HIV_1衣壳蛋白抑制剂的研究进展

收稿日期:2011-03-20
基金项目:国际合作重点项目(303DF000033);国家自然科学基金重大国际合作项目(30910103908);国家自然科学
基金面上项目(30371686,
30772629,30873133);山东省自然科学基金项目(ZR2009CM016)作者简介:李东岳(1984-),男(汉族),宁夏固原人,硕士研究生,
Tel :(0531)88382005,E-mail :dongyue@sdu.edu.cn ;*通讯作者:刘新泳(1963-),男(汉族),山东平度人,教授,博士生导师,主要从事抗病毒药物及抗
心血管疾病药物研究,
Tel :(0531)88380270,E-mail :xinyongl@sdu.edu.cn 。文章编号:1005-0108(2011)05-0397-08
HIV-1衣壳蛋白抑制剂的研究进展
李东岳,展鹏,刘新泳
*
(山东大学药学院药物化学研究所,山东济南250012)
摘要:艾滋病是由HIV-1引起的传染性和致死性疾病,目前临床使用的抗艾滋病药物容易产生耐药性和不
良反应等问题,急需开发具有全新作用机制的高效、低毒的抗艾滋病药物。衣壳蛋白在HIV-1病毒颗粒装配和成熟过程中发挥着至关重要的作用,它的稳定性直接影响HIV-1的感染能力。近年来对衣壳蛋白结构和作用机制的阐明为人类寻找艾滋病治疗新途径带来了希望。该文综述了抗艾滋病作用新靶点衣壳蛋白的结构、
功能及其抑制剂的最新研究进展。
关键词:获得性免疫缺陷综合征;人免疫缺陷病毒;衣壳蛋白;结构;功能;抑制剂;药物设计中图分类号:R914文献标志码:A
艾滋病(获得性免疫缺陷综合征)是由人免
疫缺陷病毒1型(HIV-1)引起的一种疾病,具有严重的传染性和致死性。虽然高效抗逆转录疗法
在临床中已广泛应用,但是HIV-1的耐药问题、抗病毒药物的毒性和不良反应以及长期用药的费用
等问题,使得寻求作用于病毒复制周期不同环节的新型抗HIV-1药物迫在眉睫。目前已上市的抗艾滋病药物根据其作用机制不同主要分为逆转录酶抑制剂、整合酶抑制剂、蛋白酶抑制剂、膜融合抑制剂和CCR5抑制剂等。研究显示,这些抑制剂虽然能够有效地降低病毒载量,但是并不能彻底根除病毒,而且药物的耐药性、毒性及患者的依从性等方面存在的问题常常会引起临床治疗的失败[1]
。随着人们对HIV-1病毒的深入研究,发现了一些抗艾滋病药物作用的新靶点,其中,衣壳蛋
白(capsid protein )在未成熟病毒颗粒和成熟病毒颗粒装配过程中起着非常关键的作用,
已经成为设计新型抗艾滋病药物的热点。
1HIV-1衣壳蛋白的结构及功能
HIV-1的复制周期包括吸附、融合、穿入、逆
转录、整合、mRNA 转录和早期合成、晚期合成、装配(形成未成熟的病毒颗粒)、
出芽及成熟等过程。HIV-1衣壳蛋白在形成未成熟和成熟病毒颗粒(成熟后的病毒颗粒具有传染性)过程中起着
非常关键的作用。gag 和pol 是HIV-1的结构基因。其中,
gag 编码的前体蛋白(Pr55Gag )包含4个蛋白区域[基质蛋白(MA )、衣壳蛋白(CA )、核衣壳(NC )、p6]和两个小的多肽环(SP1、SP2),示意图见图1。在病毒颗粒装配过程中,衣壳蛋白和SP1能够介导Gag 多聚化形成结构性外壳,并
将RNA 基因、gag -pol 编码的前体蛋白(Pr160Gag-Pol )及包膜(envelope ,Env )糖蛋白复合
物包裹在未成熟病毒颗粒之中[2]
。随后,未成熟病毒颗粒出芽,伴随着病毒颗粒的释放,
Gag 和Gag-Pol 前体蛋白被蛋白酶分别裂解为MA 、CA 、NC 、p6、SP1、SP2六个结构域和逆转录酶(RT )、整合酶(IN )、蛋白酶(PR )三个病毒相关酶。裂解过程引起衣壳蛋白区域的结构调整,使衣壳蛋白聚集形成圆锥形外壳,同时将RNA 基因和病毒相关酶包裹其中形成成熟的病毒颗粒。若衣壳蛋
白和圆锥形衣壳的形成受到抑制,HIV-1几乎丧失感染宿主细胞的能力
。
Figure 1
The Gag polyprotein is shown with the four protein domains and two spacer peptides
第21卷第5期2011年10月总103期中国药物化学杂志
Chinese Journal of Medicinal Chemistry
Vol.21No.5p.397Oct.2011
Sum 103
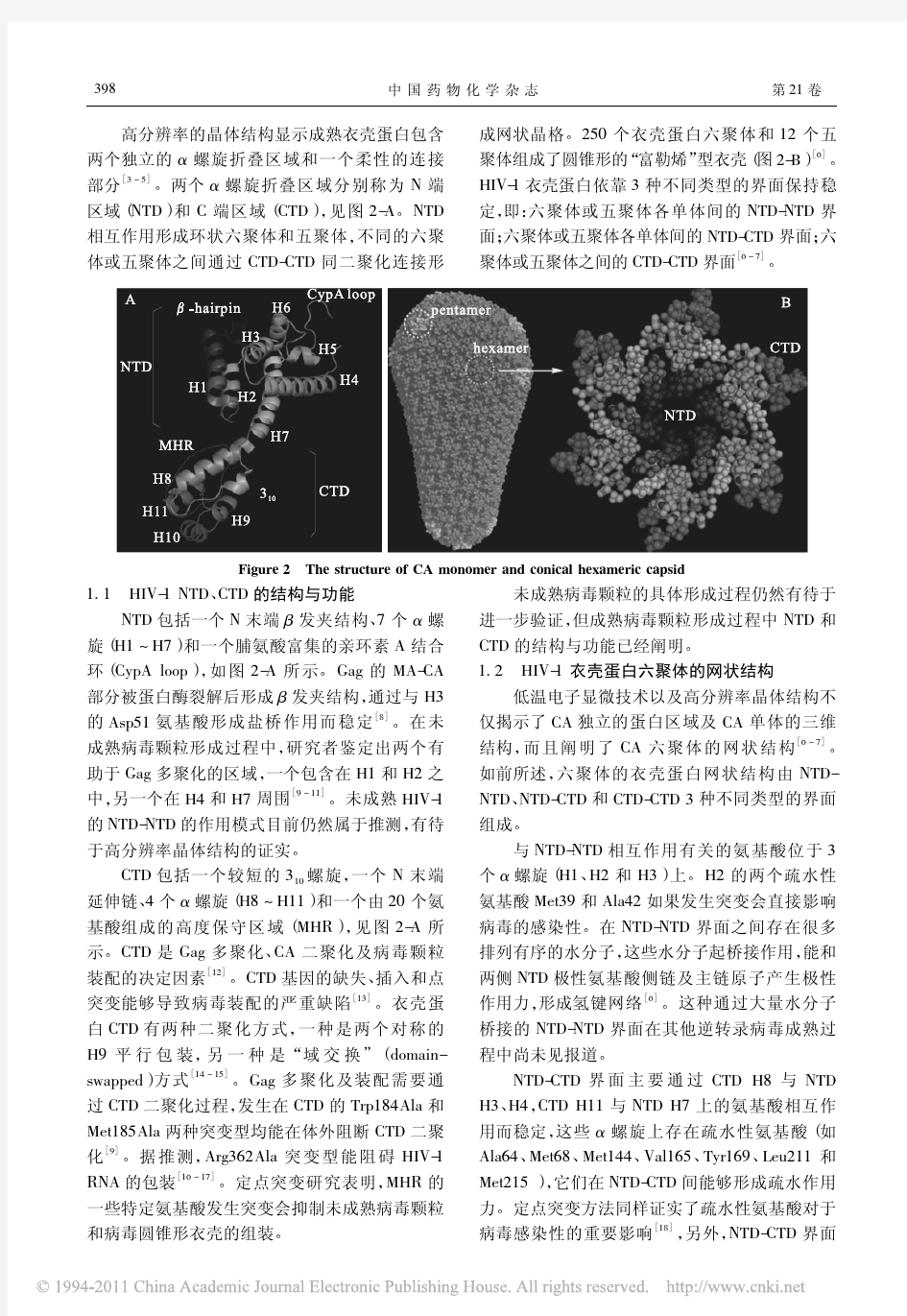
高分辨率的晶体结构显示成熟衣壳蛋白包含两个独立的α螺旋折叠区域和一个柔性的连接
部分
[3-5]
。两个α螺旋折叠区域分别称为N 端区域(NTD )和C 端区域(CTD ),
见图2-A 。NTD 相互作用形成环状六聚体和五聚体,不同的六聚
体或五聚体之间通过CTD-
CTD 同二聚化连接形成网状晶格。250个衣壳蛋白六聚体和12个五
聚体组成了圆锥形的
“富勒烯”型衣壳(图2-B )[6]。HIV-1衣壳蛋白依靠3种不同类型的界面保持稳
定,
即:六聚体或五聚体各单体间的NTD-NTD 界面;六聚体或五聚体各单体间的NTD-CTD 界面;六聚体或五聚体之间的CTD-
CTD 界面[6-7]
。Figure 2The structure of CA monomer and conical hexameric capsid
1.1HIV-1NTD 、CTD 的结构与功能
NTD 包括一个N 末端β发夹结构、7个α螺
旋(H1 H7)和一个脯氨酸富集的亲环素A 结合
环(CypA loop ),如图2-A 所示。Gag 的MA-CA 部分被蛋白酶裂解后形成β发夹结构,通过与H3
的Asp51氨基酸形成盐桥作用而稳定[8]
。在未成熟病毒颗粒形成过程中,研究者鉴定出两个有
助于Gag 多聚化的区域,
一个包含在H1和H2之中,另一个在H4和H7周围
[9-11]
。未成熟HIV-1的NTD-NTD 的作用模式目前仍然属于推测,有待
于高分辨率晶体结构的证实。
CTD 包括一个较短的310螺旋,一个N 末端延伸链、
4个α螺旋(H8 H11)和一个由20个氨基酸组成的高度保守区域(MHR ),见图2-A 所示。CTD 是Gag 多聚化、CA 二聚化及病毒颗粒装配的决定因素
[12]
。CTD 基因的缺失、插入和点
突变能够导致病毒装配的严重缺陷[13]
。衣壳蛋白CTD 有两种二聚化方式,一种是两个对称的
H9平行包装,另一种是“域交换”(domain-swapped )方式[14-15]。Gag 多聚化及装配需要通过CTD 二聚化过程,
发生在CTD 的Trp184Ala 和Met185Ala 两种突变型均能在体外阻断CTD 二聚化[9]
。据推测,
Arg362Ala 突变型能阻碍HIV-1RNA 的包装[16-17]。定点突变研究表明,MHR 的一些特定氨基酸发生突变会抑制未成熟病毒颗粒
和病毒圆锥形衣壳的组装。
未成熟病毒颗粒的具体形成过程仍然有待于
进一步验证,但成熟病毒颗粒形成过程中NTD 和CTD 的结构与功能已经阐明。1.2
HIV-1衣壳蛋白六聚体的网状结构低温电子显微技术以及高分辨率晶体结构不仅揭示了CA 独立的蛋白区域及CA 单体的三维
结构,而且阐明了CA 六聚体的网状结构
[6-7]
。如前所述,六聚体的衣壳蛋白网状结构由NTD-
NTD 、NTD-CTD 和CTD-CTD 3种不同类型的界面组成。
与NTD-NTD 相互作用有关的氨基酸位于3个α螺旋(H1、
H2和H3)上。H2的两个疏水性氨基酸Met39和Ala42如果发生突变会直接影响
病毒的感染性。在NTD-NTD 界面之间存在很多排列有序的水分子,这些水分子起桥接作用,能和
两侧NTD 极性氨基酸侧链及主链原子产生极性
作用力,形成氢键网络[6]
。这种通过大量水分子
桥接的NTD-
NTD 界面在其他逆转录病毒成熟过程中尚未见报道。
NTD-CTD 界面主要通过CTD H8与NTD
H3、H4,CTD H11与NTD H7上的氨基酸相互作用而稳定,这些α螺旋上存在疏水性氨基酸(如Ala64、Met68、Met144、Val165、Tyr169、Leu211和Met215),它们在NTD-CTD 间能够形成疏水作用力。定点突变方法同样证实了疏水性氨基酸对于
病毒感染性的重要影响[18]
,另外,
NTD-CTD 界面8
93中国药物化学杂志第21卷
也存在大量水分子介导的氢键网络[6]。
CTD-CTD同二聚化无论对成熟衣壳还是未成熟衣壳的组装都必不可少。CTD-CTD同二聚化发生在H9和H9之间,依据是发生在H9的两种氨基酸发生突变(Trp184Ala和Met185Ala)会使二聚化消失。衣壳蛋白CTD不同的X射线晶体结构研究表明,CTD二聚体拥有不同的构型,推测可能是在衣壳组装过程中CTD二聚体经历了构象改变[6]。观测显示,在未成熟病毒颗粒装配过程中,CTD“域交换”的二聚化方式并不存在于成熟衣壳中。
