第六章 DNA序列分析

第六章DNA序列分析
[本章摘要]
DNA 的序列分析有两种基本方法, Maxam-Gilbert 化学降解法和 Sanger 氏酶学法。因为测序每个反应读取的序列是有限的,做长片段 DNA 或基因组测序时,需要选用一定的策略。测序的策略有 primer walking ,随机测序,定向测序等。现在全自动测序仍然沿用 Sanger 氏酶学法,但是在标记上作了改进。 DNA 测序结果通过生物信息学分析,获取需要的信息。
第一节Maxam-Gilbert 化学降解法
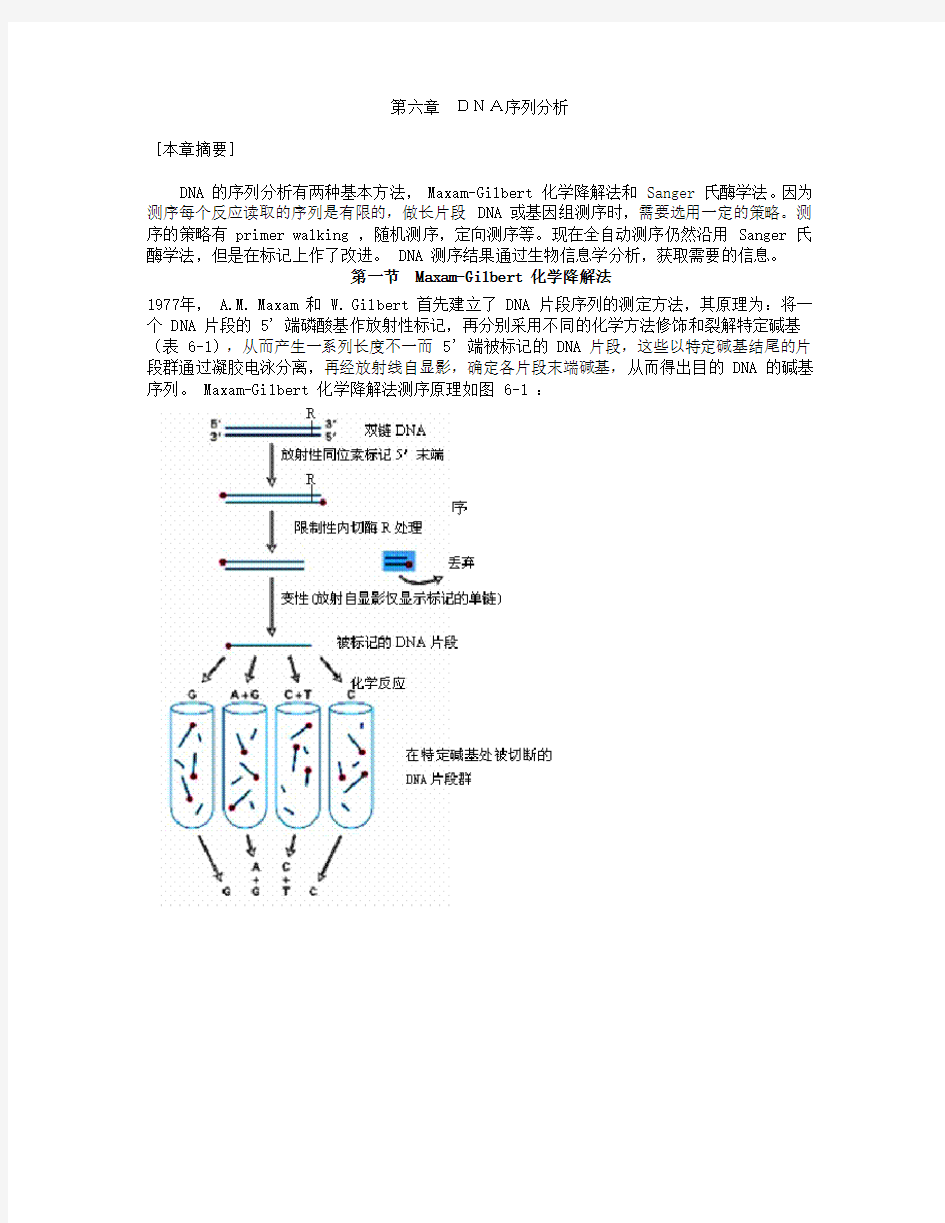
1977年, A.M. Maxam 和 W. Gilbert 首先建立了 DNA 片段序列的测定方法,其原理为:将一个 DNA 片段的 5' 端磷酸基作放射性标记,再分别采用不同的化学方法修饰和裂解特定碱基(表 6-1),从而产生一系列长度不一而 5' 端被标记的 DNA 片段,这些以特定碱基结尾的片段群通过凝胶电泳分离,再经放射线自显影,确定各片段末端碱基,从而得出目的 DNA 的碱基序列。 Maxam-Gilbert 化学降解法测序原理如图 6-1 :
表6-1:Maxam-Gilbert化学降解法测序的常用化学试剂:
碱基体
系
化学修饰试剂化学反应断裂部位
G
A + G C + T C
A > C dimethyl sulphate(硫酸二甲酯)
Piperidine formate(哌啶甲酸), pH2.0
hydrazine(肼,联氨NH2.NH2)
hydrazine + NaCl(1.5M)
90 C, NaOH(1.2M)
甲基化
脱嘌呤
打开嘧啶环
打开胞嘧啶
环
断裂反应
G
G和A
C和T
C
A和C
硫酸二甲酯[dimethyl sulphate ,DMS ,(CH3O)2SO2]是一种碱性化学试剂,可以使 DNA 链上的腺嘌呤 A 的 N2和鸟嘌呤 G 的 N7甲基化,但是鸟嘌呤 G 的 N7甲基化速度比腺嘌呤 A 的 N2甲基化速度要快 4-10 倍,并且在中性 pH 环境中, DMS 主要作用于鸟嘌呤 G ,使之甲基化,导致糖苷键断裂。
哌啶甲酸可以使 DNA 链上的嘌呤在酸的作用下发生糖苷水解,导致 DNA 链在脱嘌呤位点(G 和A)发生断裂。
肼,又称联氨 NH2.NH2,在碱性环境中作用于胞嘧啶 C 和胸腺嘧啶 T 的 C4和 C6位置,导致糖苷键断裂。如果加入高浓度的盐(1.2M NaOH),肼则主要作用于胞嘧啶 C ,使之断裂。
作为标记用的放射性同位素主要有γ-32P([γ-32P]ATP,[γ-32P]GTP. [γ-32P]TTP ,或[γ-32P]CTP),或γ-33NTP , S-35NTP 。
Maxam-Gilbert 化学降解测序法不需要进行酶催化反应,因此不会产生由于酶催化反应而带来的误差;对未经克隆的 DNA 片段可以直接测序;化学降解测序法特别适用于测定含有如 5-甲基腺嘌呤 A 或者 G , C 含量较高的 DNA 片段,以及短链的寡核苷酸片段的序列。
化学降解测序法既可以标记 5'-末端,也可以标记 3'-末端。如果从两端分别测定同一条DNA 链的核苷酸序列,相互参照测定结果,可以得到准确的 DNA 链序列。
第二节Sanger 氏酶学法
双脱氧核苷酸分子的脱氧核糖的3' 位置的-OH 缺失。当它与其他正常核苷酸混合在同一个扩增反应体系中时,在DNA 多聚酶的作用下,虽然它也能够象正常核苷酸一样参与DNA 合成,以其5' 位置的磷酸基,与上位脱氧核苷酸的3' 位置的-OH 结合,但是,由于它自身3' 位置-OH 的缺失,至使下位核苷酸的5' 磷酸基无法与之结合。如图6-2 所示。
图6-2 :双脱氧核苷酸(ddNTP)分子的结构及DNA 链合成终止反应
A.正常的DNA 合成反应;
B.ddNTP 掺入到DNA 合成反应后导致反应终止
基于双脱氧核苷酸的这种特性,Sanger 于1977 年建立了以双脱氧链终止法为基础来测定DNA 序列的方法。该方法以待测单链或双链DNA 为模板,使用能与DNA 模板结合的一段寡核苷酸为引物,在DNA 多聚酶的催化作用下合成新的DNA 链。正常情况下的DNA 多聚酶催化反应在其反应体系中只含有四种脱氧核苷酸(dATP、dCTP、dGTP和dTTP),合成与模板DNA 互补的新链。当这个反应体系中加入了一种放射性同位素32P 或35S 标记的双脱氧核苷酸(*-ddATP 或*-ddCTP 或*-ddGTP 或*-ddTTP)后,在DNA 合成过程中,标记的
*-ddNTP (例如*-ddATP)将与相应的dNTP (例如dA TP)竞争掺入到新合成的DNA 互补链中。如果是dNTP 掺入其中,DNA 互补链则将继续延伸下去;如果是*ddNTP 掺入其中,DNA 互补链的合成则到此终止。而双脱氧核苷酸的掺入是随机的,故各个新生DNA 片段的长度互不相同。过程如图6-3 所示。不同长度DNA 片段在凝胶中的移动速率不同,而聚丙烯酰胺凝胶分辨率极高,通过聚丙烯酰胺凝胶电泳能分辨出小至一个碱基长度差的DNA 片段,从而将混合产物中不同长度DNA 片段分离开,再通过放射自显影曝光,根据片段尾部的双脱氧核苷酸读出该DNA 的碱基排列顺序。作为标记用的放射性同位素主要有[α-32P]dNTP,[α-33P]dNTP 或[α-35S]NTP)。图6-4 是链终止反应产物的聚丙烯酰胺凝胶电泳的放射自显影照片。
第三节DNA 片段序列分析的策略
一、DNA 片段序列分析的策略
1.Primer walking
引物依次推进法是从目的片段的一端开始,利用载体上的序列为依据,设计第一次反应的反
