大庆加氢尾油集总动力学模型的建立_4

第3章 热裂解五集总模型的建立
3.1集总模型的建立
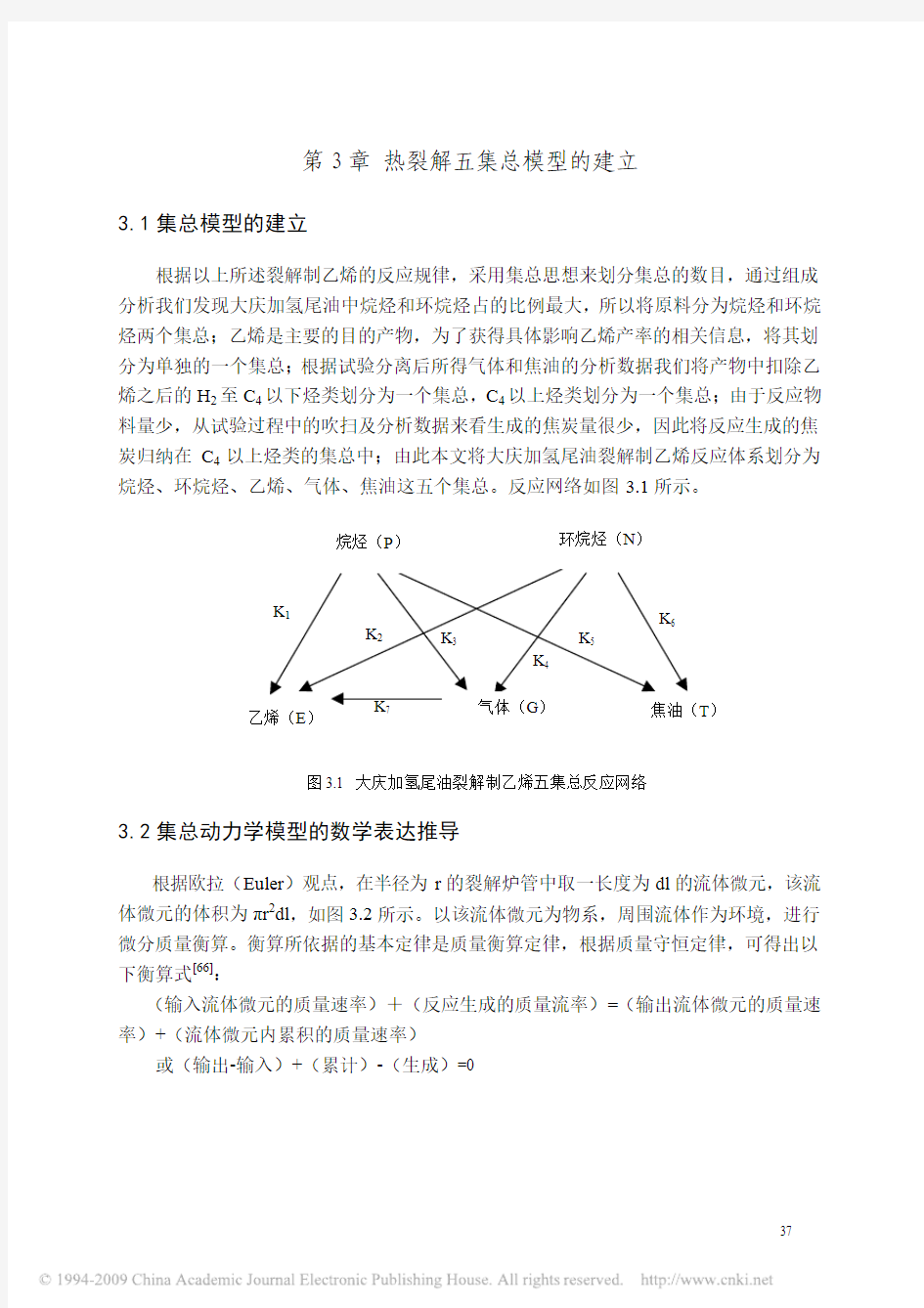
根据以上所述裂解制乙烯的反应规律,采用集总思想来划分集总的数目,通过组成分析我们发现大庆加氢尾油中烷烃和环烷烃占的比例最大,所以将原料分为烷烃和环烷烃两个集总;乙烯是主要的目的产物,为了获得具体影响乙烯产率的相关信息,将其划分为单独的一个集总;根据试验分离后所得气体和焦油的分析数据我们将产物中扣除乙烯之后的H2至C4以下烃类划分为一个集总,C4以上烃类划分为一个集总;由于反应物料量少,从试验过程中的吹扫及分析数据来看生成的焦炭量很少,因此将反应生成的焦炭归纳在C4以上烃类的集总中;由此本文将大庆加氢尾油裂解制乙烯反应体系划分为烷烃、环烷烃、乙烯、气体、焦油这五个集总。反应网络如图3.1所示。
图3.1 大庆加氢尾油裂解制乙烯五集总反应网络
3.2集总动力学模型的数学表达推导
根据欧拉(Euler)观点,在半径为r的裂解炉管中取一长度为dl的流体微元,该流体微元的体积为πr2dl,如图3.2所示。以该流体微元为物系,周围流体作为环境,进行微分质量衡算。衡算所依据的基本定律是质量衡算定律,根据质量守恒定律,可得出以下衡算式[66]:
(输入流体微元的质量速率)+(反应生成的质量流率)=(输出流体微元的质量速率)+(流体微元内累积的质量速率)
或(输出-输入)+(累计)-(生成)=0
图3.2 裂解炉管微元段示意图
上述关系即为质量守恒定律,如下将给出表达式中各项质量速率 3.2.1 各项质量速率的分析
1、输出与输入流体微元的质量流率差
设在流体微元内流体的速度为u (质量平均速度),由于裂解炉管传质存在于径向和轴向,所以在轴坐标系内,以轴向为L 轴,径向为r 轴,则速度u 在轴向与径向的分量为l u 、r u ,设组分A 的质量浓度为A ρ,则在两个坐标方向上,组分A 因流动所形成的质量通量分别为A l u ρ、A r u ρ。令组分A 在两个坐标方向上的扩散通量为l A j 、Ar j ,因此可得组分A 沿L 方向输入流体微元的总质量速率为:2()A l Al u j r ρπ+,而沿轴L 方向输出流体微元的质量流率为
22
2
[()]
[()]
()(())l l l A l A A l A A l Al A l A l