HIV-1衣壳呈圆锥形,结构高度弯曲,因此要求衣壳NTD、CTD及它们之间的3种接触界面必须具有一定的柔性。叠合4种不同的CA高分辨晶体结构发现,六元环状的NTD-NTD结构并不发生变化(亲环蛋白结合环除外),最具柔性的结构是H8和H9之间由15个氨基酸组成的肽链。CA单体间的NTD-CTD也具有柔性,能调整CA 结构,促进同二聚化形成网状圆锥形衣壳[6]。
衣壳的结构和稳定性将直接影响HIV-1的复制及感染性,因此从理论上讲,靶向衣壳蛋白抑制剂的研究对发现新型抗HIV-1药物具有重要意义。近几年,对衣壳蛋白结构生物学研究取得的巨大进展,使HIV-1衣壳蛋白已经成为引人注目的抗艾滋病作用新靶点。目前发现了多种抑制剂能有效地作用于该靶点。
2衣壳蛋白抑制剂
2.1以CA-SP1裂解位点为靶点的抑制剂
早在1994年,人们通过对天然产物进行活性筛选发现桦木酸具有弱的抗HIV活性。PA-457(1,be-virimat,DSB,MPC-4326)是桦木酸(betulinic acid)的衍生物,对野生型和临床常见耐药毒株均保持活性,在体外能非常有效地抑制HIV-1复制(IC
50
≈10nmol·L-1)[19]。PA-457只作用于HIV-1而对HIV-2无效,具有高度特异性。研究显示,PA-457作用于病毒成熟的晚期阶段。未成熟病毒颗粒出芽后,在蛋白酶的作用下Gag分步裂解。Gag首先裂解成MA-CA-SP1和NC-SP2-p6两个片段,然后分别裂解成MA、CA-SP1及NC-SP2、p6,最后裂解成MA、CA、SP1、NC、SP2、p6六个结构域(图3)[2]。放射免疫沉淀分析、蛋白质印迹技术及突变分析法均证明PA-457能特异性阻断CA-SP1的裂解,进而影响衣壳形态发生改变,抑制HIV-1复制[19]
。
Figure3The HIV-1Gag proteolytic processing cascade 初期的动物实验及Ⅰ期临床试验表明PA-457具有良好的药动学性质和安全性[20]。随后几年,PA-457的临床试验几乎停止。目前,Myriad 制药公司购买了PA-457的知识产权并正在进行Ⅱb期临床试验。一项为期14d的单一疗法试验显示,44位患者中仅有45%服用PA-457后病毒载量下降为每毫升3.16拷贝以上,而55%的患者对PA-457的反应性很低。进一步研究发现,这与含有较低保守性的SP1氨基酸残基6 8以及CA谷氨酸-缬氨酸-苏氨酸(QVT)基序(369 371)的多态性有关[21]。此外,Myriad制药公司还研制了另外两个抑制剂MPC-9055、MPC-9055,并且已经完成Ⅰ期临床试验,但它们的化学结构和具体作用机制还未见报道。
目前,研究人员已设计合成了一系列桦木酸类似物,初步得到以下构效关系:1)C-3位的3',3'-二甲基琥珀酰基是维持抗HIV-1活性的必需基团,3'S甲基取代的活性远高于3'R甲基取代,但3'R甲基仍然必不可少;C-3末端为极性基团能增强活性;将3',3'-二甲基移至4'位引起活性下降;C-3酯被酰胺基团取代后活性降低。2)C-28侧链被环状仲胺(例如哌啶)取代时代谢稳定性显著升高;C-28侧链末端为酰胺基或极性基团有利于提高活性;3位、28位双取代的类似物活性升高,甚至会超过PA-457;C-28侧链引入较短基团能够抑制HIV-2;C-28引入其他一些基团能抑制病毒侵入过程。3)C-30对整体活性影响不显著,适合引入水溶性部分(例如吗啉乙氧基);靠近C-19异丙烯基的C-30取代基有氢键供体存在时活性降低。通过对桦木酸的结构修饰及类似物的研究,得到一系列具有较高抗HIV-1活性的成熟抑制剂(如化合物2)或侵入抑制剂以及抗
993
第5期李东岳等:HIV-1衣壳蛋白抑制剂的研究进展
HIV-2活性的抑制剂[22-24]。
PF-46396(3)是通过高通量抗病毒筛选获得的小分子化合物,在MT-2细胞中抗HIV-1NL4-3和HIV-1ⅢB的EC
50
值分别为0.36μmol·L-1和0.017μmol·L-1。它的作用机制与PA-457相同,蛋白质印迹技术及突变分析法(CA:Ile201Val、SP1:Ala1Val)均证明PF-46396能特异性阻断CA-SP1的裂解。通过对临床常见变异毒株的抗病毒活性研究,观察到PF-46396的活性范围波动很大,与PA-457较为类似,可能与CA末端、SP1的多态性有关[25]。PA-457和PF-46396分属两类不同的化学结构,却具有相同的作用机制,这种结构多样性为寻找其他高效低毒的CA-SP1阻滞剂提供了广阔的空间。但是,衣壳蛋白QVT 基序及SP1的多态性能够使PA-457和PF-46396体内抗HIV-1活性下降,影响了该类抑制剂的临床使用。因此,仍需对PA-457和PF-46396的作用机制、耐受性产生的决定因素等进行深入研究
。
2.2以衣壳蛋白NTD为靶点的抑制剂
小分子化合物CAP-1(4)通过诱导契合原理
特异性结合于衣壳蛋白NTD底端的疏水口袋,该
疏水口袋只有在配体存在时才显现。HIV-1病毒
颗粒与CAP-1结合后形态大小不一,不能形成圆
锥形衣壳结构[26]。高分辨率的CAP-1/NTD复
合物晶体结构显示,CAP-1与NTD的疏水口袋结
合后诱导衣壳蛋白构象发生改变[27]。Phe32残
基能与CAP-1相互作用并产生位移,暴露出疏水
结合位点,有利于CAP-1与疏水口袋进一步的结
合。CAP-1/NTD复合物的晶体结构揭示了其作
用模式:脲基的NH部分与Val59主链氧原子形
成氢键作用;二甲胺基与Glu28、Glu29侧链相互
作用;Phe32、His62和Tyr145的疏水氨基酸侧链
发生位移并破坏了使H3/H4环构象稳定的极性
网络。因此,CAP-1是通过变构效应干扰NTD-
CTD界面的相互作用。
Prevellge[28]的研究显示,具有酰肼腙骨架且
有两个苯环取代基的化合物能抑制衣壳蛋白的组
装。杨铭等人保留了该类化合物的基本药效团,
将带有疏水或亲水性侧链的天然氨基酸引入到酰
肼腙骨架中,合成了一系列化合物,并系统地探讨
了其构效关系,得到两个最具发展前景的酰肼腙
类化合物5、6,其EC
50
值分别为0.21、
0.17μmol·L-1[29-30]。利用Autodock4.0软件
进行分子模拟,证实了该类抑制剂作用模式与
CAP-1类似,且能占据另外两个小的疏水口袋。
NTD和CypA在HIV-1装配和脱壳过程中
起着至关重要的作用。杨铭等人根据NTD、
CypA抑制剂的结构特点和作用模式设计了
NTD-CypA双靶点抑制剂[31-32],其设计思想是:
保留CAP-1能与Val59主链羰基形成氢键作用
的脲部分;保留能够进入NTD及CypA疏水口袋
的苯环部分;引入存在于CypA抑制剂结构中的
磺酰胺片段。通过对此类双靶点抑制剂的构效关
系研究,得到了具有较高HIV-1抑制活性的化合
物7。7对衣壳蛋白NTD和CypA双靶点的抑制
作用是通过紫外光谱分析、荧光亲和性分析及肽
酰-脯氨酰-顺反式异构酶抑制实验进行测定的。
Blair等人[33]通过高通量筛选得到了广谱抗004中国药物化学杂志第21卷
HIV的先导化合物PF-1385801(8),在MT-2细胞水平的抗HIV-1活性实验中,测得其EC
50
和
CC
50
值分别为4.5、61μmol·L-1,治疗指数为14。随后,他们又设计合成了活性更高的抑制剂PF-3450074(9),研究表明,PF-3450074能显著影响HIV病毒颗粒的形态及病毒体脱壳过程。它与NTD复合物的晶体结构揭示,其作用靶点位于H3、H4、H5和H7之间的口袋中,不同于CAP-1的作用位点[33]。其具体的结合模式为:PF-3450074的苯胺基与Ile73、Ala105、Thr107、Tyr130、Asn53形成疏水作用;苄基与Met66、Leu69、Val59、Ile73、Leu56形成疏水作用;吲哚环与Met66、Gln67、Lys70、Gln63形成作用力;吲哚环N原子与Gln67侧链酰胺部分通过水桥形成氢键,同时PF-3450074的酰胺基与Asn57形成非常关键的氢键作用。该研究结果为基于结构的合理药物设计奠定了基础,有利于设计合成活性更高的衣壳蛋白NTD抑制剂
。
2.3以衣壳蛋白CTD为靶点的抑制剂
CAI(10)是在对噬菌体表达的随机肽库进行系统高通量筛选过程中鉴定出的寡肽,研究表明,它在体外能抑制未成熟和成熟病毒颗粒的装配[18]。CAI作用于CTD,占据了由H8、H9、H11构成的高度疏水性口袋(图4)。变异分析证明该区域疏水性氨基酸的存在非常重要,Tyr169Ala、Leu211Ala或Leu211Ser突变均使CAI活性下降且不能形成成熟病毒颗粒。CAI主要通过两种机制发挥作用,一是竞争性抑制NTD-CTD界面的相互作用,阻断NTD与CTD结合;二是诱导CTD-CTD二聚化界面发生改变,从而影响衣壳蛋白六聚体网状结构的形成。CAI存在的问题是,虽然在体外表现出抗HIV-1活性,但是不能渗透进入细胞。为了克服CAI的缺点,研究者根据已知的CAI/CTD三维结构,利用基于结构的合理药物设计方法及烃固定(hydrocarbon stapling)技术得到了具有高度α螺旋性和细胞穿透性的CAI类似物NYAD-1(11)、NYAD-13(12),其活性比CAI 提高10倍[34-35]。NMR化学位移干扰实验证实NYAD-1作用于CTD氨基酸169 191区域,该区域拥有疏水性口袋和关键性的CTD-CTD二聚化片段。“烃固定”部分不参与疏水作用。12与11相比具有更高的水溶性