应引物进行测序,然后,依次根据前一次测序结果,设计下一次反应引物,递进测序,直至完成。引物依次推进法需要多次设计和合成引物,比较繁琐;对于重复序列较多的目的DNA ,由于引物的非特异性结合概率高,所以该方法不适合于测定含有多重复序列的DNA 片段。2.随机测序(random approach),或称作鸟枪法(shotgun strategy)
原理:将待测DNA 克隆于噬菌体载体(M13)上,然后通过一定手段将待测DNA 片断化(DNA 片断化的手段通常有超声波处理和酶切),得到两端彼此重叠的部位不同的若干小片段,再测出每一片段的序列,将各片段的序列依次排列,即能得出总的序列。原理图示如6-5:3.定向测序
定向测序的基本策略是沿着目的DNA 的同一个方向逐渐向前推进,直至目的DNA 的另一端。可采用嵌套缺失法或引物依次推进法。
嵌套缺失法是利用缺失随机突变原理,先将目的DNA 克隆到载体上,再用限制性内切酶在邻近其一端处切断载体,使之变成3' 端凹进5' 端突出的线状DNA ,再用外切酶(exonuclease Ⅲ)或者DNaseⅠ加Mn++离子,沿此末端逐步消化目的DNA ,(只能从5' 突出端开始消化,而不能从3' 突出端开始消化,这是由酶的特性所决定的)(详见后文)。于是只要控制消化时间,即可得到一组依次相差若干碱基,其一端固定于载体另一端游离的目的DNA 片断,再经末端修饰,重新连接,即可得到一群一端相同但长度不等的目的DNA 克隆。之后,用同一引物从缺失端开始测序,最后,排列和比较所有子片断的序列,即能得到目的DNA 的全部序列。
二、测序的基本方案
1.制备双脱氧链终止测序的模板
用于双脱氧链终止测序的模板可以是单链DNA ,双链DNA ,PCR 产物,BAC 或Cosmid 。单链DNA 通常为M13 噬菌体DNA(M13mpDNA),用于测序的PCR 产物DNA
量一般为200-800ng。在进行测序之前,无论选用何种测序策略,用于测序的模板DNA 均有必要用溴化乙锭-琼脂糖凝胶电泳法确定其纯度和浓度,这是保证测序成功的基本点。
2.确定测序的引物
DNA 测序的另一关键在于引物的选择和设计。作为测序引物的寡核苷酸的基本条件主要有以下几点。①引物长度18-22 碱基;②尽量避免三个以上相同碱基的重复,尤其是G 或C ;
③Tm 值约为55-60℃(至少要高于45℃);④发夹结构尽量少;⑤引物自身之间不形成二聚体结构;⑥用灭菌后的去离子水溶解合成的寡核苷酸引物,配制成5-10pmol/μl 浓度,冷冻保存。
引物浓度可以参考算式计算:
pmol/μl=100 x A260/(1.54nA + 0.75nC + 1.17nG +0.92nT)
A260:260nm 波长时的吸收值。nA:腺嘌呤A 的数目;nC:胞嘧啶C 的数目;nG:鸟嘌呤G 的数目;nT:胸腺嘧啶T 的数目。为保证引物纯度,至少应该经过聚丙烯酰胺凝胶电泳或者纯化度更高的HPLC 纯化。
三、DNA 自动测序和序列的计算机分析
1.原理
基于Sanger 双脱氧链终止法的原理,只不过不用同位素标记ddNTP ,而是用四种不同的荧光染料分别标记ddATP、ddTTP、ddCTP、ddGTP 。这四种荧光染料在激光激发下发出不同波长的荧光,因而每一种荧光代表一种碱基。延伸中的DNA 互补链分别终止于不同荧光标记的ddA TP、ddTTP、ddCTP、ddGTP 。
2.序列结果
图6-6为测序仪ABI Prism 310 Genetic analyzer 实际测得的一段DNA 序列结果。红色代表
T ;绿色代表A ;兰色代表C ;黑色代表G 。
3.序列分析
a.基因序列的信息分析
例如,两种序列的比较;基因编码领域,启动子或增强子序列的预测;蛋白质序列的氨基酸顺序的确定;
b.新基因的发现
通过分析EST(Expressed Sequence Tag)序列,结合DNA Chip 技术寻找新基因。
c.基因多型性分析
分析基因多型性特别是SNP(Single Nucleotide Polymorphism),发现并定位功能性基因,作为人类群体,进化,或者疾病关联靶标。
d.高级结构的预测
根据测序所得到的核酸一级结构的信息,可以预测核酸和蛋白质的二级结构及其三级结构,从而预测其功能。
统计基础知识第五章时间序列分析习题及答案
第五章时间序列分析 一、单项选择题 1.构成时间数列的两个基本要素是( C )(2012年1月) A.主词和宾词 B.变量和次数 C.现象所属的时间及其统计指标数值 D.时间和次数 2.某地区历年出生人口数是一个( B )(2011年10月) A.时期数列 B.时点数列 C.分配数列 D.平均数数列 3.某商场销售洗衣机,2008年共销售6000台,年底库存50台,这两个指标是( C ) (2010年10) A.时期指标 B.时点指标 C.前者是时期指标,后者是时点指标 D.前者是时点指标,后者是时期指标 4.累计增长量( A ) (2010年10) A.等于逐期增长量之和 B.等于逐期增长量之积 C.等于逐期增长量之差 D.与逐期增长量没有关系 5.某企业银行存款余额4月初为80万元,5月初为150万元,6月初为210万元,7月初为160万元,则该企业第二季度的平均存款余额为( C )(2009年10) 万元万元万元万元 6.下列指标中属于时点指标的是( A ) (2009年10) A.商品库存量 B.商品销售量 C.平均每人销售额 D.商品销售额 7.时间数列中,各项指标数值可以相加的是( A ) (2009年10) A.时期数列 B.相对数时间数列 C.平均数时间数列 D.时点数列 8.时期数列中各项指标数值( A )(2009年1月) A.可以相加 B.不可以相加 C.绝大部分可以相加 D.绝大部分不可以相加 10.某校学生人数2005年比2004年增长了8%,2006年比2005年增长了15%,2007年比2006年增长了18%,则2004-2007年学生人数共增长了( D )(2008年10月) %+15%+18%%×15%×18% C.(108%+115%+118%)-1 %×115%×118%-1 二、多项选择题 1.将不同时期的发展水平加以平均而得到的平均数称为( ABD )(2012年1月) A.序时平均数 B.动态平均数 C.静态平均数 D.平均发展水平 E.一般平均数2.定基发展速度和环比发展速度的关系是( BD )(2011年10月) A.相邻两个环比发展速度之商等于相应的定基发展速度 B.环比发展速度的连乘积等于定基发展速度
