临床试验的质量控制和质量保证; (十九)试验相关的论理学; (二十)临床试验预期的进度和完成日期;
临床试验监查报告
临床试验终期监查报告 □全面监查 □法规卷宗(Ⅰ、Ⅱ) □知情同意(Ⅲ、Ⅳ) □方案依从性与病例报告表(Ⅴ、Ⅵ、Ⅶ) □药品资料(Ⅳ、Ⅶ) □研究条件(Ⅸ、Ⅹ) □其她 跟踪监查发现得问题:□无问题 一、研究卷宗/法规资 料监查 (在研究卷宗中就 是否有如下资料) 就是否不适合未 访 视 1 附有签名得方案 与方案修正案 □□□□2 伦理委员会通过 得知情同意书样 本 □□□□ 3所有病例报告表 样本 □□□□ 4研究者手册□□□□5 研究人员履历/资 格说明 □□□□ 6 研究人员相关培 训文件 □□□□ 7 伦理委员会通过 得研究相关资料 □□□□
8实验室资格及正 常值范围 □□□□ 二、中心监察就是否不适合未 访 视 1 所有需要得文件 就是否都有 □□□□ 2 对开展临床试验 至关重要得设施 就是否都正常运 转 □□□□ 3 研究者与其工作 人员保留着与开 展试验相关得记 录 □□□□ 三、知情同意/入选情 况就是否不适合未 访 视 1已签署得原始知 情同意书都在受 试者档案里 □□□□ 2 就是否有筛选日 志 □□□□ 3 就是否有入选日 志 □□□ □ 四、研究药物就是否不适合未 访 视 1 试验药物就是否 保存在安全得地 方 □□□□ 2 试验药物就是否 由专人分发 □□□□ 3 药物运送记录及 时准确并有日期 □□□□
及签名 4 药物分发与数量 记录就是否及时准确 □ □ □ □ 5 药物就是否按照 方案分发 □ □ □ □ 6 过期得药物就是 □ □ □ □ 五、 方案依从性 就是 否 不适合 未 访视 1 筛选过程正确 □ □ □ □ 2 所有受试者都符合入选/ 排除标准 □ □ □ □ 3 不良事件就是否正确报 告,记录,评价与解决 □ □ □ □ 4 受试者到诊及过程就是 否按照方案规定顺序完成 □ □ □ □ 5 就是否按照方案给予研 究干预 □ □ □ □ 6 就是否有严重不良事件 (S AEs) □ □ □ □ 六、 病例报告表(CRFs)与源文件 就是 否 不适合 未 访视 1 能否提供CRF s与相应得源文件查阅 □ □ □ □ 2 源文件就是否足 以供CR Fs 核查 □ □ □ □ 3 CRFs 就是否完整, 清晰 ,准确 □ □ □ □ 4 CRF s就是否及 时完成与递交(如数据录入) □ □ □ □
药物临床试验总结报告的设计规范
药物临床试验总结报告的设计规范 版本号 1.0 页数11页 起草人起草日期年月日审核人审核日期年月日批准人批准日期年月日颁布日期年月日起效日期年月日 威海市立医院 药物临床试验机构
药物临床试验总结报告的设计规范 一目的 为真实地反应反映该试验的实施过程和客观地表述药物的有效性和安全性及其应用价值。 二范围 所有由本院牵头或协助参加的新药临床试验。 三内容 1报告封面 参照国家食品药品监督管理总局有关药品注册申报资料的形式要求。 2签名页 1)报告题目 2)执笔者签名。 3)主要研究者对研究试验报告的声明。 申明已阅读了该报告,确认该报告准确描述了试验过程和结果。 4)主要研究者签名和日期。(包括统计学工作者) 3报告目录 每个章节、附件、附表的页码。 4缩略语 正文中首次出现的缩略语应规范拼写,并在括号内注明中文全称。应以列表形式提供在报告中所使用的缩略语、特殊或不常用的术语定义或度量单位。 5伦理学问题 1)确认试验实施符合赫尔辛基宣言及伦理学原则。 2)伦理委员会组成及批准临床试验方案情况说明,并在附件中提供独立伦理委员会成 员表。 3)描述如何及何时获得与受试者入选相关的知情同意书,并在附件中提供原稿。 6报告摘要 报告摘要应当简洁、清晰的说明以下要点,通常不超过1500字。 1)试验题目。 2)临床批件文号。 3)主要研究者和临床试验单位。 4)试验的起止日期(第一例受试者第一次访视至最后一名受试者最后一次访视日期)。
5)试验目的及观察指标。 6)对研究药物的作用类别和主治功能的描述。 7)对试验设计作简短描述,包括试验设计类型(平行、交叉、成组序贯等)、设盲水 平(双盲、单盲或开放)、随机分组方法、对照的形式(安慰剂、阳性药对照、剂 量对照)、疗程。 8)试验人群。 9)给药方案(包括对照组) 10)评价标准(有效性和安全性评价指标)。 11)统计分析方法或模型(包括基线评价、组间比较、协变量分析、综合比较等)。 12)受试者入组情况及各组人口学资料。 13)各组疗效结果(主要和次要疗效指标)。 14)各组安全性结果(不良事件及严重不良事件)。 15)结论(有效性和安全性结论)。 