。
Figure4The structures of CAI,NYAD-1,NYAD-13and the binding mode of CAI with CA CTD 104
第5期李东岳等:HIV-1衣壳蛋白抑制剂的研究进展
2.4小分子CA-CTD抑制剂
除了寡肽CAI、NYAD-1及NYAD-13外,小分子化合物也能作用于CTD的疏水口袋,从而影响病毒颗粒的装配。Curreli等人[36]利用高通量柔性对接技术,通过对ZINC数据库中的10万个类药性分子进行虚拟筛选,得到了具有较高HIV抑制活性的先导化合物13和14,然后通过相似性搜索技术得到化合物15[36]。这3个化合物对野生型和临床上各种常见变异毒株均具有很好的抑制作用,并且抗病毒活性比化合物4略高
(IC
50≈1.60 2.16μmol·L-1)。
基于肽类研究,Abdurahman等人[37]得到了
化合物α-羟基-甘氨酰胺(α-HGA)。研究认为,
三肽甘氨酰基-脯氨酰基-甘氨酰胺(GPG-NH
2
)
被二肽基肽酶CD26裂解后产生的G-NH
2
能够
影响衣壳的组装。但随后证实G-NH
2
并不发挥
作用,而是其代谢产物α-HGA能够抑制HIV-1
的复制。α-HGA的分子量仅为90Da,推测其作
用机制可能是阻断NTD-CTD相互作用。此外,
苯并二氮杂酮类(如化合物16)[38]及聚合物类
(如没食子酸-三甘醇聚合物)[39]衣壳蛋白抑制剂
也具有较好的发展前景
。
3展望
近年来,人们对衣壳蛋白结构和功能的深入研究,逐渐揭示了衣壳蛋白独特的结构和作用机制,为基于结构的合理药物设计奠定了理论基础。衣壳蛋白本身具有多个药物结合位点,其抑制剂涵盖多种化学结构类型,例如寡肽、小分子化合物及聚合物等,这种结构多样性为设计衣壳蛋白抑制剂提供了广阔的空间。与目前上市的抗艾滋病药物相比,衣壳蛋白抑制剂无论对野生型还是临床常见变异毒株均具有较高的抑制活性。总之,衣壳蛋白作为全新的抗HIV药物作用的靶点,虽然其生物学机制还有待进一步阐明,但是通过虚拟筛选及合理药物设计等手段发现高活性及选择性的HIV-1衣壳蛋白抑制剂已经成为目前抗艾滋病药物研究的重要方向之一。
参考文献:
[1]ADAMSON C S,FREED E O.Novel approaches to inhibiting HIV-1replication[J].Antiviral Res,2010,
85(1):119-141.
[2]ADAMSON C S,SALZWEDEL K,FREED E O.Vi-rus maturation as a new HIV-1therapeutic target
[J].Expert Opin Ther Targets,2009,13(8):
895-908.[3]BERTHET-COLOMINAS C,MONACO S,NOVEL-LI A,et al.Head-to-tail dimers and interdomain flex-
ibility revealed by the crystal structure of HIV-1cap-
sid protein(p24)complexed with a monoclonal anti-
body Fab[J].EMBO J,1999,18(5):1124-1136.[4]GAMBLE T R,YOO S,VAJDOS F F,et al.Struc-ture of the carboxyl-terminal dimerization domain of
the HIV-1capsid protein[J].Science,1997,278
(5339):849-853.
[5]GITTI R K,LEE B M,WALKER J,et al.Structure of the amino-terminal core domain of the HIV-1cap-
sid protein[J].Science,1996,273(5272):231-
235.
[6]PORNILLOS O,GANSER-PORNILLOS B K,KEL-LY B N,et al.X-ray structures of the hexameric
building block of the HIV capsid[J].Cell,2009,137
(7):1282-1292.
[7]GANSER-PORNILLOS B K,CHENG A,YEAGER M.Structure of full-length HIV-1CA:a model for
the mature capsid lattice[J].Cell,2007,131(1):
70-79.
[8]von SCHWEDLER U K,STEMMLER T L,KLISH-KO V Y,et al.Proteolytic refolding of the HIV-1
capsid protein amino-terminus facilitates viral core
assembly[J].EMBO J,1998,17(6):1555-1568.[9]von SCHWEDLER U K,STRAY K M,GARRUS J E,et al.Functional surfaces of the human immunode-
204中国药物化学杂志第21卷
ficiency virus type1capsid protein[J].J Virol,
2003,77(9):5439-5450.
[10]AUERBACH M R,BROWN K R,SINGH I R.Mu-tational analysis of the N-terminal domain of molo-
ney murine leukemia virus capsid protein[J].J
Virol,2007,81(22):12337-12347.
[11]WRIGHT E R,SCHOOLER J B,DING H J,et al.Electron cryotomography of immature HIV-1virions
reveals the structure of the CA and SP1Gag shells
[J].EMBO J,2007,26(8):2218-2226.
[12]ZHANG J,LIU X,de CLERCQ E.Capsid(CA)protein as a novel drug target:recent progress in the
research of HIV-1CA inhibitors[J].Mini Rev Med
Chem,2009,9(4):510-518.
[13]IVANOV D,STONE J R,MAKI J L,et al.Mamma-lian SCAN domain dimer is a domain-swapped ho-
molog of the HIV capsid C-terminal domain[J].
Mol Cell,2005,17(1):137-143.
[14]WORTHYLAKE D K,WANG H,YOO S H,et al.Structures of the HIV-1capsid protein dimerization
domain at2.6angstrom resolution[J].Acta Crystal-
logr D,1999,55(Pt.1):85-92.
[15]IVANOV D,TSODIKOV O V,KASANOV J,et al.Domain-swapped dimerization of the HIV-1capsid
C-terminal domain[J].Proc Natl Acad Sci USA,
2007,104(11):4353-4358.
[16]GUO X F,ROY B B,HU J,et al.The R362A muta-tion at the C-terminus of CA inhibits packaging of
human immunodeficiency virus type1RNA[J].Vi-
rology,2005,343(2):190-200.
[17]MAMMANO F,OHAGEN A,HOGLUND S,et al.Role of the major homology region of human immu-
nodeficiency virus type1in virion morphogenesis
[J].J Virol,1994,68(8):4927-4936.