时间序列分析第一章王燕习题解答
时间序列分析习题解答 第一章 P. 7 1.5 习题 1.1 什么是时间序列?请收集几个生活中的观察值序列。 答:按照时间的顺序把随机事件变化发展的过程记录下来就构成一个时间序列。 例1:1820—1869年每年出现的太阳黑子数目的观察值; 年份黑子数年份黑子数年份黑子数年份黑子数年份黑子数1820 16 1830 71 1840 63 1850 66 1860 96 1821 7 1831 48 1841 37 1851 64 1861 77 1822 4 1832 28 1842 24 1852 54 1862 59 1823 2 1833 8 1843 11 1853 39 1863 44 1824 8 1834 13 1844 15 1854 21 1864 47 1825 17 1835 57 1845 40 1855 7 1865 30 1826 36 1836 122 1846 62 1856 4 1866 16 1827 50 1837 138 1847 98 1857 23 1867 7 1828 62 1838 103 1848 124 1858 55 1868 37 1829 67 1839 86 1849 96 1859 94 1869 74 例2:北京市城镇居民1990—1999年每年的消费支出按照时间顺序记录下来,就构成了一个序列长度为10的消费支出时间序列(单位:亿元)。 1686,1925,2356,3027,3891,4874,5430,5796,6217,6796。 1.2 时域方法的特点是什么? 答:时域方法特点:具有理论基础扎实,操作步骤规范,分析结果易于解释的优点,是时间序列分析的主流方法。 1.3 时域方法的发展轨迹是怎样的? 答:时域方法的发展轨迹: 一.基础阶段: 1. G.U. Yule 1972年AR模型 2. G.U.Walker 1931年 MA模型、ARMA模型 二.核心阶段:G.E.P.Box和G.M.Jenkins 1. 1970年,出版《Time Series Analysis Forecasting and Control》 2. 提出ARIMA模型(Box-Jenkins模型) 3. Box-Jenkins模型实际上主要运用于单变量、同方差场合的线性模型 三.完善阶段: 1.异方差场合: a.Robert F.Engle 1982年 ARCH模型
2019第4章时间序列分析
校级精品课程《统计学》 习题
第四章时间序列 一、单项选择题 1. 时间序列是( ) A. 分配数列 B.分布数列 C.时间数列 D.变量数列 2. 时期序列和时点序列的统计指标( )。 A. 都是绝对数 B.都是相对数 C.既可以是绝对数,也可以是相对数 D.既可以是平均数,也可以是绝对数 3. 时间序列是( )。 A .连续序列的一种 B .间断序列的一种 C. 变量序列的一种 D.品质序列的一种 4. 最基本的时间序列是( )。 A. 时点序列 B.绝对数时间序列 C.相对数时间序列 D.平均数时间序列 5. 为便于比较分析,要求时点序列指标数值的时间间隔( )。 A. 必须连续 B.最好连续 C.必须相等 D.最好相等 6. 时间序列中的发展水平( )。 A. 只能是总量指标 B.只能是相对指标 C. 只能是平均指标 D.上述三种指标均可 7. 在平均数时间序列中各指标之间具有( )。 A.总体性 B.完整性 C.可加性 D.不可加性 8. 序时平均数与一般平均数相比较( )。
A. 均抽象了各总体单位的差异 B. 均根据同种序列计算 C. 序时平均数表明现象在某一段时间内的平均发展水平,一般平均数表明现象在规定时间内总体的一般水平 D. 严格说来,序时平均数不能算作平均数 9. 序时平均数与一般平均数的共同点是( )。 A.两者均是反映同一总体的一般水平 B.都是反映现象的一般水平 C.两者均可消除现象波动的影响 D.都反映同质总体在不同时间的一般水平 10. 时期序列计算序时平均数应采用( )。 A.加数算术平均法 B.简单算术平均法 C.简单算术平均法 D.加权算术平均数 11. 间隔相等连续时点序列计算序时平均数,应采用( )。 A.简单算术平均法 B.加数算术平均法 C.简单序时平均法 D.加权序时平均法 12. 由间断时点序列计算序时平均数,其假定条件是研究现象在相邻两个时点之 间的变动为( )。 A.连续的 B.间断的 C.稳定的 D.均匀的 13. 时间序列最基本速度指标是( )。 A.发展速度 B.平均发展速度 C.增减速度 D.平均增减速度 14. 用水平法计算平均发展速度应采用( )。 A.简单算术平均 B.调和平均 C.加权算术平均 D.几何平均 15. 计算速度指标应采用( )。
第六章 时间序列分析 补充作业 参考答案