7报告正文 1)前言 一般包括:受试药品研究背景;研究单位和研究者;目标适应症和受试人群、 治疗措施;受试者样本量;试验的起止日期;国家食品药品监督管理局批准临床试验的文号;制定试验方案时所遵循的原则、设计依据;申报者与临床研究单位之间有关特定试验的协议或会议等予以说明或描述。简要说明临床试验经过及结果。 2)试验目的 应提供对特定试验目的的陈述(包括主要、次要目的)。注意具体说明本项试验的受试因素、受试对象、研究效应,明确试验要回答的主要问题,明确药品的临床定位。 3)试验方法 (1)试验设计 a)总体研究设计和计划的描述(包括临床试验的流程图)。如试验过程中方案有 修正,应说明原因、更改内容及依据。 b)对试验总体设计的依据、合理性进行讨论,具体内容应视设计特点进行有针对 性的阐述。如采用单盲或开放试验设计,应说明理由。 c)提供样本含量的具体计算方法、计算过程以及计算过程中所用到的统计量的估 计值及其来源依据。 d)描述期中分析计划。
会议情况总结报告如何写
会议总结报告怎么写 一、会议报告的性质 (一)会议报告的概念 会议报告,是在重要会议和群众集会上,主要领导人或相关代表人物发表的指导性讲话。它是一种书面文字材料,又是会议文件的重要组成部分和贯彻会议精神的依据,还是供查阅的历史资料。它包括政治报告、工作报告、动员报告、总结报告、典型发言、开幕词、闭幕词等。会议报告具有宣传、鼓动、教育作用。这些作用是通过报告人的报告和听众的接受来实现的。因而,有时为了让更多的人知道报告内容,广播电台、电视台可进行现场转播,报刊也可登载。如党的十七大报告。 (二)会议报告的特点 1.理论性和逻辑性。会议报告是领导人在大型会议上或重要场合作的有关政治、经济、文化和局势等方面的报告,是以领导或领导代表的身份站在决策集团角度上所发表的讲话。它在广泛深入调研、充分占有材料的基础上,纵览全局,找准焦点,围绕实际工作中出现的问题,尤其是那些迫切需要解决的,带有普遍性的,人民群众最关心、最直接、最现实利益问题进行透彻分析,细致研究,从而抓住问题的关键,
对症下药,达到推动各项工作健康发展的目的。所以,在分析研究中,它必须依据有关方针政策,结合实际地对所提建议、对策、问题等进行认真研究,反复推敲,从理论和实际的结合上把握哪些是最有价值、最需要解决的问题,它充分考虑所提意见的针对性、正确性、合理性、可靠性,使意见和措施能真正有助于解决实际问题。因而,会议报告既注重事实分析,又必须从理论的高度上进行归纳概括,进而指导实践,有较强的理论性和逻辑性。 2.双向性和交流性。会议报告依据讲话稿直面听众公开发表讲话,具有直接性、当众性、范围广、影响大的特点,在领导活动中具有特殊的地位和作用。正是由于这种面对面的宣讲传播形式,就使主体和客体之间在时间、空间上的结合比较紧密,“报告”的成功与否,不决定于形式,即过程的结束,很大程度取决于主体对客体的“磁性”交流强度,即吸引力的大校这种报告的吸引力既决定于报告的文采或领导的演讲口才,又决定于听众是否接受。而且更关键的还取决于报告内容是否为受众认可,是否反映了实际情况。所以,会议报告实际上是一种在时间、空间上获得统一的、由报告主体和受众客体双向结合的交流形式。 3.切实性和针对性。会议报告的核心,是对实际问题的分析和解决。它一般要总结成绩经验、说明现状和存在问题,部署工作,规划未来等。它要求在分析的基础上提出解决问题的意见或对策,具有很强的
药物临床试验基本流程(总结)
药物临床试验基本流程 1.临床试验启动阶段 1.1获得药物临床试验批件(有效期3年) 1.1制作研究者手册 理化性质、药理、药化、药效、毒理以及已有的临床研究资料,制作研究者手册。 研究者手册是临床试验开始前的资料汇编。研究者手册的内容一般包括:目录、序言、化学和物理性质、临床前研究、药理学研究、药代动力学研究、毒理学研究、已有临床资料、药品使用信息。 1.2筛选主要研究者 CFDA 网站中浏览临床药理基地名录,筛选医院(一般来说不是整个医院而是 某个科室),然后选择合适的主任级医生; 联系主任医生(看是否有意向,在给予任何资料前,签保密协 议,然后发放可行性问卷,收集可行性问卷,确认完整无误,回复感谢信, 感谢其参与问卷调查) 如果可行,可安排访视,具体考察该基地是否有能力执行临床试验; (电话预约研究者,给予方案和研究者手册,agenda 确认要讨论的具体问题) Agenda:研究人员的分配 方案的可行性 能否入够病人及如何保证其依从性 药品管理 档案管理 研究者职责 主要研究者资质的确认 相关研究人员的资质 硬件设施考察 伦理委员会的访视(承不承认中心伦理、伦理开会频率、送审清单、伦理委员会人员清单) 书写访视报告; 再次拜访,与其讨论方案、试验时间、试验费用等问题; 1.3试验文件准备 会同CRO 