[18]BARTONOVA V,IGONET S,STICHT J,et al.Re-sidues in the HIV-1capsid assembly inhibitor bind-
ing site are essential for maintaining the assembly-
competent quaternary structure of the capsid protein
[J].J Biol Chem,2008,283(46):32024-32033.[19]LI F,GOILA-GAUR R,SALZWEDEL K,et al.PA-457:a potent HIV inhibitor that disrupts core con-
densation by targeting a late step in Gag processing
[J].Proc Natl Acad Sci USA,2003,100(23):
13555-13560.
[20]MARTIN D E,BLUM R,WILTON J,et al.Safety and pharmacokinetics of bevirimat(PA-457),a no-
vel inhibitor of human immunodeficiency virus mat-
uration,in healthy volunteers[J].Antimicrob Agents
Chemother,2007,51(9):3063-3066.
[21]MCCALLISTER S,LALEZARI J,RICHMOND G,et al.HIV-1Gag polymorphisms determine treatment
response to bevirimat(PA-457)[EB/OL].[2011-
06-24].http://www.natap.org/2008/ResisWk-
sp/ResisWksp_23.htm.
[22]QIAN K D,YU D L,CHEN C H,et al.Anti-AIDS agents.78.Design,synthesis,metabolic stability as-
sessment,and antiviral evaluation of novel betulinic
acid derivatives as potent anti-human immunodefi-
ciency virus(HIV)agents[J].J Med Chem,2009,
52(10):3248-3258.
[23]QIAN K D,KUO R Y,CHEN C H,et al.Anti-AIDS agents81.Design,synthesis,and structure-activity
relationship study of betulinic acid and moronic acid
derivatives as potent HIV maturation inhibitors[J].J
Med Chem,2010,53(8):3133-3141.
[24]DANG Z,LAI W H,QIAN K D,et al.Betulinic acid derivatives as human immunodeficiency virus type2
(HIV-2)inhibitors[J].J Med Chem,2009,52
(23):7887-7891.
[25]BLAIR W S,CAO J,FOK-SEANG J,et al.New small-molecule inhibitor class targeting human mmu-
nodeficiency virus type1virion maturation[J].An-
timicrob Agents Chemother,2009,53(12):5080-
5087.
[26]TANG C,LOELIGER E,KINDE I,et al.Antiviral inhibition of the HIV-1capsid protein[J].J Mol
Biol,2003,327(5):1013-1020.
[27]KELLY B N,KYERE S,KINDE I,et al.Structure of the antiviral assembly inhibitor CAP-1complex
with the HIV-1CA protein[J].J Mol Biol,2007,
373(2):355-366.
[28]PREVELIGE P J.Small molecule inhibitors of HIV-1 capsid assembly:WO,2007048042[P].2007-
04-26.
[29]JIN Y X,TAN Z W,HE M Z,et al.SAR and mo-lecular mechanism study of novel acylhydrazone
compounds targeting HIV-1CA[J].Bioorg Med
Chem,2010,18(6):2135-2140.
[30]TIAN B H,HE M Z,TANG S X,et al.Synthesis and antiviral activities of novel acylhydrazone deri-
vatives targeting HIV-1capsid protein[J].Bioorg
Med Chem Lett,2009,19(8):2162-2167.
[31]LI J B,TAN Z W,TANG S X,et al.Discovery of dual inhibitors targeting both HIV-1capsid and hu-
man cyclophilin A to inhibit the assembly and un-
coating of the viral capsid[J].Bioorg Med Chem,
304
第5期李东岳等:HIV-1衣壳蛋白抑制剂的研究进展
2009,17(8):3177-3188.
[32]CHEN K,TAN Z W,HE M Z,et al.Structure-acti-vity relationships(SAR)research of thiourea driva-
tives as dual inhibitors targeting both HIV-1capsid
and human cyclophilin A[J].Chem Biol Drug De-
sign,2010,76(1):25-33.
[33]BLAIR W S,PICKFORD C,IRVING S L,et al.HIV capsid is a tractable target for small molecule
therapeutic intervention[J].PLoS Pathoge,2010,6
(12):e1001220.
[34]ZHANG H,ZHAO Q,BHATTACHARYA S,et al.
A cell-penetrating helical peptide as a potential
HIV-1inhibitor[J].J Mol Biol,2008,378(3):
565-580.
[35]BHATTACHARYA S,ZHANG H T,DEBNATH A K,et al.Solution structure of a hydrocarbon stapled
peptide inhibitor in complex with monomeric C-ter-
minal domain of HIV-1capsid[J].J Biol Chem,
2008,283(24):16274-16278.
[36]CURRELI F,ZHANG H,ZHANG X,et al.Virtual screening based identification of novel small-mole-
cule inhibitors targeted to the HIV-1capsid[J].
Bioorg Med Chem,2011,19(1):77-90.
[37]ABDURAHMAN S,VEGVARI A,LEVI M,et al.Isolation and characterization of a small antiretroviral
molecule affecting HIV-1capsid morphology[J].
Retrovirology,2009,6:34.
[38]FADER L D,BETHELL R,BONNEAU P,et al.Discovery of a1,5-dihydrobenzo[1,4]diazepine-2,
4-dione series of inhibitors of HIV-1capsid assem-
bly[J].Bioorg Med Chem Lett,2011,21(1):
398-404.
[39]DOMENECH R,ABIAN O,BOCANEGRA R,et al.Dendrimers as potential inhibitors of the dimerization
of the capsid protein of HIV-1[J].Biomacromole-
cules,2010,11(8):2069-2078.
Recent progress in the development of
HIV-1capsid protein inhibitors
LI Dong-yue,ZHAN Peng,LIU Xin-yong*
(Institute of Medicinal Chemistry,School of Pharmacy,Shandong University,Ji'nan250012,China)