第六章 时间序列分析 补充作业 参考答案 1、解: (1)、各季平均每月总产值 一季度平均每月总产值:)(34003 3600 340032001 210万元=++= ++++= n a a a a a n 二季度平均每月总产值:)(38503 3900385038001 210万元=++=++++= n a a a a a n 三季度平均每月总产值:)(42003 4400420040001 210万元=++=++++= n a a a a a n 四季度平均每月总产值:)(33.463334800460045001 210万元=++=++++= n a a a a a n (2)、全年平均每月总产值: )(83.40204 33 .46334200385034001210万元=+++=++++= n a a a a a n 或: )(83.402012 4800 46004500440042004000390038503800360034003200万元=+++++++++++= a 2、解: 2006年平均存款余额: ) (21.9612 5.115435313 2102 10052100903290971297952221 1221110万元==+++?++?++?++?+=+++++=∑=-n i i n n n f f a a f a a f a a a 3、解: 年份 2001 2002 2003 2004 2005 2006 0a 1a 2a 3a 4a 5a 发展水平(万元) 500 550 625 775 968.75 1023 逐期增长量(万元) —— 50 75 150 193.75 54.25 累计增长量(万元) —— 50 125 275 468.75 523 平均增长量(万元) —— 50 62.5 91.67 117.19 104.6 环比发展速度(%) —— 110 113.64 124 125 105.6 定基发展速度(%) 100 110 125 155 193.75 204.6 环比增长速度(%) —— 10 13.64 24 25 5.6 定基增长速度(%) 0 10 25 55 93.75 104.6 增长1%的绝对值(万元) —— 5 5.5 6.25 7.75 9.69
时间序列分析 第一章 时间序列分析简介
input time monyy7. price; format time monyy5. ; cards; jan2005 101 feb2005 82 mar2005 66 apr2005 35 may2005 31 jun2005 7 ; run; proc print data=example1_1; run; 实验结果: 实验分析:该程序的到了一个名为sasuser.example1_1的永久数据集。所谓的永久数据库就是指在该库建立的数据集不会因为我们退出SAS系统而丢失,它会永久的保存在该数据库中,我们以后进入SAS系统还可以从该库中调用该数据集。 3.查看数据集 data example1_1; input time monyy7. price; format time monyy5. ; cards; jan2005 101 feb2005 82 mar2005 66 apr2005 35 may2005 31 jun2005 7 ; run; proc print data=example1_1; run; 实验结果:
2.序列变换 data example1_3; input price; logprice=log(price); time=intnx('month','01jan2005'd,_n_-1); format time monyy.; cards; 3.41 3.45 3.42 3.53 3.45 ; proc print data=example1_3; run; 实验结果: 实验分析:在时间序列分析中,我们得到的是观测值序列xt,但是需要分析的可能是这个观察值序列的某个函数变换,例如对数序列lnxt。在建立数据集时,我们可以通过简单的赋值命令实现这个变换。再该程序中,logprice=log(price);是一个简单的赋值语句,将price的对数函数值赋值给一个新的变量logprice,即建立了一个新的对数序列。 3.子集 data example1_4; set example1_3; keep time logprice; where time>='01mar2005'd; proc print data=example1_4; run; 实验结果:
高通量测序生物信息学分析(内部极品资料,初学者必看)
基因组测序基础知识 ㈠De Novo测序也叫从头测序,是首次对一个物种的基因组进行测序,用生物信息学的分析方法对测序所得序列进行组装,从而获得该物种的基因组序列图谱。 目前国际上通用的基因组De Novo测序方法有三种: 1. 用Illumina Solexa GA IIx 测序仪直接测序; 2. 用Roche GS FLX Titanium直接完成全基因组测序; 3. 用ABI 3730 或Roche GS FLX Titanium测序,搭建骨架,再用Illumina Solexa GA IIx 进行深度测序,完成基因组拼接。 采用De Novo测序有助于研究者了解未知物种的个体全基因组序列、鉴定新基因组中全部的结构和功能元件,并且将这些信息在基因组水平上进行集成和展示、可以预测新的功能基因及进行比较基因组学研究,为后续的相关研究奠定基础。 实验流程: 公司服务内容 1.基本服务:DNA样品检测;测序文库构建;高通量测序;数据基本分析(Base calling,去接头, 去污染);序列组装达到精细图标准 2.定制服务:基因组注释及功能注释;比较基因组及分子进化分析,数据库搭建;基因组信息展 示平台搭建 1.基因组De Novo测序对DNA样品有什么要求?