u j r u j u j r dl r u j dl l
ρπρρππρ?+?+++
=++
??
于是可得,组分A 沿L 方向输出与输入流体微元的质量流率差为
2
2
2
[()]
(())()()()l l l l
A l A A l A l A l A l
A A l u j r u j d u j r j u r dl
l l
ρπρρπρπ?+=++
?+???=+??l (输出-输入)
同理,组分A 沿r 方向输出与输入流体微元的质量流率差为
2
()(
)A r Ar
u j r dl r r
ρπ??=+??r (输出-输入) 在2个方向上输出与输入流体微元的总质量流率差为
2
()()()l
A A l A r Ar j u u j r dl r l r l
ρρπ????=+++????(输出-输入) (3-1)
2、流体微元内累积的质量速率
设组分A 的质量浓度(,,)A f l r t ρ=,则在流体微元中任一瞬时组分A 的质量为
2A A M r dl πρ= (3-2) 质量累积速率为
2A A M r dl t t
ρ
π??=?? 3、反应生成的质量流率
流体在裂解管内高温下发生裂解反应,设单位体积流体中组分A 的生成质量速率为A r ,当A 为生成物时,A r 为正,当A 为反应物时,A r 则为负。由此可得,流体微元内由于裂解反应生成的组分A 的质量速率为
2=A r r dl π反应生成的质量速率 (3-3) 3.2.2 传质微分方程
将式(3-1)、(3-2)、(3-3)代入质量守恒定律表达式中,得
()()0l A A l A r Ar A
A j u u j r r l r l t ρρρ?????++++?=????? 展开可得
()0l A l r A A Ar A
A l r A j u u j u u r l r l r r l t ρρρρ???????++++++?=??????? (3-4)
由随体导数的定义式
A A A A l r D u u Dt t l r ρρρρ???=++??? 因此得 (
)0l
A l r A Ar A A j u u D j r l r Dt r l
ρρ????++++?=???? (3-5)
式中的扩散通量可由费克第一定律给出,即
l A A j D
l ρ?=?? A Ar j D r ρ
?=??