、申办者、主要研究者,共同制定临床试验方案、病例报告表、知情同意书样本、原始文件;CRO 制定临床试验中其他用表; 1.4其他研究者筛选 从临床基地名录中选择其他可能的研究者,可根据首研者的推荐以及公司曾经合作的情况进行综合判断; 准备资料:方案、研究者手册、知情同意书样本、病例报告表; 与其谈论方案,要求提供其医院所能提供的病例数、时间进度和经费预算; 选定合适研究者,征得其医院管理部门的同意; 1.5召开多中心临床方案认论会(研究者会议),讨论试验方案、CRF 等 注:是否需要召开研究者会议要看具体情况 1.6取得伦理委员会批件 注:IRB (Independent Review Board )/IEC (Independent Ethics Committee )——IRB 为美国的伦理委员会,IEC/EC 为欧盟的伦理委员会 按照GCP 的要求,所有临床试验必须得到伦理委员会的批准。在实际进行临床试验时,首先必须取得牵头单位伦理委员会的批准,其他参加单位是否要过伦理根据各家单位的具体要求而定。 伦理委员会将对有关批件、药检报告、研究者手册、知情同意书样本、试验方案(protocal )、病例报告表进行审批; 1.7试验药品准备 督促申办方进行试验用药品的送检; 生物统计师设计随机分组方案; 根据随机分组方案,设计药品标签; 设计应急信件; 药品包装:为每一个受试者准备好一盒药物,药盒上贴好标签,并装入相应的应 先拜访研究者还是医院相关部门视具体情况而定,没有统一标准 1.7及1.8可在研究者会议后开始准备,同时申请伦理委员会批件
会议总结英文范文模板【实用】
导语:会议的重要性是根据内容来定的,那么会议的总结怎么写,以下为大家介绍会议总结英文模板,欢迎大家阅读参考! 总结会议 Concluding a Business Meeting Action公司第三季会议在各部门负责人一一报告,以及与会者热烈讨论之后,即将落幕。会议主持人Jennifer,此时再度上台,为会议做个总结。 That wraps up the last item on the agenda. Before we close, are there any questions? Fine. In summary, I think we agree that this quarter's domestic sales figures show a marked trend in microwave sales. Sam, I'd like you to follow up on that, please. Let's see where this is heading. Jane, thank you for the prehensive PR status report. I understand there are still a few problems to be worked out, but we all trust in your ability. Ladies and gentlemen, the new design is satisfactory. Let's keep Action ahead of the game. Finally, I appreciate your ments and suggestions about the Canadian sales outlook. This is our first opportunity to promote our products in North America, so we certainly want to cover every detail. Well, then, that covers everything. I make a motion to close the third quarterly meeting of Action Appliances. Is there a second? 短语解说 wrap up 完成;结束 这是个口语用法,意同"conclude"、"finish"和"close"。主词可以是人或物。 I'd like to quickly wrap up this meeting, so we can go home. 我想赶快把这个会议结束,那我们就可以回家。 marked trend 明显的趋势 "marked"是过去分词当形容词,有‘醒目的、明显的’意思;"trend"是指事物或社会现象变动的趋势。 Judging from the graph, you can see a marked trend upward on the Japanese stock exchange. 依照这张图表,你们可以看出日本股市明显地上涨。 follow up on (something) 进一步了解 "follow"原是「跟随」的意思;这个词组是指注意某事发展,并进一步深入地了解它。介系词"on"之后所跟的名词词组即代表需要了解的事。 We can take care of that problem when we follow up. 等我们更进一步地了解问题后,就容易解决了。