Abstract:The acquired immunodeficiency syndrome(AIDS)is a pandemic and mortal disease mainly caused by human immunodeficiency virus type1(HIV-1).Although current treatment regimen can control the virus to undetectable levels,clinically treatment failure can often be caused by the occurrence of drug re-sistance,unexpected side effects and so on.Therefore,it is necessary to develop novel antiretroviral drugs that offering sustained successful treatment of HIV-1infection and low toxicity via new mechanisms.The HIV-1capsid protein whose stability can affect the viral infectivity plays crucial roles in the processes of viral assembly and maturation.The progress that has been made in the structure and mechanism of action of capsid protein in recent years brings hope to HIV-1therapy.Recent research in the structure and functions of HIV-1capsid protein,as well as related inhibitors are reviewed.
Key words:acquired immunodeficiency syndrome;human immunodeficiency virus;capsid protein;structure;function;inhibitors;drug design
404中国药物化学杂志第21卷
丝氨酸蛋白酶抑制剂的研究进展教学提纲
丝氨酸蛋白酶抑制剂的研究进展
丝氨酸蛋白酶抑制剂的研究进展 梁化亮 (生物与食品工程学院,江苏常熟 215500) Progress on antimicrobial peptide [摘要]蛋白酶抑制剂(PIs)是一类能抑制蛋白酶水解酶的催化活性的蛋白或多肽,广泛存在于生物体内,在许多生命活动过程中发挥必不可少的作用。根据活性位点氨基酸种类不同可将蛋白酶抑制剂分为四大类型:丝氨酸蛋白酶抑制剂、巯基蛋白酶抑制剂、天冬氨酸蛋白酶抑制剂和金属蛋白酶抑制剂。其中尤以丝氨酸蛋白酶及其抑制剂在体内一些重要生理活动中起关键性的调控作用。其能对蛋白酶活性进行精确调控,包括分子间蛋白降解,转录,细胞周期,细胞侵入,血液凝固,细胞凋亡,纤维蛋白溶解作用,补体激活中所起的作用。[关键词]丝氨酸蛋白酶抑制剂分类临床应用防御
1 丝氨酸蛋白酶抑制剂 免疫系统是由组织,细胞,效应分子构成,并逐渐进化形成用于阻挠病原微生物的侵入攻击,限制它们扩散进入宿主内环境。这其中起到主要作用的是宿主产生的蛋白酶抑制剂,广泛存在于生物体内的蛋白酶抑制剂在机体内与相应的蛋白酶形成一个动态的系统,在生物体系以及一系列的生理过程中起着调控作用[1],是生物体内免疫系统的重要组成部分。它不仅能使侵入体内的蛋白酶失活并且能将其清除,使附着在宿主表面的病原细菌无法附着生存。其中丝氨酸蛋白酶及其抑制剂在体内一些重要生理活动中起关键性的调控作用[2]。 丝氨酸蛋白酶抑制剂(serine protease inhibitor)泛指具有抑制丝氨酸蛋白酶水解活性的一类物质,广泛存在于动物、植物、微生物体中[3]。在动物体中,丝氨酸蛋白酶抑制剂是维持体内环境稳定的重要因素,一旦平衡失调即导致多种疾病,任何影响其活性的因素也会造成严重的病理性疾病。它们最基本的功能是防止不必要的蛋白水解,调节丝氨酸蛋白酶的水解平衡。作为调控物,丝氨酸蛋白酶抑制剂参与机体免疫反应,对生物体内的血液凝固、补体形成、纤溶、蛋白质折叠、细胞迁移、细胞分化、细胞基质重建、激素形成、激素转运、细胞内蛋白水解、血压调节、肿瘤抑制以及病毒或寄生虫致病性的形成等许多重要的生化反应和生理功能有重要的影响[4]。鉴于其重要的生理功能,丝氨酸蛋白酶抑制剂一直倍受研究者的关注,目前已分离得到多种天然丝氨酸蛋白酶抑制剂,同时如何将其更好地应用于食品、医药领域也成为近来研究热点。 1.1 丝氨酸蛋白酶抑制剂分类
常见蛋白酶抑制剂
当前位置:生物帮 > 实验技巧 > 生物化学技术 > 正文 蛋白酶及蛋白酶抑制剂大全 日期:2012-06-13 来源:互联网 标签: 相关专题:解析蛋白酶活性测定聚焦蛋白酶研究新进展 摘要: 破碎细胞提取蛋白质的同时可释放出蛋白酶,这些蛋白酶需要迅速的被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解。以下列举了5种常用的蛋白酶抑制剂和他们各自的作用特点,因为各种蛋白酶对不同蛋白质的敏感性各不相同,因此需要调整各种蛋白酶的浓度 恩必美生物新一轮2-5折生物试剂大促销! Ibidi细胞灌流培养系统-模拟血管血液流动状态下的细胞培养系统 广州赛诚生物基因表达调控专题 蛋白酶抑制剂 破碎细胞提取蛋白质的同时可释放出蛋白酶,这些蛋白酶需要迅速的被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解。以下列举了5种常用的蛋白酶抑制剂和他们各自的作用特点,因为各种蛋白酶对不同蛋白质的敏感性各不相同,因此需要调整各种蛋白酶的浓度。由于蛋白酶抑制剂在液体中的溶解度极低,尤其应注意在缓冲液中加人蛋白酶抑制剂时应充分混匀以减少蛋白酶抑制剂的沉淀。在宝灵曼公司的目录上可查到更完整的蛋白酶和蛋白酶抑制剂表。 常用抑制剂 PMSF 1)抑制丝氨酸蛋白酶(如胰凝乳蛋白酶,胰蛋白酶,凝血酶)和巯基蛋白酶(如木瓜蛋白酶); 2)10mg/ml溶于异丙醇中; 3)在室温下可保存一年; 4)工作浓度:17~174ug/ml(0.1~1.0mmol/L); 5)在水液体溶液中不稳定,必须在每一分离和纯化步骤中加入新鲜的PMSF。 EDTA 1)抑制金属蛋白水解酶; 2)0.5mol/L水溶液,pH8~9;
丝氨酸蛋白酶抑制物在2型糖尿病合并颈动脉粥样硬化患者中的意义
丝氨酸蛋白酶抑制物在2型糖尿病合并颈动脉粥样硬化患者中的意 义 目的分析血清丝氨酸蛋白酶抑制物(vaspin)在2型糖尿病(T2DM)合并颈动脉粥样硬化(CAS)患者中的意义。方法根据有无CAS,将197例T2DM 患者分为CAS T2DM组(A组)和单纯T2DM组(B组)与正常对照组(NC 组)比较血清vaspin水平。结果T2DM患者vaspin高于NC组(P<0.05);B 组vaspin较A组高(P<0.05)。结论vaspin在T2DM合并CAS患者血管病变中有保護作用。 标签:Vaspin;血管病变;糖尿病,2型 [Abstract] Objective To analyze the significance of serine protease inhibitor in patients with type 2 diabetes and carotid artherosclerosis. Methods 197 cases of T2DM patients were divided into the CAS T2DM group (A group)and simple T2DM group (B group)according to whether there was CAS,and the serum vaspin level was compared with that in the normal control group. Results The vaspin in the T2DM patients was higher than that in the NC group(P<0.05),and the vaspin in the group B was higher than that in the group A(P<0.05). Conclusion The vaspin has a protection effect in the vascular lesion of patients with T2DM and CAS. [Key words] Vascular;Vascular lesion;Diabetes;Type 2 2型糖尿病(type 2 diabetes mellitus,T2DM)患者死亡原因中大血管病变占59%[1]。丝氨酸蛋白酶抑制物(vaspin)参与了糖尿病大血管病变的发生发展的过程。该研究探讨T2DM患者血清vaspin与颈动脉粥样硬化(carotid atherosclerosis,CAS)的关系,并探讨其机制。 1 资料与方法 1.1 一般资料 选取2016年1—5月于保定市第一中心医院内分泌一科住院治疗的T2DM 患者197例(T2DM组),均符合WHO1999年推荐的T2DM诊断与分型标准。其中男性107例,女性90例。按照有无CAS分为T2DM合并CAS组(A组)111例和单纯T2DM组(B组)86例,其中A组男56例,女55例,B组男51例,女35例。排除标准:其他类型糖尿病者;合并严重其他系统疾病者;糖尿病急性并发症者;应激状态者;近期有创伤、手术者。正常对照组(NC组)70名,为同期健康体检者,NC组男45名,女25名。 1.2 方法
视神经脊髓炎与其特异性抗体_抗水通道蛋白4抗体_综述_