将其带入(3-4)中,可得
2222
()()l r A A A
A A u u D D r l r Dt l r ρρρρ????++=++???? (3-6) 此式即为裂解炉管内的传质微分方程,该式是以质量为基准推导的,若以摩尔为基准推导,同样可得
2222(
)()ml mr A A A
A A u u Dc c c c D R l r Dt l r ????++=++???? (3-7) 符号意义及单位:
A c :A 组分的浓度(mol/m 3)
u ml ,u mr :A 组分在径向,轴向上的摩尔平均流速(m/h )
A r :A 组分的反应速率(mol/(g·h )) dl :裂解管内取的微元段(m ) t :反应时间(s )
l A j :轴向扩散通量(mol/(m 2·h ))
由于物料在裂解炉管中的径向流速很大,管路长径比大,裂解反应速率很快,因此对裂解反应作以下假设:
1)设裂解炉管内流体流动为活塞流 2)轴向,径向扩散忽略不计
3)反应气体处于稳态,且各集总之间的反应均为一级反应 4)在每一流体微元内,径向各组分的物性是恒定的 由此,裂解炉管内的传质微分方程可化为以下形式:
A
ml
A A c u R kc l ?==?? 根据程序处理做以下转换
A
A
ml
A c c u R k l
ρ
ρρ
ρ
?
==??即
A
v
A A a G R k a l
ρ?==?? (3-8)
v G :v ml G u ρ= 体积流量(g/m 2·h ) A a :A
A c a ρ
=
A 组分的浓度(mol/g )
ρ:密度(g/m 3)
根据理想气体状态方程PV=nRT 得
PM
RT
ρ=
(3-9) M :混合气体的平均分子质量(g/mol )
P :压力(Pa )
由混合物质平均分子质量定义可得 1
j j
j
j
a M M a
a
=
=
∑
∑
∑
(3-10)
由于所取系统为油水混合物,因此以1g 混合烃为参考,根据气烃比sh ,可得到水
的量,因此上式变为
1
1
sh
18(1+sh)
j
j
j
j
a M
M a a a a a a
a
=
=
=
+++++
∑∑∑烷烃环烷烃乙稀气体焦油 因为v W
G S
=
(3-11) W :装置每小时处理量,这里是指总共处理量,包含水(g/h )
S :裂解炉管界面积(m 2)
因此由(3-8)(3-9)(3-10)(3-11)得
P 1
sh 18(1+sh)
P
sh
18(1+sh)
A A A
V V A
V a PM ka ka x G RT G RT a a
a a a ka a a a a a G RT
?=?=?+++++=
+++++
烷烃环烷烃乙稀气体焦油烷烃环烷烃乙稀气体焦油()
由此根据图所示反应网络可得五集总模型的动力学方程式为:
()135P P v j a P
k k k a l
G RT a ?=?++?∑
()246N N v j a P k k k a l
G RT a ?=?++?∑
127N G
E P P N G v j E E E M M a M P k a k a k a l G RT a M M M ???=++?????∑
347G N G
P P N G v j G G E a M M M P k a k a k a l G RT a M M M ???=+??????∑ 56N
T P P N v j
T T M a M P k a k a l G RT a M M ???=+?????