2021年会议总结报告模板
会议总结报告模板 会议总结报告模板 根据省市教研室要求,我校组织本校语文组老师参加了省教研室组织的教研会议,收获良多,特总结如下: 一、倾听专家报告和经验介绍,受到了启发和震动。 1、关于提高教师解读文本和执教语文课堂的能力。 会议上,有关专家提到了应该加强对经典篇目的解读,这对我们是有益的启发。 经典篇目由于长期被教育名家研读、分析,似乎已经没有多少可以挖掘的东西了。 但是,随着时代的变迁和教育教学理念的更新,对传统篇目的理解和分析会出现更多更新的角度和切入点,对文本的解读是没有止境的,如能找到合适的突破口,老课文也可以上出新意。 2、学生不是教师完成讲课任务的道具。
很多时候,我们讲课的时候更多地想到的是如何顺利地完成,而不是学生在课堂上能得到多少东西。 这个问题牵涉到教育最终为了谁这个终极命题。 专家认为,真正适应学生的教育才是最好的教育,真正适应学生的课堂才是最好的课堂。 那么,以此为标准,不管是教师的讲授还是学生的学习,都应以能不能促进学生的更好成长为标准。 3、让中青年教师重视自身的成长。 教师是成人,在工作之前受过多年的教育,参加工作后也获得了不少有益的教学经验,但这并不表示教师就不需要学习和成长了。 一个真正的教师应该始终是一个学习者,他有学习的渴望,不断试图超越自己,在不断地反思和否定中获得更好的成长。 但在实际教学工作中,由于缺乏有效的指导和激励,很多教师喜欢靠经验教学,迷信老办法。
不注重学习和研究,这是非常危险的,因为他极可能因为年龄的增长而逐渐和学生越走越远,最终丧失学生的信任和喜爱,那么,他的教学也就必然走向了失败。 4、加强双基,强调多读多写。 在平时的教学中,我们的讲授占据了很大的比重,仿佛不讲,这节课就没法进行,就白上了。 这显然是认识上的不足,而且没有从长远的角度来看问题。 只要我们稍微考查一下成年人的语文能力就会发现,恰恰是我们在课堂上喋喋不休反复强调的东西,是在我们成年后最没有价值,最容易被忘掉的东西。 在一个成年人身上,读写几乎是不可分的,而且也是最有价值的。 一个人爱读书,写一点东西并不是什么难事,反过来说,一个擅长写的人,几乎也就是因为多读了几本书。
临床试验总结报告的体例格式和内容要求
临床试验总结报告体例和内容要求 1.题目封面 封面题页应包括如下内容: ——试验题目 ——试验药物/研究产品的名称 ——试验用药品的适应症 ——如不能在题目中表明、则简要描述(以1~2句话)设计(平行、交叉、双盲、随机)比较(安慰剂、活性成分、剂量/反应),间隔,剂量和病例数。 ——申办者姓名 ——原始规划与计划的确认(编码或数目,确认日期) ——试验预期进度与进程安排 ——研究开始数据(受试的第一位病人,或任何其它数据) ——后期研究终末数据(最终一个受试者的名称、或终末研究者完成的数据) ——试验研究完整的数据(最后的受试者) ——参加者或合作者或主办者责任医务官等人士的名称和职责。 ——药业公司名称/主办方签名(在公司/主办者中负责研究报告的人,公司/主办方联络者的姓名,电话和传真号码,以备在回顾研究报告提出问题时,在此页码或应用的字母有所表明)。 ——表明该项研究是否在优良临床试验管理规范(GCP)条件下进行的包括应达到的基本文件、研究设施、人员知识结构,统一培训等。 ——报告的资料(通过题目和资料,确定与该项研究相同的任何其它较早报告) 2.内容概述 应提供—个临床试验的主要内容提要(通常限于3页内容),该提要应包括试验题目、研究人员、研究出版物名称、试验时间、试验目的、试验方法、试验研究样本、诊断及进入研究的主要标准、试验的产品、剂量、给药方式及批号、治疗持续时间、参考的治疗、剂量、给药方式及批号、评价标准(有效性、安全性)、统计方法、总结——结论(效能结论、安全性结论、结论)及报告日期。该提要应包括表明结果的数字资料,而不仅仅是文字和P 值(见附件l,研究内容概述)。 3.内容目录 内容表格应包括: ——页码或其它每一个部分的局部资料,包括归纳性表格、图和图表。 ——附录、列表和任何能提供的病例报告形式的汇总和定位。 4.名词、术语缩写和定义 应提供报告中使用的—系列缩写符、特殊的或不常用的术语,或测量单位定义,应拼写
临床试验总结报告的撰写