(1) 对于细菌真菌,样品来源一定要单一菌落无污染,否则会严重影响测序结果的质量。基因组完整无降解(23 kb以上), OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;每次样品制备需要10 μg样品,如果需要多次制备样品,则需要样品总量=制备样品次数*10 μg。 (2) 对于植物,样品来源要求是黑暗无菌条件下培养的黄化苗或组培样品,最好为纯合或单倍体。基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。 (3) 对于动物,样品来源应选用肌肉,血等脂肪含量少的部位,同一个体取样,最好为纯合。基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。 (4) 基因组De Novo组装完毕后需要构建BAC或Fosmid文库进行测序验证,用于BAC 或Fosmid文库构建的样品需要保证跟De Novo测序样本同一来源。 2. De Novo有几种测序方式 目前3种测序技术 Roche 454,Solexa和ABI SOLID均有单端测序和双端测序两种方式。在基因组De Novo测序过程中,Roche 454的单端测序读长可以达到400 bp,经常用于基因组骨架的组装,而Solexa和ABI SOLID双端测序可以用于组装scaffolds和填补gap。下面以solexa 为例,对单端测序(Single-read)和双端测序(Paired-end和Mate-pair)进行介绍。Single-read、Paired-end和Mate-pair主要区别在测序文库的构建方法上。 单端测序(Single-read)首先将DNA样本进行片段化处理形成200-500bp的片段,引物序列连接到DNA片段的一端,然后末端加上接头,将片段固定在flow cell上生成DNA簇,上机测序单端读取序列(图1)。 Paired-end方法是指在构建待测DNA文库时在两端的接头上都加上测序引物结合位点,在第一轮测序完成后,去除第一轮测序的模板链,用对读测序模块(Paired-End Module)引导互补链在原位置再生和扩增,以达到第二轮测序所用的模板量,进行第二轮互补链的合成测序(图2)。 图1 Single-read文库构建方法图2 Paired-end文库构建方法
第六章时间序列分析题库1-0-8
第六章时间序列分析 题库1-0-8
问题: [单选]下列数列中属于时间数列的是() A.学生按学习成绩分组形成的数列 B.一个月内每天某一固定时点记录的气温按度数高低排列形成的序列 C.工业企业按产值高低形成的数列 D.降水量按时间先后顺序排列形成的数列
问题: [单选]评比城市间的社会发展状况,将各城市每人分摊的绿化面积按年排列的时间数列是属于。 A.时期数列 B.时点数列 C.相对指标时间数列 D.平均指标时间数列 相对指标时间数列是指将同一相对指标的数值按其发生的时间先后顺序排列而成的数列。题中,平均每人分摊绿化面积是一个强度相对指标,将其按年排列的时间数列属于相对指标时间数列。
问题: [单选]已知某商业集团2008-2009年各季度销售资料,如表5-1所示。 表5-1 则表5-1中,属于时期数列的有。 A.A.1、2、3 B.1、3、4 C.2、4 D.1、3 1、3的每个数值反映的是现象在一段时期内发展过程的绝对数之和,故属于时期指标数列;2的每个数值反映的是现象在某一时间上所达到的绝对水平,故属于时点指标数列;4是把同一相对指标在不同时间上的数值按时间先后顺序排列而形成的数列,故属于相对指标数列。 (天津11选5 https://www.360docs.net/doc/ed11602666.html,)
问题: [单选]下列对时点数列特征的描述,错误的一项是。 A.时点数列中的指标数值可以相加 B.时点数列中指标数值的大小与计算时间间隔长短无关 C.时点数列中各指标数值的取得,是通过一次性调查登记而来的 D.时点数列属于总量指标时间数列 A项,时点数列中的指标数值不能相加,相加没有意义。
全基因组重测序数据分析
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排 突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使 得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组 学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基 因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需 要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。
第六章 时间序列分析
第六章时间序列分析 重点: 1、增长量分析、发展水平及增长量 2、增长率分析、发展速度及增长速度 3、时间数列影响因素、长期趋势分析方法 难点: 1、增长量与增长速度 2、长期趋势与季节变动分析 第一节时间序列的分析指标 知识点一:时间序列的含义 时间序列是指经济现象按时间顺序排列形成的序列。这种数据称为时间序列数据。 时间序列分析就是根据这样的数列分析经济现象的发展规律,进而预测其未来水平。 时间数列是一种统计数列,它是将反映某一现象的统计指标在不同时间上的数值按时间先后顺序排列所形成的数列。表现了现象在时间上的动态变化,故又称为动态数列。 一个完整的时间数列包含两个基本要素: 一是被研究现象或指标所属的时间; 另一个是该现象或指标在此时间坐标下的指标值。 同一时间数列中,通常要求各指标值的时间单位和时间间隔相等,如无法保证相等,在计算某些指标时就涉及到“权”的概念。 研究时间数列的意义:了解与预测。 [例题·单选题]下列数列中哪一个属于时间数列(). a.学生按学习成绩分组形成的数列 b.一个月内每天某一固定时点记录的气温按度数高低排列形成的序列 c.工业企业按产值高低形成的数列 d.降水量按时间先后顺序排列形成的数列 答案:d 解析:时间序列是一种统计数列,它是将反映某一现象的统计指标在不同时间上的数值按时间先后顺序排列所形成的数列,表现了现象在时间上的动态变化。 知识点二:增长量分析(水平分析)
一.发展水平 发展水平是指客观现象在一定时期内(或时点上)发展所达到的规模、水平,一般用y t (t=1,2,3,…,n) 。 在绝对数时间数列中,发展水平就是绝对数; 在相对数时间数列中,发展水平就是相对数或平均数。 几个概念:期初水平y 0,期末水平y t ,期间水平(y 1 ,y 2 ,….y n-1 ); 报告期水平(研究时期水平),基期水平(作为对比基础的水平)。 二.增长量 增长量是报告期发展水平与基期发展水平之差,增长量的指标数值可正可负,它反映的是报告期相对基期增加或减少的绝对数量,用公式表示为: 增长量=报告期水平-基期水平 根据基期的不同确定方法,增长量可分为逐期增长量和累计增长量。 1.逐期增长量:是报告期水平与前一期水平之差,用公式表示为: △ = y n - y n-1 (i=1,2,…,n) 2.累计增长量:是报告期水平与某一固定时期水平(通常是时间序列最初水平)之差,用公式表示为: △ = y n - y (i=1,2,…,n)(i=1,2,…,n) 二者关系:逐期增长量之和=累计增长量 3.平均增长量 平均增长量是时间序列中的逐期增长量的序时平均数,它表明现象在一定时段内平均每期增加(减少)的数量。 一般用累计增长量除以增长的时期数目计算。 (y n - y )/n [例题·单选题]某社会经济现象在一定时期内平均每期增长的绝对数量是()。 a.逐期增长量 b.累计增长量 c.平均增长量 d.增长速度 答案:c 解析:平均每期增长的绝对数量是平均增长量。 知识点三:增长率分析(速度分析) 一.发展速度