收稿日期:2007 09 03;修订日期:2007 10 30 作者简介:梁松岚(1975 ),女(汉族),黑龙江省人,主治医师,在读博士。 王维治(1946 ),男,山东省人,教授(主任医师),博士生导师,主要从事神经内科临床及神经免疫学研究。通讯地址:哈尔滨医科大学附属第二医院神经科,哈尔滨150086。联系电话:(0451)89661609。E mail:lun ar0941@https://www.360docs.net/doc/b8160538.html, 。(通讯作者) 视神经脊髓炎与其特异性抗体 抗水通道蛋白4抗体(综述) 梁松岚,王维治,梁庆成 (哈尔滨医科大学附属第二医院神经内科,黑龙江哈尔滨150086) 摘要: 视神经脊髓炎(NM O)是累及视神经和脊髓的脱髓鞘疾病。最近,它的特异性抗体 抗水通道蛋白4(A Q P4)抗体被发现。A Q P4主要分布于中枢神经系统(CN S),参与胶质细胞与脑脊液(CSF)以及血液之间水的调节和运输。NM O 患者的A Q P4蛋白显著减少甚至丧失,血液及CSF 中存在抗 A Q P4抗体。实验证实,抗 AQ P4抗体对NM O 的诊断具高度敏感性和特异性,其抗体滴度水平有助于判断疗效和预后。关键词:视神经脊髓炎;抗水通道蛋白4抗体 中图分类号:R744 5+2 文献标识码:A 文章编号:1006 2963(2008)02 0109 03 视神经脊髓炎(neur omyelitis optica,NMO )是一种严重的神经系统疾病,以视神经炎和横贯性脊髓炎为特征,可导致失明和截瘫。50%的患者患病5年内失去视觉功能,不能独立行走。以前曾认为NMO 是多发性硬化(MS )的一个亚型,其实两者在遗传背景、发病机制、病理改变等方面都存在不同。在治疗方面,NM O 主要应用免疫抑制剂治疗,免疫调节剂[干扰素、醋酸格拉太咪尔(g lati r am er acetate)]则推荐用于治疗M S [1] 。当严重进展性脊髓炎应用皮质醇治疗无效时,血浆交换对NMO 患者比对MS 患者更有益。但目前临床尚无特异性诊断标志物用于区分这两种疾病,许多以NMO 症状为早期表现的患者最终被诊断为M S,如能早期明确诊断则对改善NMO 和M S 预后非常有益。 最近,NMO 的疾病特异性血清抗体已被发现。Lenno n [2]等在NM O 患者血清中发现了一种称为NM O IgG 的自身抗体,可作为NMO 特异性的标志。它主要结合在病变的微血管、软脑脊膜、软脑脊膜下以及Virchow Ro bin 间隙(VRS)。NMO Ig G 作为NMO 的特异性自身抗体,它的靶抗原为水通道蛋白4(aquaporin 4,AQP4),这提示NMO 也是一种自身免疫性通道病。 1 水通道蛋白(AQP)基因的克隆和AQP4 蛋白的表达分布 AQP 广泛存在于动植物和微生物中。1991 年Ag re 完成了CH IP28的cDNA 克隆,并于次年成功地转染非洲蟾卵母细胞,显示了其选择性水通透的功能,即第一个水通道蛋白(AQP1)被克隆。迄今已发现11种水通道蛋白(AQP0 AQ P10)[3]。 AQP4是脑中重要的水通道蛋白,参与脑组织与血液、脑组织与CSF 间的水转运和渗透压调节。在CNS,AQP4主要存在于构成血 脑脊液屏障(BCB)的星形胶质细胞终足上,是星形胶质细胞质膜内在蛋白质。AQP4大量存在于视神经和脊髓,同时也遍及脑的各个部分。经免疫组化检测显示,在脑和脊髓接触毛细血管和软脑膜的星形胶质细胞终足上、下丘脑视上核胶质板、室管膜细胞的基底外侧膜均有AQP4的明显表达[4] 。血管周围的胶质细胞突起是水分子流动的主要部位。AQ P4通道可使水分子跨细胞膜移动,生理条件下AQ P4参与CSF 的形成和吸收,参与BCB 对水分子的转运调节,并调节细胞外间隙钾离子浓度;病理条件下,A QP4的表达发生变化,参与各种原因引起的脑水肿。 2 AQP4蛋白是NMO IgG 的特异性靶抗原 现已证实NM O Ig G 是NM O 相关疾病,包括复发型脊髓炎和复发性视神经炎的敏感和特异性标记。Lennon 等[1]运用间接免疫荧光法检测发现,NM O IgG 能与脑内软脑膜和微血管成分、肾髓质的集合管和胃黏膜壁细胞特异性结合,而且
蛋白酶抑制剂的研究进展
蛋白酶抑制剂的研究进展 郭川 微生物专业,200326031 摘要:自然界共发现四大类蛋白酶抑制剂:丝氨酸蛋白酶抑制剂、巯基蛋白酶抑制剂、金属蛋白酶抑制剂和酸性蛋白酶抑制剂,本文就各大类蛋白酶抑制剂的结构特点,活性部位的研究概况及其在各领域应用的原理及进展。 关键词:蛋白酶抑制剂;结构;应用 天然的蛋白酶抑制剂(PI)是对蛋白水解酶有抑制活性的一种小分子蛋白质,由于其分子量较小,所以在生物中普遍存在。它能与蛋白酶的活性部位和变构部位结合,抑制酶的催化活性或阻止酶原转化有活性的酶。在一系列重要的生理、病理过程中:如凝血、纤溶、补体活化、感染、细胞迁移等,PI发挥着关键性的调控作用,是生物体内免疫系统的重要组成部分。从Kunitz等最早分离纯化出一种PI至今,已有多种PI被发现,根据其作用的蛋白酶主要分以下几类:抑制胰蛋白酶、胰凝乳蛋白酶等的丝氨酸蛋白酶抑制剂,抑制木瓜蛋白酶、菠萝蛋白酶等的巯基蛋白酶抑制剂,抑制胃蛋白酶、组织蛋白酶D等的羧基蛋白酶抑制剂、抑制胶原酶、氨肽酶等的金属蛋白酶抑制剂等。而根据作用于酶的活性基团不同及其氨基酸序列的同源性,可将自然界发现的PI分为四大类:丝氨酸蛋白酶抑制剂、巯基蛋白酶抑制剂(半胱氨酸蛋白酶抑制剂)、金属蛋白酶抑制剂和酸性蛋白酶抑制剂[1]。 1 结构与功能 1.1丝氨酸蛋白酶抑制剂(Serine Protease Inhibitor,Serpin) 丝氨酸蛋白酶抑制剂是一族由古代抑制剂趋异进化5亿年演变而来的结构序列同源的蛋白酶抑制剂。Sepin为单一肽链蛋白质。各种serpin大约有30%的同源序列,疏水区同源性高达70%。血浆中的serpin多被糖基化,糖链经天东酰胺的酰胺基与主链相连。位于抑制性serpin表面、距C端30~40个氨基酸处的环状结构区RSL(reactive site loop)中,存在能被靶酶的底物识别位点识别的氨基酸P1[2];近C端与P1相邻的氨基酸为P1’,依此类推,即肽链结构表示为N端-P15~P9~P1-P1’~P9’~P15’-C端。在对靶酶的抑制中。Serpin 以RSL中的类底物反应活性位点与靶酶形成紧密的不易解离的酶-抑制剂复合物,同时P1-P1’间的反应活性位点断裂。几种perpin氨基酸序列比较发现,serpins各成员的抑制专一性是由P1决定的,且被抑制的酶特异性切点一致。如抗凝血酶,抑制以Arg羧基端为敏感部位的丝氨酸蛋白酶,其中P1为Arg[2]。 1.2巯基蛋白酶抑制剂(Cytsteine Proteinase Inhiitor,CPI) 对于丝氨酸蛋白酶抑制剂(SPI)已有大量研究,巯基蛋白酶抑制剂(CPI)的研究则相对要晚一些。而动物和微生物来源的CPI已有一些研究,发现它们在结构上具有同源性,Barrett等将CPI统称为胱蛋白超家族,并按分子内二硫键的有无与数量,分子量大小等将此家族分为3个成员(F1、F2、F3)。在3个家族中,大多数F1和F3的CPI中都有Glu53-Val54-Val55-Ala56-Gly57保守序列,其同源序列在其它CPI中也被发现,如F2中的Gln-X-Val-Y-Gly和CHα-ras基因产物中的Gln-Val-Val肽段。人工合成的Glu-Val-Val-Ala-Gly 短肽也显示对木瓜蛋白酶有抑制活性,因此可以认为这一保守区段在抑制活性中起着全部或部分的关键作用[3]。对植物来源的CPI研究的不多,已有报道的有水稻、鳄梨和大豆。水稻巯基蛋白酶抑制剂(Oryzacystatin,OC) 具有102个氨基酸残基,有典型的Glu-Val-Val-Ala-Gly保守序列,应与动物CPI同源进化而来。从OCI没有二硫键来看,它应归为F1成员,但从序列比较看,则更接近F3。对OCIGlu---Gly保守序列进行点突变试验表明,突变使其抑制活性大幅度下降,其中当Glu被Pro替代时则活性全无,由此说明,这一段保守序列在OCI的抑制活性中,同动物CPI一样必不可少。除Glu---Gly保守区域外,OCI序列中其
粗粮含蛋白酶抑制剂
粗粮含蛋白酶抑制剂。荞麦、燕麦、莜麦、高粱面、红薯等粗粮中,含有抗营养素蛋白酶抑制剂。其中,荞麦、莜麦含量最高。粗粮发酵以后,酵母菌大大降低蛋白酶抑制剂的活性,所以粗粮发酵后蒸窝头、贴饼子等食用为好。 各种粗 粮 甘蓝含有硫苷。卷心菜、紫甘蓝、荠菜、萝卜、洋葱、花菜等十字花科蔬菜中,含有抗营养素——硫苷。硫苷降解的某些产物能抑制甲状腺素的合成和对碘的吸收。硫苷具有两面性,虽然它有副作用,但对子宫癌、乳腺癌等多种癌有显著的抑制作用。硫苷对热敏感,将蔬菜炒熟后,可去除其中的大部分硫苷。理想的做法是,将其一半生吃一半熟吃,这样既可保留防癌成分,又有利于其他营养成分的吸收。 黄瓜等含有抗坏血酸氧化酶。黄瓜、西葫芦、莴笋、水芹、花菜、南瓜等食物中,含有抗营养素——抗坏血酸氧化酶。抗坏血酸氧化酶会破坏蔬菜和水果中维生素C的含量。所以食用黄瓜时不必切开,生吃即可。西葫芦、莴笋、水芹、花菜等蔬菜宜大火快炒,最好不要加醋。 蛋白质抑制剂:这是大豆和其它豆类中存在的一种特殊蛋白质,可以抑制体内胰蛋白酶等十几种消化酶的流活性,其代表为胰蛋白酶抑制剂,它能抑制蛋
白酶对蛋白质的消化吸收。它需经蒸发气加热30分钟或高压蒸气加热15~2 0分钟才能被破坏。 皂角素:大豆中含有的皂角素,对消化道粘膜有强烈的刺激性,人吃了没有煮熟的大豆或豆浆,常会产生恶心、呕吐、腹痛、腹泻等症状,就是由于皂角素没有完全破坏所引起。皂角素需加热至100度才破坏,因此食用豆类或豆浆必须煮开10~20分钟后才能食用。 凝血素:也是一种特殊蛋白质,称为植物血球凝集素,可使人体细胞凝集,但加热即可被破坏,或在体内经蛋白酶作用也可使其失去活性,不致被肠道吸收后引起凝血。 棉子糖合成酶:众所周知,多吃大豆后肚子容易胀气。其原因是大豆中含有一种棉子糖合成酶,它进入人体后,可以合成大量低聚糖,如棉子糖、水苏糖等。这些糖不能被子人体吸收,大部分在肠中被细菌分解利用,同时产生大量二氧化碳、氢和甲烷。但大豆充分加熟后,此酶即被破坏,产气也随之减少;加工成豆制品或发酵制品也可去除这种酶,故吃豆腐、腐乳等豆制品就不会胀气。 植酸:这是一种含磷化合物,一般植物性食品中都含有。但大豆中含量很高,大豆中占60%~80%的磷都是以植酸形式存在,植酸可与蛋白质、无机盐及矿物元素钙、磷、铁、锌等结合而影响其消化吸收。大豆中的锌很难吸收,就是受了植酸的影响,可利用发芽米分解植酸,提高大豆中铁、锌、钙、镁等矿物元素的生物利用率。