临床试验总结报告的撰写 定义:是反映药物临床研究设计、实施过程,并对试验结果 作出分析、评价的总结性文件,是正确评价药物是否 具有临床实用价值(有效性和安全性)的重要依据, 是药品注册所需的重要技术资料。 基本准则:真实、完整地描述事实 科学、准确地分析数据 客观、全面地评价结局 结构与内容:药品名称:资料项目编号:33-Ⅱ ****II期临床试验研究报——以***为对照药评价***治疗***安全性有效性的分层区组 随机、双盲双模拟、平行对照、多中心临床研究 研究机构名称:***(负责单位)(盖章) ***(参加单位)(盖章)研究机构地址及电话: **省**市**** **** 主要研究者: *** 主任医师(签名): 试验起止日期:****年**月-****年**月 原始资料保存地点:***医院 联系人姓名:*** 联系人电话:**** 申报单位:***(盖章) 报告签名 报告题目: 主要研究者声明及签名 我已详细阅读了该报告,该报告客观、准确描述了 试验过程和结果。 ***医院 ***医师(签名):年月日 研究负责人签名 ***医院 ***医师(签名):年月日
统计分析负责人签名 ***医院 ***医师(签名):年月日 申办者声明及签名 我们对该临床试验的全过程进行了监查,试验按临床试方案进行,我们已详细阅读了该报告,该报告客观、准确描述了试验过程和结果。 ***公司 负责人:***(签名):年月日 监查员:***(签名):年月日 执笔者签名 ***医院 ***医师(签名):年月日 报告目录 缩略语 论理学声明 报告摘要 试验目的 试验方法 讨论 结论 参考文件 附件 缩略语 缩写中文全称英文全称 ALT 丙氨酸氨基转换酶alannine transaminase RBC 红细胞red blood cell WBC 白细胞white blood cell N 中性粒细胞neutrophilic granulocyte L 淋巴细胞lymphocyte PLT 血小板blood platelet
会议总结报告范文大全
会议总结报告范文大全 我们每个人都参加过大大小小的会议,会议是管理沟通的重要方式,会议是群体或组织中相互交流意见的一种形式,也是工作中最常见的一种沟通方式。会议有着发布信息、解决问题、做出决策、分配任务等作用。 管理学家彼得·德鲁克说:“我们之所以开会,是因为要想完成某一项具体工作,单凭一个人的知识和经验不够,需要结合几个人的学识和经验。”在信息急剧膨胀、竞争愈加激烈的商业社会,企业尤其需要通过会议互相合作、分享信息。在很多企业,会议往往决定了是否做某项工作以及何时做,换句话说,会议控制了工作流程。开会提供各方沟通的机会,但沟通本身并不是开会的目的,会议的目的是通过沟通达成共识。成功的会议有几个特征:会议时间恰如其分,与会各方达成共识,会议结果具体有效,会议决定得到落实。 会议是我们日常工作非常重要的一个环节,占用了我们的很多时间和精力。然而会议沟通却常常无法发挥它应有的作用、达到它的原先的目的。 美国两家著名的管理顾问机构的“白领回忆状况”表明,近八成的职场人士认为自己参加的会议有三分之二是在浪费时间,超过5
成职场人在开会时没有集中注意力、6成人对主题不感兴趣而不发言、6成人没有为会议做任何准备。 将大家对会议的抱怨进行总结,我们会发现很多会议存在以下四点不良的现象: (一)会议前没有明确的讨论主题和重点,当有临时的一些想法产生时,自然大大延长了会议时间。 (二)没有强制的会议时间,虽然在开会前,大都会预估一个会议时间,但在会议过程中,大多数人不会太在意时间。 (三)参加会议的人员没有提前做准备,导致会议上不能提出有见解的想法,因此很难得到有效的会议结论。 (四)会后没有追踪,很多会上形成的决议和计划,大家并没有按此执行,导致反复开会和沟通,工作无法正常推进。 这样的会议状况,让很多人一提起会议,都或多或少有厌烦的情绪。与会人员在一个缺乏架构、无法沟通、冗长的会议中,不仅对所达成的决议漠不关心和抱怨,甚至对组织也会产生某种不认同的心态。甚至有人为此总结出了七不开会议:
最新药物临床试验工作总结-(1)
药物临床试验机构工作总结 根据江苏省三级综合医院医疗技术水平标准(2017版)和江苏省省级临床重点专科评分标准的相关要求,我院自2017年1月份启动创建国家药物临床试验机构。依照《药物临床试验质量管理规范》(2003版)的标准有序开展各项工作。以质量控制为抓手,注重内涵建设;加强机构办管理人员及各专业科室相关人员的培训;强化临床试验相关的伦理审查;规范伦理委员会的管理;大大提高了临床试验质量和管理水平。现对2017年的工作总结如下: 一、建立健全组织管理机构 我院自2017年1月份启动国家药物临床试验机构创建工作,3月份召开了创建动员大会。先后成立了国家药物临床试验工作领导小组、组织管理机构、机构伦理委员会和机构工作实施方案。机构办公室配置机构药房,资料档案室、质控小组。根据各临床专业的综合实力及科研技术水平遴选了17个临床专业研究团队。 二、制定规章制度、标准操作规程 机构办公室严格按照《药物临床试验质量管理规范》(2003版)