第五章 时间序列的模型识别
第五章时间序列的模型识别 前面四章我们讨论了时间序列的平稳性问题、可逆性问题,关于线性平稳时间序列模型,引入了自相关系数和偏自相关系数,由此得到ARMA(p, q)统计特性。从本章开始,我们将运用数据开始进行时间序列的建模工作,其工作流程如下: 图5.1 建立时间序列模型流程图 在ARMA(p,q)的建模过程中,对于阶数(p,q)的确定,是建模中比较重要的步骤,也是比较困难的。需要说明的是,模型的识别和估计过程必然会交叉,所以,我们可以先估计一个比我们希望找到的阶数更高的模型,然后决定哪些方面可能被简化。在这里我们使用估计过程去完成一部分模型识别,但是这样得到的模型识别必然是不精确的,而且在模型识别阶段对于有关问题没有精确的公式可以利用,初步识别可以我们提供有关模型类型的试探性的考虑。 对于线性平稳时间序列模型来说,模型的识别问题就是确定ARMA(p,q)过程的阶数,从而判定模型的具体类别,为我们下一步进行模型的参数估计做准备。所采用的基本方法主要是依据样本的自相关系数(ACF)和偏自相关系数(PACF)初步判定其阶数,如果利用这种方法无法明确判定模型的类别,就需要借助诸如AIC、BIC 等信息准则。我们分别给出几种定阶方法,它们分别是(1)利用时间序列的相关特性,这是识别模型的基本理论依据。如果样本的自相关系数(ACF)在滞后q+1阶时突然截断,即在q处截尾,那么我们可以判定该序列为MA(q)序列。同样的道理,如果样本的偏自相关系数(PACF)在p处截尾,那么我们可以判定该序列为AR(p)序列。如果ACF和PACF 都不截尾,只是按指数衰减为零,则应判定该序列为ARMA(p,q)序列,此时阶次尚需作进一步的判断;(2)利用数理统计方法检验高阶模型新增加的参数是否近似为零,根据模型参数的置信区间是否含零来确定模型阶次,检验模型残差的相关特性等;(3)利用信息准则,确定一个与模型阶数有关
最新地震处理教程——1 第一章 时间序列分析基础
第一章时间序列分析基础 一维傅里叶变换 首先观察一个实验。将弹簧的一端固定并悬垂,另一端挂一重物。向下拉重物使弹簧拉伸某一距离,比如说0.8个单位,使其振动。现假定弹簧是弹性的,那么它将无休止地上下运动。若将运动起始的平衡位置定为时间零,那么重物的位移量将随着时间函数在极限[+0.8—-0.8]之间变化。如果有一装置能给出位移振幅随时间函数变化的轨迹,就会得到一条正弦波曲线。其相邻两峰值间的时间间隔为0.08秒(80毫秒)。我们称它为弹簧的周期,它取决于所测弹簧刚度的弹性常数。我们说弹簧在一个周期时间内完成了一次上下振动。在1秒的观测时间内记下其周期数,我们发现是12.5周,这个数被称为弹簧振动的频率。你一定会注意到,1/0.08=12.5,这就是说频率为周期的倒数。 我们取另一个刚性较大的弹簧,并重复上面的实验。不过这次弹簧的振幅峰值位移为0.4个单位。它的运动轨迹所显示的是另一条正弦曲线。量其周期和频率分别为0.04秒和25周/秒,为了记下这些测量结果,我们做每个弹簧峰值振幅与频率的关系图,这便是振幅谱。 现在取两个相同的弹簧。一个弹簧从0.8个单位的峰值振幅位移开始松开,并使其振动。这时注意弹簧通过零时平衡位置的时间,就在它通过零时的一刹那,请你将另一弹簧从0.8个单位的同样峰值振幅位移处松开。这样由于起始的最大振幅相同,所以两个正弦时间函数的振幅谱也应该一样。但肯定两者之间是有差别的,特别是当第1个正弦波达到峰值振幅时,另一个的振幅为零。两者的区别为:第2个弹簧的运动相对于第1个弹簧的运动有一个等于四分之一周期的时间延迟。四分之一周期的时间延迟等于90°相位滞后。所以除振幅谱之外,我们还可以作出相位延迟谱,至此,这个实验做完了。那么我们学到了什么呢?这就是弹簧的弹性运动可以用正弦时间函数来描述,更重要的是,可以用正弦波的频率、峰值振幅及相位延迟来全面地描述正弦波运动。这个实验告诉我们弹簧的振动是怎样随时间和频率函数变化的。 现在设想有一组弹簧,每个弹簧的正弦运动都具有特定的频率、峰值振幅和相位延迟。所有弹簧的正弦响应如图1所示。我们可以把该系统的运动“合成”为一个总的波动,来代替该组中的各单个分量的运动。这一合成是直接把所有记录道相加,其结果得到一个与时间相关的信号,在图1中由第一道表示。我们通过这种合成可以把这一运动由频率域变换到时间域。这一变换是可逆的:即给定时间域信号,我们可以把它变换到频率域的正弦分量。在数学上,这种双向过程是由傅里叶变换完成的。在实际应用中,标准的运算是所谓快速傅氏变换。通过傅氏正变换可以把与时间相关的信号分解成它的频率分量,而所有的频率分量合成为时间域信号又是通过反傅氏变换来实现的。图2概括了信号的傅氏变换。振幅谱和相位谱(严格地讲是相位延迟谱)是图1中所显示的正弦波最简单的表示形
时间序列分析基于R——习题答案
第一章习题答案 略 第二章习题答案 2.1 (1)非平稳 (2)0.0173 0.700 0.412 0.148 -0.079 -0.258 -0.376 (3)典型的具有单调趋势的时间序列样本自相关图 2.2 (1)非平稳,时序图如下 (2)-(3)样本自相关系数及自相关图如下:典型的同时具有周期和趋势序列的样本自相关图
2.3 (1)自相关系数为:0.2023 0.013 0.042 -0.043 -0.179 -0.251 -0.094 0.0248 -0.068 -0.072 0.014 0.109 0.217 0.316 0.0070 -0.025 0.075 -0.141 -0.204 -0.245 0.066 0.0062 -0.139 -0.034 0.206 -0.010 0.080 0.118 (2)平稳序列 (3)白噪声序列 2.4 ,序列LB=4.83,LB统计量对应的分位点为0.9634,P值为0.0363。显著性水平=0.05 不能视为纯随机序列。 2.5 (1)时序图与样本自相关图如下
(2) 非平稳 (3)非纯随机 2.6 (1)平稳,非纯随机序列(拟合模型参考:ARMA(1,2)) (2)差分序列平稳,非纯随机 第三章习题答案 3.1 ()0t E x =,2 1 () 1.9610.7 t Var x ==-,220.70.49ρ==,220φ= 3.2 1715φ=,2115 φ= 3.3 ()0t E x =,10.15 () 1.98(10.15)(10.80.15)(10.80.15) t Var x += =--+++ 10.8 0.7010.15 ρ= =+,210.80.150.41ρρ=-=,3210.80.150.22ρρρ=-= 1110.70φρ==,2220.15φφ==-,330φ= 3.4 10c -<<, 1121,1,2 k k k c c k ρρρρ--?=? -??=+≥? 3.5 证明: 该序列的特征方程为:32 - -c 0c λλλ+=,解该特征方程得三个特征根: 11λ=,2λ=3λ=
应用时间序列分析 第5章