∑
3.3集总动力学模型参数的求取
石油大学在求解催化裂化五集总动力学参数时提出了Marquardt 法的两种改进算法,并成功的取得了应用,使得求解参数时降低了算法对初值得要求,并且在保证收敛稳定的同时加快了迭代收敛速度,因此,在求解我们所建立的热裂解五集总动力学参数时我们也采用了Marquardt 法的改进算法,并根据自身反应特点编写了计算程序。 3.3.1 Marquardt 改进算法求取集总反应速率常数的原理
当模型中被估参数与函数值为非线性依从关系时,就形成非线性最小二乘问题。非线性问题很难处理,往往连解的存在性和唯一性都难以确定,因此,如果通过函数变换
能使被估参数以线性形式出现,就应该优先采用变换,把原问题变成可简单有效处理的线性最小二乘问题。
非线性最小二乘问题与最小值问题很相似,但又有区别。非线性最小二乘问题的主要特点是:第一,由于建模用比较简单的模型函数,所以它的导数总能求得,有利于构造比较完善的专用算法;第二,2χ(即二乘)函数的最小值点总处在浅坦、平缓的凹区中,因此不要盲目企求高精度解,通常精度是2-3位有效数字。
对于非线性参数估计来说,不能保证所得结果是全局最小二乘残差对应的估计。因此,获得满足要求的不同结果的现象并不少见。
Marquadt 算法,是专门为参数估计的最小二乘准则而设计的。Marquadt 算法认为:第一,关于被估参数的最小二乘(或2χ)函数,在接近最小值点时,呈抛物线形态,且可用Taylor 级数的前三项足够好的近似,宜取较小步长;第二,在远离最小点时,变化陡峭,适于最速下降法处理,步长宜取大。 其基本数学描述是:被估参数采用式
min 0a a a δ≈+更新,在此12()a a H δχ?=??。Hessian 矩阵近似表达和迭代计算为:
2
21(,)12[]n
m ij m m i
f a x H y a =?=??∑
,(1)H H I k ?+i 。这里(1)I k +是元素为(1)k +的对角阵。远离最小点时,k 取大;反之取小[67]。
由此根据Marquardt 算法原理,构造如下目标函数[46]:
()()[]()()[]k ,t C ,t y y k ,t C ,t y y )k (S cal obs T
cal obs ???=∑ (3-12)
式中 k :反应速率常数
y :观察变量 C :浓度向量
T :数学符号,表示向量矩阵的转置 cal :计算值
obs :实测值
T n )C ,,C ,C ,C (y 432= (3-13)
在k 的初值点对y cal 进行Taylor 展开,并求S (?k )对?k 的偏导数为零,得
(
)[])k ,t (y y C y k C k k C C y C y k C )
(cal
obs T
k
cal T k k k cal T k cal T k )()()()()()(0000000?????=?????????∑∑?
(3-14)
令C y G ??=
,k
C
??=Λ G 可通过y 和C 计算,按定义有
n
n n
n
n
n
M M M M M M M M M M G 1
4321100000
10000010000010??????=
(3-15)
而敏感矩阵Λ中的各元素可以用差分法求得
()()[]j p j j i p j j i ij k k ,...,k k ,...,k ,k C k ,...,k k ,...,k ,k C ???Λ22121??+= (3-16)
于是,方程(6-32)可写成b k *A =?