和《药物临床试验机构资格认定标准》的具体要求,起草制定了药物临床试验管理制度20条、各类人员职责14条、药物临床试验标准操作规程89条、药物临床试验设计规范5条并汇编成书100本,汇编印发应知应会手册300册。 三、强化药物临床试验培训,保证试验过程规范,结果科学可靠 积极组织专业人员参加院、内外GCP及相关技术规范培训:2017年的3-6月份有关院领导带领办公室成员先后到省、市级具有国家药物临床试验机构资质的6家医院调研参观学习;4-5月份组织两次38人次参加的江苏省药物临床试验法规、技术及实务操作和伦理审查技术高级培训班;7月份邀请有关药物临床试验知名专家,在我院举办由中国药理学会药物临床试验专业委员会主办的国家级药物临床试验质量管理规范培训班,约240人参加。目前共有129人次取得了国家级、省级药物临床试验培训证书。在创建期间组织院内培训8次约300人次,涉及内容包括对17个临床专业科室的研究者、质量管理员、药品管理员的专题培训及药物临床试验安全性评价与不良事件处理、进行心肺复苏、呼吸机、除颤仪等设备使用的SOP进行培训。四、 五、完成试验设施设备的配备 2017年8月份完成了机构办公室、伦理委员会办公室及各申报专
I期临床试验总结报告样本
SFDA临床试验批准号:2002XXXXXX号 XXX注射液 Ⅰ期临床人体耐受性试验总结报告 试验单位:XXXXXXXX医院 试验负责:XXX 试验设计:XXX,XXXX 试验日期:2003年X月X日至X月X日 申办单位:XXXXX有限公司 (以下仅提供报告格式,其数据和文字均为虚构,如有巧合,纯属偶然;数据前后有矛盾之处,也请原谅) XXX注射Ⅰ期临床人体耐受性试验总结报告 摘要 单次给药的耐受性试验:30名健康受试者,根据体重和性别随机分配到7个剂量组(组1、组2、组3、组4、组5、组6、组7)。组1、组2每组2人;组3、组4、组5每组6人;组6、组7每组4人。参照费氏递增法(改良Fibonacci法)递增给药,各组分别静脉滴注XXX注射液5、10、20、30、40、50、60ml,滴速20-30滴/min。每个受试者只接受一个相应的剂量,从小剂量开始,每个剂量应用后未见不良反应,才可用下
一个剂量。观察给药后健康人体对XXX注射液的反应和耐受性。 多次(累积)给药的耐受性试验:12名健康受试者根据体重和性别随机分配到甲、乙两个剂量组,每组6人。分别静脉滴注XXX注射液20、10 ml,滴速20-30滴/min,连续5d。 结论:(1)每次按10 ml/次给药,有可能引血胆红素轻度增高或尿胆原阳性,但受试者无明显不适,因此推荐II期临床试验为10 ml/次;15 ml/次有可能引起血胆红素轻度增高或尿胆原阳性,但风险不会太大,可在严密监护下尝试。20 ml/次虽然引起的不良反应尚属轻度,但发生的频度较高,慎用。(2)10 ml/次就有可能引起轻度不良反应,主要为血胆红素轻度增高或尿胆原阳性,但受试者无自觉症状。20 ml/次可引起的不良反应有所增加。这种血胆红素轻度增高或尿胆原阳性,且两次重复结果相近,推测是药物引起的轻度溶血所致。其他主要不良反应为口干(2例)、头晕(1例)、皮疹(1例)。 根据国家药品监督管理局XXX号批文的要求,按照《新药审批办法》,《药品临床试验管理规范》(GCP),《中药新药临床研究的技术要求》,以及《中药新药临床研究指导原则》和XXX的化学组成、功能主治、药效学、毒理学研究资料,于2003年7月17日~2003年9月20日,对XXX有限公司申报的XXX注射液中药二类新药,进行Ⅰ期临床人体耐受性试验,总结报告如下。 1 研究目的 选择健康人为受试者,从安全的初始剂量开始,观察人体对XXX注射液的耐受性,为制定本品的Ⅱ期临床试验给药方案提供依据。 2 临床资料与研究方法 病例选择 入选标准 1.健康志愿者。 2.年龄在18~50岁,男女各半。 3.体重在标准体重的±10%范围内[标准体重(kg)=×(身高cm-80)]。 4.无烟酒嗜好。 5.心、肝、肾、血液等检查指标均在正常范围。 6.知情同意,志愿受试。 排除标准 1.妊娠期、哺乳期妇女。 2.具有原发性心、肝、肾、血液学疾病者。 3.精神或法律上的残疾患者。 4.有酒精、药物滥用病史。 5.过敏体质(对两种或两种以上药物或食物、花粉过敏史者)。 6.3个月内参加过其他药物临床试验者。 7.正在应用其他预防和治疗的药物者。 8.研究者认为不能入组的其他原因。 剔除标准 对已被选入本临床研究,属于以下情况之一者,作为剔除病例。 1.不符合纳入标准,或符合排除标准者