佛山科学技术学院 应用时间序列分析实验报告 实验名称第五章非平稳序列的随机分析 一、上机练习 通过第4章我们学习了非平稳序列的确定性因素分解方法,但随着研究方法的深入和研究领域的拓宽,我们发现确定性因素分解方法不能很充分的提取确定性信息以及无法提供明确有效的方法判断各因素之间确切的作用关系。第5章所介绍的随机性分析方法弥补了确定性因素分解方法的不足,为我们提供了更加丰富、更加精确的时序分析工具。 5.8.1 拟合ARIMA模型 【程序】 data example5_1; input x@@; difx=dif(x); t=_n_; cards; 1.05 -0.84 -1.42 0.20 2.81 6.72 5.40 4.38 5.52 4.46 2.89 -0.43 -4.86 -8.54 -11.54 -1 6.22 -19.41 -21.61 -22.51 -23.51 -24.49 -25.54 -24.06 -23.44 -23.41 -24.17 -21.58 -19.00 -14.14 -12.69 -9.48 -10.29 -9.88 -8.33 -4.67 -2.97 -2.91 -1.86 -1.91 -0.80 ; proc gplot; plot x*t difx*t; symbol v=star c=black i=join; proc arima; identify var=x(1); estimate p=1; estimate p=1 noint; forecast lead=5id=t out=out; proc gplot data=out; plot x*t=1 forecast*t=2 l95*t=3 u95*t=3/overlay; symbol1c=black i=none v=star; symbol2c=red i=join v=none; symbol3c=green I=join v=none;
进化基因组学研究进展
进化基因组学研究进展 刘超 (山东大学生命科学学院济南250100) 摘要:进化基因组学是利用基因组数据研究差异基因功能、生物系统演化、从 基因在水平探索生物进化的学科。随着近年来基因组数据的不断增加,进化基因组学得到了长足的发展。进化基因组学主要包括从基因组水平理解和诠释生物进 化和新基因分析研究探索两方面的内容。本文介绍了进化基因组学研究的主要内容和较为常用的方法,以及近年来在细菌、酵母、果蝇进化基因组学方面的研究进展。 关键词:进化基因组学系统进化比较基因组学新基因 前言 随着基因测序技术的不断进步以及基因组学的飞速的发展,人们积累了大量的基因组学数据,利用所得的大量的基因组数据与进化生物学相结合,在基因组水平研究生物进化机制,随即产生了进化基因组学(Evolutional Genomics)。 近年来进化基因组学取得了长足的进展,在研究差异基因功能、生物系统演化、从基因在水平探索生物进化的终极方式等方面有重大突破,对人类理解生命现象和过程有重要作用。 1进化基因组学研究内容 研究系统进化学通常包括两个关键步骤:一方面,在不同物种中鉴定同源性特佂,另一方面利用构建系统进化树的方法比较这些特征,进而重新构建这些物种的进化历史[1]。针对这两个关键步骤,传统系统进化学,常采用基于形态学 数据和单个基因研究的同源性状鉴定和重建系统进化树(常包括距离法、最大简约法、概率法)[1]的方法来研究。在目前拥有丰富基因组数据的条件下,我们 可以分析基因组数据,利用进化基因组学研究系统进化。
目前进化基因组学的研究内容主要集中于两个方面:(1)在比较不同生物的基因数据的基础上,从基因组水平理解和诠释生物进化;(2)通过对新基因的分析研究探索基因进化过程的规律两个方面[2](如图1)。在进行全基因组进化分析方面,进化基因组学主要集中于构建系统进化树、研究基因组进化策略、研究生物功能变化和进化机制、进化和生态功能基因组学[2]、基因注释的等方面;在新基因方面主要分析基因产生机制和新基因固定及其动力学研究。 图1 进化基因组学主要研究内容 目前进化基因组学的研究有力的解决了一些基础性的进化问题,但也出现了一些未来需要急需解决的挑战。例如生物进化的本质和目前重建系统进化树方法 的限制[1]。 2研究进化基因组学的方法 研究进化基因组学的方法主要包括利用基因组数据分析和研究新基因的产 生和演化两种。 2.1利用基因组数据进行系统进化分析 利用基因组数据进行系统进化分析,常有基于基因序列的方法和基于全基因特征的方法。(如图2)
时间序列分析方法第章谱分析完整版
时间序列分析方法第章 谱分析 HEN system office room 【HEN16H-HENS2AHENS8Q8-HENH1688】
第六章 谱分析 Spectral Analysis 到目前为止,t 时刻变量t Y 的数值一般都表示成为一系列随机扰动的函数形式,一般的模型形式为: 我们研究的重点在于,这个结构对不同时点t 和τ上的变量t Y 和τ Y 的协方差具有什么样的启示。这种方法被称为在时间域(time domain)上分析时间序列+∞∞-}{t Y 的性质。 在本章中,我们讨论如何利用型如)cos(t ω和)sin(t ω的周期函数的加权组合来描述时间序列t Y 数值的方法,这里ω表示特定的频率,表示形式为: 上述分析的目的在于判断不同频率的周期在解释时间序列+∞∞ -}{t Y 性质时所发挥的重要程度如何。如此方法被称为频域分析(frequency domain analysis)或者谱分析(spectral analysis)。我们将要看到,时域分析和频域分析之间不是相互排斥的,任何协方差平稳过程既有时域表示,也有频域表示,由一种表示可以描述的任何数据性质,都可以利用另一种表示来加以体现。对某些性质来说,时域表示可能简单一些;而对另外一些性质,可能频域表示更为简单。 § 母体谱 我们首先介绍母体谱,然后讨论它的性质。 6.1.1 母体谱及性质 假设+∞∞-}{t Y 是一个具有均值μ的协方差平稳过程,第j 个自协方差为: 假设这些自协方差函数是绝对可加的,则自协方差生成函数为: 这里z 表示复变量。将上述函数除以π2,并将复数z 表示成为指数虚数形式)ex p(ωi z -=,1-=i ,则得到的结果(表达式)称为变量Y 的母体谱: 注意到谱是ω的函数:给定任何特定的ω值和自协方差j γ的序列+∞∞-}{j γ,原则上都可以计算)(ωY s 的数值。 利用De Moivre 定理,我们可以将j i e ω-表示成为: 因此,谱函数可以等价地表示成为: 注意到对于协方差平稳过程而言,有:j j -=γγ,因此上述谱函数化简为: 利用三角函数的奇偶性,可以得到: 假设自协方差序列+∞∞-}{j γ是绝对可加的,则可以证明上述谱函数
时间序列分析第五章作业
时间序列分析第五章作业 班级:09数学与应用数学 学号: 姓名: 习题5.7 1、 根据数据,做出它的时序图及一阶差分后图形,再用ARIMA 模型模拟该序列的发展,得出 预测。根据输出的结果,我们知道此为白噪声,为非平稳序列,同时可以得出序列t x 模型 应该用随机游走模型(0,1,0)模型来模拟,模型为:,并可以预测到下一天 的收盘价为296.0898。 