其中:ΛΛ=∑G G A T
T ;[]))k ,t (C ,t (y y G b )
(cal
obs T T 0?=
∑Λ; 解出?k 后,将k k k '?+=作为初值继续迭代直到收敛为止。
为了改善速率常数收敛的稳定性和降低对初值的要求,在矩阵A 的对角线各元素上加一个阻尼因子d 。但是,引入阻尼因子d 后,在降低对初值的要求,保证收敛的同时,一旦出现S N+1大于S N ,需不断增大阻尼因子d ,d 值增大,则△k 减小,收敛减慢。最佳步长策略法是对Marquardt 法的一种改进,它是在速率常数的迭代式中引入一个步长因子λ,使'k =k+λ?k ,进一步降低算法对初值的要求。最佳步长λ可根据S/=0λ??导出的一元三次方程求得。但是,引入步长因子λ后,由于λ小于1,使得k 值的收敛速度减慢,特别是λ值越接近其真值时,d 值越大,λ越小,收敛速度越缓慢。而且,求解最佳步长λ本身涉及一元三次方程的求解,需对敏感矩阵反复计算才能得到,无疑增加了算法的复杂性。
因此,根据对算法的分析,Marquardt 法的两种改进如下:
1、根据最佳步长策略法的原理,引入步长因子λ,但在迭代过程中并不是通过计算求得最佳步长因子,而是给定步长因子的初值,并随着迭代过程中目标函数的变化调节其大小。当S N+1大于S N 时,λ减半,当S N+1小于S N 时,λ值则不变。这样既控制计算过程收敛,又保证迭代速度,记此法为Marquardt +法。
2、首先采用Marquardt +法使初值进入Maquardt 法的收敛区间,然后以Marquardt 法计算,并调节阻尼因子d 的大小,加速收敛。即当目标函数S N+1大于S N 时,采用
Marquardt+计算,并使阻尼因子d 加倍;反之,则采用Marquardt 法计算,减半d 。在两种算法的转换过程中,使阻尼因子返回预先设定值。记此法为Marquardt ++法。Marquardt ++算法求取反应速率常数的计算程序框图见图3.3。
计算过程中还考察了阻尼因子d 和步长因子K 对求解过程的影响,对就加氢尾油裂解五集总模型反应动力学参数估计,d 和K 的取值分别以1×10-13和0.5较为适宜。
图3.3 Marquardt算法求取反应速率常数的计算程序框图
3.3.2 集总动力学参数的求取方法
如第1章所述,求取集总模型反应速率常数数值解的方法有分层法、分步法和一步
法三种。
对于简单的集总动力学模型,反应速率常数较少,可以采用一步法直接求取。而对于复杂的集总动力学模型,反应速率常数很多,往往需要采用分层法或分步法求取。
如果采用分层法求取反应速率常数,那么不仅实验和参数优化估计的工作量大,而且下一层的反应状况与总的反应状况差别较大,所以要用下一层的反应速率常数来回归上一层的反应速率常数,则必然存在一定的计算误差。欧阳福生等人[68,69]的研究也表明分层法的计算误差要大于一步法。
分步法所需的实验工作量远小于分层法,其计算工作量也比较小,而且通过适当的集总合并,使得反应速率常数的初值范围变宽,求取变得简单。但是对于具有不同催化剂失活函数的模型,或者是具有不同反应级数的模型,集总的合并和拆分会带来一定的误差。
同分层法和分步法相比,一步法所需的实验工作量最小,但是一次性估计所有的反应速率常数,计算工作量比较大,而且对反应速率常数的初值范围要求比较严格。
在本论文的研究过程中,采用一步法和分布法相结合的办法。对于大庆加氢尾油热裂解五集总动力学模型,先采用分布法求得反应速率常数的大概范围,然后再用一步法求取所有的反应速率常数。求取不同反应温度下的反应速率常数之后,求取了三个温度T下满足精度要求的速率常数k后,通过阿罗尼乌斯方程进而求得指前因子A和活化能Ea。
由阿罗尼乌斯方程:k=Aexp(-Ea/RT),可推导得:
lnk=-Ea/R*1/T+lnA
本研究中,参数的求取属于约束优化问题,即所有的参数都要大于零,指前因子和活化能均为正值,且有一定的数值范围。
3.4 石脑油五集总动力学模型
3.4.1五集总动力学模型参数的求取
根据Marquardt算法原理,编写了专门用于求取集总模型反应速率常数的Matlab程序。结合大庆石脑油在820℃、840℃和840℃的试验数据分别回归求取了反应速率常数见表3-1,以及指前因子和活化能见表3-2。