会议报告总结范文
会议报告总结范文 会议是很多公司和单位的一种传达方式,会议怎么写报?怎么写总结 ,大家不妨来看看小编推送的会议报告 总结范文,希望给大家带来帮助! 根据省市教研室要求, 我校组织本校语文组老师参加了省教研室组织的教研 会议,收获良多,特总结如下: 一、倾听专家报告和经验介绍,受到了启发和震动。 1、关于提高教师解读文本和执教语文课堂的能力。会议上,有关专家提到 了应该加强对经典篇目的解读, 这对我们是有益的启发。 经典篇目由于长期被教 育名家研读、分析,似乎已经没有多少可以挖掘的东西了。但是,随着时代的变 迁和教育教学理念的更新, 对传统篇目的理解和分析会出现更多更新的角度和切 入点,对文本的解读是没有止境的,如能找到合适的突破口,老课文也可以上出 新意。 2、学生不是教师完成讲课任务的道具。很多时候,我们讲课的时候更多地 想到的是如何顺利地完成, 而不是学生在课堂上能得到多少东西。 这个问题牵涉 到教育最终为了谁这个终极命题。 专家认为, 真正适应学生的教育才是最好的教 育,真正适应学生的课堂才是最好的课堂。那么,以此为标准,不管是教师的讲 授还是学生的学习,都应以能不能促进学生的更好成长为标准。 3、让中青年教师重视自身的成长。教师是成人,在工作之前受过多年的教 育, 参加工作后也获得了不少有益的教学经验, 但这并不表示教师就不需要学习 和成长了。一个真正的教师应该始终是一个学习者,他有学习的渴望,不断试图 超越自己,在不断地反思和否定中获得更好的成长。但在实际教学工作中,由于 缺乏有效的指导和激励,很多教师喜欢靠经验教学,迷信老办法 。不注重学习和研究,这是非常危险的,因为他极可能因为年龄的增长而逐 渐和学生越走越远,最终丧失学生的信任和喜爱,那么,他的教学也就必然走向 了失败。 4、加强双基,强调多读多写。在平时的教学中,我们的讲授占据了很大的 比重,仿佛不讲,这节课就没法进行,就白上了。这显然是认识上的不足,而且 没有从长远的角度来看问题。 只要我们稍微考查一下成年人的语文能力就会发现, 恰恰是我们在课堂上喋喋不休反复强调的东西, 是在我们成年后最没有价值, 最
1/6
临床试验GCP汇总
临床试验G C P汇总集团档案编码:[YTTR-YTPT28-YTNTL98-UYTYNN08]
第九章 一、数据管理的目的是什么? 答:把试验数据迅速、完整、无误的纳入报告,所有涉及数据管理的各种步骤均需记录在案,以便对数据质量及试验实施进行检查。 二、什么叫盲态审核? 答:盲态审核(blindreview)是指在最后一份病例报告表输入数据库后,第一次揭盲之前对数据保持盲态的预分析审核,以便对统计分析计划作最后的决定。 三、什么叫质疑表?由谁传送?能不能通过电话传达质疑表的内容? 答:质疑表(?querylist,queryform)是数据管理员再次检查病例报告表发现任何问题后,及时通知检查员,要求研究者作出回答的文件。检查员、研究者、数据管理员之间的各种疑问及解答的交换都由质疑表完成。质疑表应保存备查。质疑表必须由检查员亲自传送给研究者,不能通过电话传达。 四、什么叫结果锁定? 答:结果锁定指在盲态审核认为所建立的数据库正确无误后,由主要研究者、药物注册申请人、生物统计学专业人员和保存盲底的有关人员对数据库进行锁定。此后,对数据库的任何改动只有在以上几方人员同意(可书面形式)的情况下才能进行。 五、临床试验统计分析中常用那两种分析方法? 答:1.PP分析(合格病例分析)对完成治疗方案,且依从性好的病例分析。 2.ITT分析(意向性分析)对所有的病例进行分析,包括合格、脱落,出现不良反应的病例的分析。 六、什么是ITT?什么是FAS? 答:ITT是Intention-To=Treat的缩写,即意向性。指所有经随机化分组,分配了随机号的全部病例,也称为愿意治疗人群。意向性分析时其中未能观察到全部治疗过程的病例资料,用最后一次观察数据接转到试验最终结果,对疗效和不良事件发生二进行意向性分析。ITT的误差较大。 FAS是全分析集(FullAnalysisSet)指尽可能接近符合ITT原则的理想的受试者人群。它应包括几乎所有的随机化后的受试者。只有在导入期中被排除而未入组或者入组后没有任何的随访数据才能从FAS人群中排除。即只要服用了一次药,做了一次有效检测的受试者都应入FAS。 七、什么是PP? 答:PP是Pet-Protocol的缩写,即符合方案集。指所有符合试验方案、依从性好(如接受治疗、主要指标可以测定等)、试验期间未服禁止用药、完成CRF规定填写内容的病例,对其疗效统计分析。 八、ITT和PP有何关系?