水通道蛋白
水通道蛋白 水通道蛋白(Aquaporin),又名水孔蛋白,是一种位于细胞膜上的蛋白质(内在膜蛋白),在细胞膜上组成“孔道”,可控制水在细胞的进出,就像是“细胞的水泵”一样。 水通道是由约翰霍普金斯大学医学院的美国科学家彼得·阿格雷所发现,他与通过X射线晶体学技术确认钾离子通道结构的洛克斐勒大学霍华休斯医学研究中心的罗德里克·麦金农共同荣获了2003年诺贝尔化学奖。 水分子经过Aquaporin时会形成单一纵列,进入弯曲狭窄的通道内,内部的偶极力与极性会帮助水分子旋转,以适当角度穿越狭窄的通道,因此Aquaporin的蛋白构形为仅能使水分子通过之原因 水通道蛋白的发现 编辑 Agre等(1988)在分离纯化红细胞膜上的Rh多肽时,发现了一个28 kD的疏水性跨膜蛋白,称为形成通道的整合膜蛋白28(channel-forming inte—gral membrane protein,CHIP28),1991年完成了其cDNA克隆(Verkman,2003)。但当时并不知道该蛋白的功能,在进行功能鉴定时,将体外转录合成的CHIP28 mDNA 注入非洲爪蟾的卵母细胞中,发现在低渗溶液中,卵母细胞迅速膨胀,并于5 min 内破裂。为进一步确定其功能,又将其构于蛋白磷脂体内,通过活化能及渗透系数的测定及后来的抑制剂敏感性等研究,证实其为水通道蛋白。从此确定了细胞膜上存在转运水的特异性通道蛋白,并称CHIP28为Aquaporinl(AQPl)。 水通道蛋白分类 编辑 AQP0 AQP0最初称之为主体内在蛋白(major intrinsic protein,MIP),在晶状体纤维中细胞中表达丰富,与晶状体的透明度有关.AQpo的突变可能导致晶状体水肿和白内障。小鼠缺乏AQPO将患先天性白内障[61]。 AQP1 AQP1是1988年发现的,开始将这种蛋白称为通道形成整合蛋白(CHIP),是人的红细胞膜的一 种主要蛋白。它可以使红细胞快速膨胀和收缩以适应细胞间渗透性的变化。AQP1蛋白也存在于
常见蛋白酶抑制剂
蛋白酶及蛋白酶抑制剂大全 标签: 相关专题:解析蛋白酶活性测定聚焦蛋白酶研究新进展 摘要: 破碎细胞提取蛋白质的同时可释放出蛋白酶,这些蛋白酶需要迅速的被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解。以下列举了5种常用的蛋白酶抑制剂和他们各自的作用特点,因为各种蛋白酶对不同蛋白质的敏感性各不相同,因此需要调整各种蛋白酶的浓度 恩必美生物新一轮2-5折生物试剂大促销! Ibidi细胞灌流培养系统-模拟血管血液流动状态下的细胞培养系统 广州赛诚生物基因表达调控专题 蛋白酶抑制剂 破碎细胞提取蛋白质的同时可释放出蛋白酶,这些蛋白酶需要迅速的被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解。以下列举了5种常用的蛋白酶抑制剂和他们各自的作用特点,因为各种蛋白酶对不同蛋白质的敏感性各不相同,因此需要调整各种蛋白酶的浓度。由于蛋白酶抑制剂在液体中的溶解度极低,尤其应注意在缓冲液中加人蛋白酶抑制剂时应充分混匀以减少蛋白酶抑制剂的沉淀。在宝灵曼公司的目录上可查到更完整的蛋白酶和蛋白酶抑制剂表。 常用抑制剂 PMSF 1)抑制丝氨酸蛋白酶(如胰凝乳蛋白酶,胰蛋白酶,凝血酶)和巯基蛋白酶(如木瓜蛋白酶); 2)10mg/ml溶于异丙醇中; 3)在室温下可保存一年; 4)工作浓度:17~174ug/ml(0.1~1.0mmol/L); 5)在水液体溶液中不稳定,必须在每一分离和纯化步骤中加入新鲜的PMSF。 EDTA 1)抑制金属蛋白水解酶; 2)0.5mol/L水溶液,pH8~9; 3)溶液在4℃稳定六个月以上;
4)工作浓度:0.5~1.5mmol/L. (0.2~0.5mg/ml); 5)加入NaOH调节溶液的pH值,否则EDTA不溶解。 胃蛋白酶抑制剂(pepst anti n) l)抑制酸性蛋白酶如胃蛋白酶,血管紧张肽原酶,组织蛋白酶D和凝乳酶; 2)1mg/ml溶于甲醇中; 3}储存液在4℃一周内稳定,-20℃稳定6个月; 4)1作浓度:0.7ug/ml(1umol/L) 5)在水中不溶解。 亮抑蛋白酶肽(leupeptin) 1)抑制丝氨酸和巯基蛋白酶,如木瓜蛋白酶,血浆酶和组织蛋白酶B; 2)lOmg/ml溶于水; 3)储存液4℃稳定一周,-20℃稳定6个月; 4)工作浓度0.5mg/ml。 胰蛋白酶抑制剂(aprotinin) 1)抑制丝氨酸蛋白酶,如血浆酶,血管舒缓素,胰蛋白酶和胰凝乳蛋白酶; 2)lOmg/ml溶于水,pH7~8 3}储存液4℃稳定一周,-20℃稳定6个月; 4)工作浓度:0.06~2.0ug/ml(0.01~0.3umol/L); 5)避免反复冻融: 6)在pH>12.8时失活。 蛋白酶抑制剂混合使用 35ug/ml PMSF…………………………………丝氨酸蛋白酶抑制剂 0.3mg/ml EDTA…………………………………金属蛋白酶抑制剂 0.7ug/ml胃蛋白酶抑制剂(Pepstatin)…………酸性蛋白酶抑制剂 0.5ug/ml亮抑蛋白肽酶(Leupeptin)……………广谱蛋白酶抑制剂
丝氨酸蛋白酶抑制剂的研究进展
丝氨酸蛋白酶抑制剂的研究进展 梁化亮 (生物与食品工程学院,常熟 215500) Progress on antimicrobial peptide [摘要]蛋白酶抑制剂(PIs)是一类能抑制蛋白酶水解酶的催化活性的蛋白或多肽,广泛存在于生物体,在许多生命活动过程中发挥必不可少的作用。根据活性位点氨基酸种类不同可将蛋白酶抑制剂分为四大类型:丝氨酸蛋白酶抑制剂、巯基蛋白酶抑制剂、天冬氨酸蛋白酶抑制剂和金属蛋白酶抑制剂。其中尤以丝氨酸蛋白酶及其抑制剂在体一些重要生理活动中起关键性的调控作用。其能对蛋白酶活性进行精确调控,包括分子间蛋白降解,转录,细胞周期,细胞侵入,血液凝固,细胞凋亡,纤维蛋白溶解作用,补体激活中所起的作用。 [关键词]丝氨酸蛋白酶抑制剂分类临床应用防御
1 丝氨酸蛋白酶抑制剂 免疫系统是由组织,细胞,效应分子构成,并逐渐进化形成用于阻挠病原微生物的侵入攻击,限制它们扩散进入宿主环境。这其中起到主要作用的是宿主产生的蛋白酶抑制剂,广泛存在于生物体的蛋白酶抑制剂在机体与相应的蛋白酶形成一个动态的系统,在生物体系以及一系列的生理过程中起着调控作用[1],是生物体免疫系统的重要组成部分。它不仅能使侵入体的蛋白酶失活并且能将其清除,使附着在宿主表面的病原细菌无法附着生存。其中丝氨酸蛋白酶及其抑制剂在体一些重要生理活动中起关键性的调控作用[2]。 丝氨酸蛋白酶抑制剂(serine protease inhibitor)泛指具有抑制丝氨酸蛋白酶水解活性的一类物质,广泛存在于动物、植物、微生物体中[3]。在动物体中,丝氨酸蛋白酶抑制剂是维持体环境稳定的重要因素,一旦平衡失调即导致多种疾病,任何影响其活性的因素也会造成严重的病理性疾病。它们最基本的功能是防止不必要的蛋白水解,调节丝氨酸蛋白酶的水解平衡。作为调控物,丝氨酸蛋白酶抑制剂参与机体免疫反应,对生物体的血液凝固、补体形成、纤溶、蛋白质折叠、细胞迁移、细胞分化、细胞基质重建、激素形成、激素转运、细胞蛋白水解、血压调节、肿瘤抑制以及病毒或寄生虫致病性的形成等许多重要的生化反应和生理功能有重要的影响[4]。鉴于其重要的生理功能,丝氨酸蛋白酶抑制剂一直倍受研究者的关注,目前已分离得到多种天然丝氨酸蛋白酶抑制剂,同时如何将其更好地应用于食品、医药领域也成为近来研究热点。 1.1 丝氨酸蛋白酶抑制剂分类 目前,典型的丝氨酸蛋白酶抑制剂基于其序列、拓扑结构及功能的相似性,至少可分为18个家族[5],如表1-1所示。不同家族抑制剂的空间结构也不同。通常这类抑制剂是β片层或混合了α螺旋和β片层的蛋白质,也可能是α螺旋或富含二硫键的不规则蛋白质。但它们都拥有规的反应活性位点环的构象,从而使这些非相关的蛋白质具有相似的生物学功能[6]。因此典型的丝氨酸蛋白酶抑制剂最明确最广泛地代表了蛋白质的趋同进化。 1.2 Serpins Serpins是一类分子量较大的丝氨酸蛋白酶抑制剂超家族,氨基酸残基数为
常见蛋白酶抑制剂
当前位置:生物帮〉实验技巧 > 生物化学技术>正文 蛋白酶及蛋白酶抑制剂大全 日期:2012—06-13 来源:互联网 标签: 相关专题:解析蛋白酶活性测定聚焦蛋白酶研究新进展 摘要: 破碎细胞提取蛋白质得同时可释放出蛋白酶,这些蛋白酶需要迅速得被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解、以下列举了5种常用得蛋白酶抑制剂与她们各自得作用特点,因为各种蛋白酶对不同蛋白质得敏感性各不相同,因此需要调整各种蛋白酶得浓度 恩必美生物新一轮2-5折生物试剂大促销!?Ibidi细胞灌流培养系统-模拟血管血液流动状态下得细胞培养系统 广州赛诚生物基因表达调控专题 蛋白酶抑制剂 破碎细胞提取蛋白质得同时可释放出蛋白酶,这些蛋白酶需要迅速得被抑制以保持蛋白质不被降解、在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解、以下列举了5种常用得蛋白酶抑制剂与她们各自得作用特点,因为各种蛋白酶对不同蛋白质得敏感性各不相同,因此需要调整各种蛋白酶得浓度。由于蛋白酶抑制剂在液体中得溶解度极低,尤其应注意在缓冲液中加人蛋白酶抑制剂时应充分混匀以减少蛋白酶抑制剂得沉淀。在宝灵曼公司得目录上可查到更完整得蛋白酶与蛋白酶抑制剂表。 常用抑制剂 PMSF 1)抑制丝氨酸蛋白酶(如胰凝乳蛋白酶,胰蛋白酶,凝血酶)与巯基蛋白酶(如木瓜蛋白酶); 2)10mg/ml溶于异丙醇中; 3)在室温下可保存一年; 4)工作浓度:17~174ug/ml(0。1~1.0mmol/L); 5)在水液体溶液中不稳定,必须在每一分离与纯化步骤中加入新鲜得PMSF。 EDTA 1)抑制金属蛋白水解酶; 2)0.5mol/L水溶液,pH8~9; 3)溶液在4℃稳定六个月以上;