由表3-1可知,原料发生一次裂解的反应速率常数k1~k6远远大于二次反应的速率常数k7,说明在大庆石脑油裂解过程中,主要是原料油的一次裂解反应,二次反应所占的比重较小。
由表3-2可以看出,各集总间反应活化能的数值比较大,生成乙烯和气体的活化能小于生成焦油的活化能。
从表3-1中可以看出随着温度的升高,各反应速率常数明显增加,而从表3-2中可
以看到一次反应的活化能大于二次反应的活化能,所以提高温度有利于提高一次反应对二次反应的相对速度,提高乙烯收率。
表3-1 大庆石脑油裂解五集总反应速率常数
反应温度,℃
反应速率常数/h-1
820 840 860
K197841 159803 254939
K276000 119239 185791
K3161635 261497 387603
K4111654 176615 260873
K547174 86196 143176
K653681 90218 143843
K713128 23063 29651
表3-2 大庆石脑油裂解五集总模型参数
反应速率常数指前因子/ g·mol-1·s-1活化能/KJ·mol-1
K1 3.02×1015 221.80
K2 2.22×1016 237.65
K3 1.22×1015 209.95
K4 3.86×1015 217.07
K5 2.64×1016 244.61
K6 7.03×1017 275.54
K7 6.45×1013 202.36
3.4.2 模拟计算值与试验值的对比
为了检验所建集总动力学模型的计算准确性,计算了820℃时的集总组分产率,并与实验值进行对比,其相对误差列入表3-3。可见所建模型具有较好的计算准确度,相对误差在10%以内的数据占71.11%,在20%以内的占95.56%;特别是乙烯、气体两个集总,计算值与实验值的相对误差都比较小。
乙烯是重油裂解的目的产物,为此利用程序分别求取了820℃、840℃和860℃三个温度下的乙烯产率,并与实验值进行比较,其相对误差见表3-4。可见,所建模型对目的产物的计算准确度较高,所有产率计算值的相对误差都在18%以内,其中相对误差在10%以内的数据占92.6%。
表3-3 820℃时计算的集总产物产率与实验值的相对误差(%)
序号烷烃环烷烃乙烯气体焦油
1 0.0825 0.098 -0.0989 -0.08928 -0.1072
2 0.113
3 0.1647 -0.08752 -0.07
4 -0.0219
3 0.0727 0.156 -0.053
4 -0.03348 -0.007
4 -0.0596 -0.0131 0.07
5 0.0495 0.014
5 -0.099 -0.00785 0.024
6 0.0148 0.0799
6 -0.1309 -0.01298 0.01406 0.0176 0.0327
7 -0.1875 -0.11185 0.1736 0.1228 0.1187
8 -0.05332 0.053 -0.024 -0.01146 0.0659
9 -0.3544 -0.21763 0.0846 0.0573 0.103804
注:表中的“-”表示模型的计算值小于实验值。
表3-4 不同温度下乙烯产率的计算值与实验值的相对误差(%)
乙烯收率
温度℃820 840 860
1 -0.0989 -0.1295 -0.0771
2 -0.08752 -0.066 -0.06347
3 -0.053
4 -0.023 -0.00372
4 0.07
5 0.0175 0.0127
5 0.024
6 -0.019 -0.0014
6 0.01406 0.0252 0.0191
7 0.1736 0.0946 0.07416
8 -0.024 0.0689 0.0444
9 0.0846 0.0595 0.0464
3.5 加氢尾油五集总动力学模型
3.5.1五集总动力学模型参数的求取
加氢尾油在820℃、840℃和840℃的试验数据进行回归求取得反应速率常数见表3-5,指前因子和活化能数据见表3-6。
由表3-5可以看出,由于加氢尾油的裂解深度较大,在高温、高转化率条件下,烷烃和环烷烃生成乙烯的反应速率常数比较接近,并且随着反应温度的升高,各集总的反应速率常数的增加值并不如石脑油那样快,而是缓慢上升,这说明了在高转化率条件下,升温所带来的乙烯收率的增加并不明显,反而会加大焦油的产量,这与试验所得的数据