会议总结报告基本格式
会议总结报告基本格式 会议总结报告基本格式 会议报告的基本格式和写法 会议报告一般由标题、称谓、开头、主体、结尾五个部分构成。 1、标题。写法有两种。一种是直接写成“X X X同志在X X X工作会议上的报告”的形式。另一种是不出现报告人的姓名、会议名称和“报告”字样,而另拟写一体现会议主要精神的标题,如“当前的经济形势和今后的经济建设方针”。标题的下行写报告的时间,再下一行写报告人的姓名。 2、称谓。报告是面对面进行的,它有明确的报告对象,称谓可根据报告对象的身份而定,要恰当合体。写法大致有两种情况:一是只写在报告的开头;二是除开头的称呼外,在报告的进程中适当穿插使用,作用是提示听众注意。每次称呼的出现,都标志着讲话进入了一个新层次。 3、开头。会议报告开头的写法多种多样:有的开门见山,揭示题旨;有的提出问题,巧设悬念;有的交代背景,介绍情况;有的讲述一个故事,吸引听众。不管采用哪种写法,总的要求是要开门见山,接触正题,提出全文的中心论点或主要议题,说明报告的意图,以便听众抓住要领,并造成一种气氛,控制住听众的情绪,使他们全神贯注地听取报告。
如:云南省第八次党代会报告就是开门见山,开始就把报告的主旨明确地点了出来,吸引住听众的注意力。十七大报告亦如此。 4、主体。是报告的主要内容的集中表述,它决定着一篇报告的成败。报告主体,要紧扣论题或主旨,展开分析论述。既要有深刻的理论分析,又要有典型的例证,从各个方面,多种角度,透辟地阐明报告的主题。 报告主体部分的结构形式主要有三种:(1)递进式。即层层深入地讲述。其特点是各层都以前边一层的意思为论述的基础,各层之间形成步步深入、层层递进的逻辑关系。(2)并列式(有书称条陈式)。即从几个方面来阐述。其特点是对报告主旨所包含的几项主要内容,分别进行阐述。几个层次之间的关系是并列的,它们分别从不同的方面来论证报告主旨。但并列式结构,并不是随意罗列,各层意思谁先谁后,也有一定的依据:或按性质的强弱,或按问题的主次,或按时间的先后等等。(3)对比式。把两种不同意见、不同方面的情况对照起来加以阐述。在实践中,以其中的一种形式为主,两三种结构形式结合使用,也是长篇报告经常采用的结构形式。但是,不管采取哪种结构,都必须集中于一个中心、一个主旨讲深讲透。这样,才能使听众得到一个完整、清晰、深刻的印象。 5、结尾。即报告的结束语。这部分从内容上看要注意
最新药物临床试验机构筹建工作总结及整改措施
药物临床试验机构筹建工作总结及整改措施我院xx年开始药物临床试验机构的筹建及申请工作,医院高度重视,建立了组织机构和伦理委员会,建立了各项管理制度,制定了各项试验设计规范、SOP等,研究人员进行了临床试验技术和GCP培训,并与xx年向国家药品食品监督管理局提交了资格认定申请,受理编号:XX XXXX年XX月XX日国家药监局专家专家对我院药物临床试验机构xx专业资格认定进行了现场检查,对我院的工作给予充分的肯定,并给出了综合评定意见,认为我院:专业技术力量较强,医疗设备齐全,专业病原病种充足,能满足药物临床试验的需要,医院领导重视药物临床试验机构的建设,成立了管理机构并制定了相关的管理制度、设计规范和SOP,机构人员接受过不同层次的GCP和药物临床试验技术培训,基本具备开展药物临床试验的条件。 但SFDA药品认证管理中心专家组也指出了检查中我们尚存在的一些问题。针对这些问题,医院领导组织有关人员进行了认真的讨论和研究,并做出如下整改: 1. 机构选派部分研究和管理人员到XX临床试验基地进行进修学习,学习结束后,重新制定和完善了部分管理制度、技术设计
规范和SOP,使其符合本专业的特点及GCP要求并具有可操作性。 2. 机构和专业分别选派具有相应的专业技术职称,参见过GCP 培训,并有参加过药物临床试验的经历并掌握临床试验相关技术的及相关法律的人员,设专业质控员和机构办公室质控员,分别从专业和实验全过程对药物临床实验进行监督检查,实现并强化药物临床实验的三级质控,完善药物临床试验质量保证体系与制度,确保我院承担的国家药物临床试验过程规范,结果真实可靠,保证受试者和申办单位的合法权益。 3. 选派机构主要研究和管理人员参加了国家级的GCP培训并取得了培训证书。同时在院内继续进行GCP知识和试验技术知识培训,并从认定工作结束后开始定期派机构管理人员对GCP知识和试验技术知识掌握情况进行抽查和督促,使所有试验相关人员熟悉GCP知识和试验技术知识, 4. 进一步完善了药物临床试验资料保存条件,设立专职人员负责药物临床试验资料的保存和管理,添置资料保存柜,更换较大的适合药物临床试验资料保存的资料保存室。 XXXX 医院