各代码: data example5_1; input x@@; difx=dif(x); t=_n_; cards ; 304 303 307 299 296 293 301 293 301 295 284 286 286 287 284 282 278 281 278 277 279 278 270 268 272 273 279 279 280 275 271 277 278 279 283 284 282 283 279 280 280 279 278 283 278 270 275 273 273 272 275 273 273 272 273 272 273 271 272 271 273 277 274 274 272 280 282 292 295 295 294 290 291 288 288 290 293 288 289 291 293 293 290 288 287 289 292 288 288 285 282 286 286 287 284 283 286 282 287 286 287 292 292 294 291 288 289 ; proc gplot ; plot x*t difx*t; symbol v =star c =black i =join; proc arima data =example5_1; identify Var =x(1) nlag =8 minic p = (0:5) q = (0:5); estimate p =0 q =0 noint; forecast lead =1 id =t out =results; run ; proc gplot data =results; plot x*t=1 forecast*t=2 l95*t=3 u95*t=3/overlay ; symbol1 c =black i =none v =star; symbol2 c =red i =join v =none; symbol3 c =green i =join v =none l =32; run ; 时序图:
统计学__第11章 时间序列分析
图例7 一、循环变动及其测定目的二、循环变动的测定方法(一)直接法(二)剩余法循环变动分析循环变动分析-意义循环变动分析―形式直接法剩余法操作步骤用移动平均法,得到TC的估计,由Y/TC,得到仅含季节变动的序列,计算季节指数对原序列建立趋势方程,得趋势项T的估计值原始序列Y/TS得CI的数据对CI进行移动平均得到C的估计注:剔除趋势求季节指数,如果没有特别要求就先采用移动平均法求其趋势,然后求季指回总目录回本章目录平稳时间序列概述平稳时间序列定义常见时间序列模型严平稳回总目录回本章目录平稳时间序列所谓平稳时间序列,指如果序列二阶矩有限 , 且满足如下条件:对任意整数为常数;对任意整数自协方差函数仅与时间间隔有关,和起止时刻无关。即则称序列为宽平稳(或协方差平稳,二阶矩平稳)序列当时,自协方差函数就是方差回总目录回本章目录平稳序列图形上来看就是:(1)序列围绕常数的长期均值波动,称为是均值回复(Meaning Reversion) (2)在每一时刻,方差对均值的偏离基本相同,波动程度大致相等。回总目录回本章目录最简单的宽平稳序列是白噪声,常记为,它是构成其他序列的基石,一般白噪声的定义如下:对任意对任意对不同的时刻自回归模型(AR:Auto-regressive);滑动平均模型(MA:Moving-Average);自回归滑动平均模型(ARMA:Auto-regressive Moving-Average)。回总目录回本章目录常见时间序列模型 P阶自回归模型AR(P)模型回总目录回本章目录其中
称为自回归系数,为白噪声序列上式称为是p阶自回归模型,简记为AR(p) 当满足一定条件时,序列是平稳的零均值时间序列满足如下形式 q阶滑动平均模型MA(q)模型回总目录回本章目录其中称为滑动平均系数,为白噪声序列上式称为是q阶滑动平均模型,简记为MA(q) 当阶数q有限时,序列是平稳的零均值时间序列满足如下形式自回归滑动平均模型(ARMA)模型回总目录回本章目录其中称为自回归系数,称为滑动平均系数,为白噪声序列上式称为是p阶自回归模型-q阶滑动平均模型,简记为AMMA(p,q). 当p=0, AMMA(p,q)--MA(q) 一般ARMA模型的数学形式为当满足一定条件时,序列是平稳的.从以上定义中可以看出,AR模型和MA模型即为ARMA模型的特例当q=0, AMMA(p,q)--MA(p) 回总目录回本章目录 ARMA模型的识别相关函数定阶法信息准则定阶法严平稳回总目录回本章目录相关函数定阶法采用ARMA模型对现有的数据进行建模,首要的问题是确定模型的阶数,即相应的p,q的值,对于ARMA模型的识别主要是通过序列的自相关函数以及偏自相关函数进行的。序列的自相关函数度量了与之间的线性相关程度,用表示,定义如下其中表示序列的方差 * * 第十一章时间序列分析时间序列把某种现象发展变化的指标数值按一定时间顺序排列起来形成的数列,称为时间序列(数列),有时也称为动态数列。任何一个时间序列都具有两个基本要素:
基因组学总结
一、前言 继20世纪50年代Watson和Crick揭示了遗传信息携带者DNA的双螺旋结构后,近50年来分子生物学的发展势如破竹。60年代中期遗传信息传递的中心法则的初步确定;70年代基因重组理论和技术的崛起;以及近二三十年来基因的表达和调控及相关的发育分子生物学的进展;蛋白质翻译后加工、折叠、组装、转运,生物大分子相互识别、信号转导的深入研究等;一个个里程碑工作接踵而来。人类基因组计划业已完成,不久完整的人类基因组序列将呈现在人们面前。一个崭新的时代——后基因组时代已经来临。 基因即DNA分子上有遗传效应的特定核苷酸序列的总称,基因组即细胞或生物组的全部遗传物质,遗传物质即基因的编码序列,大量的非编码序列同样含有遗传物质。1985年美国科学家率先提出了人类基因组计划(HGP:Human Genome Plan),1990年正式启动。这是一项规模宏大的跨国跨学科的科学探索工程,其宗旨在于测定人类染色体中所包含的30亿个碱基对组成的核苷酸序列,从而绘制人类基因组图谱,并且辨认其载有的基因及其序列,从而达到破译人类遗传信息的目的。该项计划是继曼哈顿计划和阿波罗登月计划之后人类历史上的一个伟大工程。2001年人类基因组工作草图的发表被认为是人类基因组计划成功的里程碑,2005年人类基因组计划的测序工作已经基本完成,同时制作出了遗传图谱、物理图谱、序列图谱和基因图谱四张图谱。 二、人类基因组计划的成功完成对人类的意义 1、对人类各个领域的贡献 a 对人类疾病基因研究的贡献:人类疾病相关的基因是人类基因组中结构和功能完整性至关重要的信息。对于单基因病,采用“定位克隆”和“定位候选克隆”的全新思路,导致了亨廷顿氏舞蹈症、遗传性结肠癌和乳腺癌等一大批单基因遗传病致病基因的发现,为这些疾病的基因诊断和基因治疗奠定了基础。对于心血管疾病、肿瘤、糖尿病、神经精神类疾病(老年性痴呆、精神分裂症)、自身免疫性疾病等多基因疾病是目前疾病基因研究的重点。健康相关研究是HGP的重要组成部分,1997年相继提出:“肿瘤基因组解剖计划”“环境基因组学计划”。 b 对医学的贡献:基因诊断、基因治疗和基于基因组知识的治疗、基于基因组信息的疾病预防、疾病易感基因的识别、风险人群生活方式、环境因子的干预。 c 对生物技术的贡献:对研发基因工程药物和诊断研究试剂产业有巨大推动。 d 对细胞、胚胎、组织工程的贡献:胚胎和成年期干细胞、克隆技术、器官再造。 f 人类基因组计划的完成,在社会经济、生物进化等方面都有重要影响。 2、基因检测在个体化医学方面的应用 人类基因组计划和一系列的实验完成之后积累的大量的数据资料,科学家们面临的挑战就是如何利用这些数据的巨大潜力去改善人类的健康状况并使人类更好的生存,探索出一条造福人类健康的崭新途径。 大部分表型都是由遗传因素(基因及其产物)和非遗传因素(环境因素)交互作用,HGP的研究成果以及基因组学的研究,有助于我们了解遗传因素在人类健康和疾病中的角色,精确确定非遗传因素,并迅速将新发现用于疾病的预防、诊断和治疗。例如鉴定基因及其路径在健康和疾病中的角色,测定它们与环境因素之间的关系,预测药物反应,疾病的早期诊断,疾病在分子水平上的精确分类等。因此基因组学的进展将推动人们发展相应基因组研究方法,对人类基因组可遗传变异进行更为深入细致全面描述和分析。目前科学家们建立起一套人类基因常见差异的细目,包括核苷酸多态性(SNPs),小的缺失和插入,以及其它结构上的