是相符的。
表3-5 大庆加氢尾油裂解五集总反应速率常数
反应温度,℃
反应速率常数/h-1
820 840 860
K14000000 4500000 4534610
K23800000 4300000 4320388
K35900000 6000000 5959458
K44400000 4600000 4570821
K51500000 2000000 2016478
K62500000 300000 3010323
K785000 85000 175821
表3-6 大庆加氢尾油裂解五集总模型参数
反应速率常数指前因子/ g·mol-1·s-1活化能/KJ·mol-1
K1 1.47×108 32.597
K2 1.61×108 33.335
K3 7.9×106 2.619
K4 1.3×107 9.915
K5 7.4×109 76.90
K6 5.2×108 48.28
K7 6.93×1013 187.63
3.5.2 模拟计算值与试验值的对比
为了检验所建集总动力学模型的计算准确性,计算了820℃时的集总组分产率,并与实验值进行对比,其相对误差列入表3-7。由表中数据可以看出,所建集总模型对产物产率模拟较好,但对所求烷烃、环烷烃的产物误差较大,这与在高温、高汽烃比条件下,裂解深度的增加及原料组分的加重有一定关系。
乙烯是重油裂解的目的产物,计算了820℃、840℃和860℃三个温度下的乙烯产率,并与实验值进行比较,其相对误差见表3-8。由表中数据可以看出,所建模型对目的产物的计算准确度很高,所有产率计算值的相对误差都在1%以内。
表3-7 820℃时计算的集总产物产率与实验值的相对误差(%)
序号烷烃环烷烃乙烯气体焦油
1 0.3537 0.553 -0.0389 -0.0263
2 -0.0033
2 0.232 0.411 -0.0461 -0.03295 0.174
3 0.5226 1.41 -0.01739 -0.0137 -0.108
4 0.2157 0.344 -0.0243 -0.0229 0.063
5 0.25
6 0.53
7 -0.0114 -0.01125 -0.0224
6 0.228 0.332 -0.048 -0.0396 -0.021
7 0.047 0.213 0.0613 0.05836 -0.048
8 0.186 0.299 0.009 0.0085 -0.105
9 -0.066 -0.0086 0.0188 0.0287 0.0228 注:表中的“-”表示模型的计算值小于实验值。
表3-8 不同温度下乙烯产率的计算值与实验值的相对误差(%)
乙烯收率
温度℃820 840 860
1 -0.0389 -0.01734 -0.01157
2 -0.0461 0.0008 0.01494
3 -0.01739 0.02925 0.0439
4 -0.0243 0.0081 -0.02008
5 -0.0114 0.019
6 0.0131
6 -0.048 0.033 0.0353
7 0.0613 -0.0152 -0.03357
8 0.009 0.0083 0.0065
9 0.0188 0.0281 0.0338
结 论
1、通过对石脑油实验数据的分析,可以看出石脑油所得数据符合高温、短停留时间、高汽烃比的裂解规律,程序模拟各集总所得动力学参数符合烃类裂解规律,并且对各产物的模拟值与实验数据误差较小,说明程序对各集总产物的预测较准。
2、通过对加氢尾油的实验数据分析,可知加氢尾油所得数据同样符合高温、短停留时间、高汽烃比的裂解规律,然而对汽烃比对乙烯收率的影响进行分析时发现裂解规律有所变化,分析实验数据及操作条件,发现可能是由于汽烃比的变化加大了摩尔流量,相应的减小了停留时间导致了乙烯收率规律出现了异常,为此,在以后的进一步试验将采取固定摩尔流量的方法来进行试验。
3、建立了石脑油、加氢尾油五集总动力学模型,分别求取了820、840和860℃时各集总间的反应速率常数、活化能和指前因子。模型的计算值与实验值比较接近,说明该集总模型具有较好的计算准确度。