水通道蛋白综述与展望
水通道蛋白水通道-从原子结构到临床医学 生物膜的透水性在生理学上是一个长期存在的问题,但负责此类蛋白质的蛋白质仍然未知,直到发现水通道蛋白1(AQP1)水通道蛋白。AQP1由渗透梯度驱动的水选择性渗透。人类AQP1的原子结构最近被定义。四聚体的每个亚基含有允许水分子单文件通过但中断氢键通过质子所需的单独水孔。已经鉴定了至少10种哺乳动物水通道蛋白,并且它们被水(水通道蛋白)或水加甘油(水甘油聚糖)选择性渗透。表达位点与临床表型密切相关,从先天性白内障到肾源性尿崩症。在植物,微生物,无脊椎动物和脊椎动物中发现超过200个水通道蛋白家族成员,并且它们对这些生物体的生理学的重要性正在被揭开。 在20世纪20年代发现脂质双层提供了当沐浴在较低或较高pH或含有毒性浓度的Ca2 +或其他溶质的细胞外液中时细胞如何维持其最佳细胞内环境的解释。从1950年代开始发现离子通道,交换剂和共转运体为溶质的跨膜运动提供了分子解释。然而,长期以来,假定水的输送是由于通过脂质双层的简单扩散。来自具有高膜渗透性的多个实验系统的观察,例如两栖膀胱和哺乳动物红细胞,表明通过脂质双层的扩散不是水跨越膜的唯一途径。虽然提出了各种解释,但直到10年前发现AQP1才能知道分子水 - 特异性转运蛋白(Preston 等,1999)。
现在人们普遍同意扩散和通道介导的水分运动都存在。通过所有生物膜以相对较低的速度发生扩散。水通道蛋白水通道发现于上皮细胞的一部分10至100倍的水渗透能力。值得注意的是,水通道蛋白水通道的选择性非常高,甚至质子(H3O +)被排斥。在大多数组织中,扩散是双向的,因为水进入细胞并从细胞释放,而水通道蛋白介导的体内水流则由渗透或液压梯度引导。扩散的化学抑制剂是未知的,扩散发生在高Ea(Arrhenius活化能)。相比之下,大多数哺乳动物水通道蛋白受汞的抑制,Ea等同于大量溶液中水的扩散(?5 kcal mol_1)。 水通道蛋白的发现说明了偶发性在生物学研究中的重要性,并且引起了上游流体运输过程中水如何穿过生物膜的范式的完全转变。这个话题对正常生理学以及影响人类的多种临床疾病的病理生理学非常重要。水通道蛋白在几乎每一种生物体中被鉴定出来,包括高等哺乳动物,其他脊椎动物,无脊椎动物,植物,真细菌,原细菌和其他微生物,表明这种新认可的蛋白质家族参与了整个自然界的不同生物过程。 一、发现AQP1 红细胞Rh血型抗原不知道参与水运(Heitman&Agre,2000),但是Rh的研究导致了水通道蛋白的偶
丝氨酸蛋白酶 (2)
丝氨酸蛋白酶 摘要:丝氨酸蛋白酶是一种种类丰富的酶类【1】,之所以以此命名是因为在酶的催化活性位点上包含丝氨酸在内的丝氨酸、组氨酸、天冬氨酸组成的催化三联体。有些丝氨酸蛋白酶类如凝血酶类蛋白酶,其中包括凝血酶,组织纤维蛋白溶酶原激活剂、血纤维蛋白溶酶,它们参与凝血的发生以及炎症应答反应;也有些如胰蛋白酶类的丝氨酸蛋白酶类的参与消化的酶类,包括胰蛋白酶、弹性蛋白酶、胰凝乳蛋白酶;还有一些表达在神经系统中的丝氨酸蛋白酶类,这些酶类与神经系统正常的维持或是介导病理情况的发生。其实丝氨酸蛋白酶类在执行功能的时候也受到许多因素的限制,如受一些抑制剂的影响等,这些物质对蛋白酶功能的执行起到重要的作用。 关键词:丝氨酸蛋白酶催化机制功能调节 酶的功能 已知所有的蛋白分解酶类丝氨酸蛋白酶占到了其中的三分之一,这些酶又可以细分成很多种类有胰蛋白酶、胰凝乳蛋白酶、弹性蛋白酶、凝血酶、纤溶酶、组织纤溶酶原激活剂、神经源类的丝氨酸蛋白酶等。这些酶类具有消化凝血、纤溶、消化、受精、生长发育、凋亡、免疫等方面都有重要的作用。 酶的催化位点 由于丝氨酸蛋白酶的种类很多根据其催化的特点以及种树亲疏性可以分成不同的类别,不同的组织器官,不同的生物种系中酶的分布与种类是不同的(见表格)。但是其催化特点通常都是其反应的催化三联体,丝氨酸的亲核攻击,即丝氨酸的羟基攻击酰胺键的羰基碳,但是在生物进化的长时间了这种催化活性结构也发生了改变。如在有些酶中其催化三联体不在是固定的丝氨酸、天冬氨酸、组氨酸,而是只有丝氨酸与天冬氨酸或是组氨酸的一种组成催化活性位点,也有的如组氨酸成对出现于丝氨酸组合形成新的催化结构,但是无论怎样其上的丝氨酸残基是固定保守的。 酶的活化 对于丝氨酸蛋白酶类的活化,一般来说是通过对酶前体【2】的加工使其形成具有催化活性的酶,或者是通过一些辅助因子的协同作用使其由闭合的非活化状态转成活性状态,也有通过信号的捕获诱发一系列的级联反应从而活化蛋白,或是通过一些关键因子的作用使得构想发生改变来实现活化等等。通常来说酶的状态一种是抑制非活化状态,另一种是活化的活性状态,但是在一些研究中酶具有新的状态,而这种状态与酶原或是缺少辅因子而显示无活性的酶的状态是不同的,虽然这种状态下的酶也没有活性,但是其结构上出现一些特有的变化,在对凝血酶的研究中发现,这种状态称为E*【3】,其伴有一些氨基酸链陷入酶的催化活性部位从而破坏其中的氧离子空穴,致使没得活性受阻,因此对于这种酶的活化一定有其他的方式,研究发现当E*状态下在远离活性部位连接一种配体时会将这种氨基酸陷入活性位点的状况扭转过来,从而恢复酶的活性位点,并在其他因子的作用下得到活化。 酶的催化机制 对于丝氨酸蛋白酶类的催化活性,有的是通过前体酶原的活化,比如胰蛋白酶类
蛋白酶抑制剂选择指南
蛋白酶抑制剂选择指南 1 蛋白酶抑制剂选择指南 抑制剂 工作浓度 分子量 抑制蛋白酶种类 稳定性 AEBSF终浓度1mM MW:239.5不可逆的丝氨酸蛋白酶抑制剂,抑制胰蛋白酶,糜 蛋白酶,纤溶酶,凝血酶及激肽释放酶. 可溶于水,其pH7的水溶液在4o C可保持稳定1-2个月,在pH>8的情况下会发生缓慢水解 Aprotinins 抑肽酶终浓度2ug/ ml MW:6512 可逆的丝氨酸蛋白酶抑制剂,可抑制纤溶酶,激肽 释放酶,胰蛋白酶,糜蛋白酶,但不抑制凝血酶和 Factor Xa。 非常稳定,当pH>12.8时失去活性,可溶于 水(10mg/ml),-20o C下可长期保存 Bestatin终浓度10uM MW:308.4 可逆的丙氨酰-氨基肽酶抑制剂, 工作液可保存一天,1mM的甲醇贮存液在 -20o C可保存至少一个月 E-64 Protease Inhibitor终浓度10uM MW:357.4 不可逆的半胱氨酸酸蛋白酶抑制剂,抑制半胱氨酸 酸蛋白酶而不会影响其他酶的半胱氨酸残基,与小 分子量的巯基醇如beta-巯基乙醇不会产生反应, 具有高度特异性。工作液在正常pH值下可保持稳定数天,1mM的水溶液在-20o C可保存几个月 EDTA, 4Na终浓度10mM MW:380.2 金属蛋白酶的可逆性螯合物,可能同时影响其他金 属依赖性生物过程。其水溶液很稳定,其贮存液(pH8.5的0.5M 水溶液)在4o C可保存数月 Leupeptin, 半硫酸盐 亮抑酶肽(亮肽素) 终浓度100uM MW:493.6 可逆的丝氨酸及半胱氨酸蛋白酶制剂,可抑制胰蛋 白酶样蛋白酶及一些半胱氨酸蛋白酶如:Lys-C内 切蛋白酶,激肽释放酶,木瓜蛋白酶,凝血 酶,Cathepsin B及胰蛋白酶。 工作液的稳定期为数小时,贮存液(10mM 水溶液)在4o C时稳定期为一周,-20o C时 稳定期为一个月 Pepstatin A 终浓度1uM MW:685.9 可逆的天冬氨酸蛋白酶,可抑制胃蛋白 酶,Cathepain B&L,血管紧张肽原酶(renin)及以1mg/ml溶于甲醇,搅拌过夜可以 1mg/ml溶于乙醇,333mg/ml溶于6N的
蛋白酶抑制剂基因及转基因植物研究进展
蛋白酶抑制剂基因及转基因植物研究进展 摘要: 植物蛋白酶抑制剂是除Bt之外又一个愈来愈研究较多的抗虫基因资源,其分布广泛,在豆科、茄科、禾本科、葫芦科及十字花科等植物中存在较多。植物蛋白酶抑制剂抗虫基因主要通过2种途径获得并在多种植物中进行转化,获得抗虫转基因植株。植物蛋白酶抑制剂在基因工程中的应用已有很大的发现进展。 关键字:蛋白酶抑制剂基因作用机理转基因 正文: 一蛋白酶抑制剂作用机理 广泛存在于植物组织中的蛋白酶抑制剂是一种多肽物质, 对许多昆虫有防 卫作用。该基因及其编码区域较小、没有内含子。研究表明, 这些蛋白酶抑制剂在植物对危害昆虫以及病原体侵染的夭然防御系统中担当着重要角色。昆虫饲喂实验发现, 某些纯化的蛋白酶抑制剂具有明显的抗虫作用。利用蛋白酶抑制剂基因来提高植物的抗虫能力, 已成为植物基因工程研究的一个热门领域。在植物中发现有三类蛋白酶抑制剂: 丝氨酸蛋白酶抑制剂, 琉基蛋白酶抑制剂和金属蛋白酶抑制剂。其中对丝氨酸类蛋白酶抑制剂的研究最为透彻, 目前在植物中至少已经发现有6 个家族, 其中的弧豆胰蛋白酶抑制剂, 马铃薯蛋白酶抑制剂兀的抗虫效果最为理想。蛋白酶抑制剂的杀虫机理蛋白酶抑制剂杀虫的机理在于: 它能与昆虫消化道内的蛋白消化酶相互作用形成酶抑制剂复合物( E l ) 阻断或减弱消 化酶的蛋白水解作用。所以, 一旦昆虫摄食进蛋白酶抑制剂, 就会影响外来蛋白的正常消化, 同时, 蛋白酶抑制剂和消化酶形成E l 复合物, 能刺激消化酶的过 量分泌, 通过神经系统的反馈, 使昆虫产生厌食反应。由于蛋白酶抑制剂抑制了昆虫的进食及消化过程, 不可避免地将导致昆虫缺乏代谢中必需的氨基酸, 最终造成昆虫的非正常发育或死亡。 二植物蛋白酶抑制剂基因作用机理及获得的途径 蛋白酶抑制剂基因的作用机理及其应用蛋白酶抑制剂( P l ) 是自然界含量 最为丰富的蛋白种类之一, 存在于所有生命体中。国内外有关抗虫的植物蛋白酶抑制剂基因的获得大多通过2种途径。一种通过从植物不同部位的组织或细胞中提取抗虫活性蛋白,然后分析其起作用的活性核苷酸序列,继而克隆和转化到寄主细胞,进行筛选和选育抗虫树种。利用该方法获得抗虫树种的研究越来越多,该方法中最为关键的环节是蛋白酶抑制剂提取和活性测定方法的选择和建立。植物蛋白酶抑制剂分离和纯化的策略主要依据不同植物中的蛋白酶抑制剂生理生
AQP1 抑制剂4
关于乙酰唑胺与碳酸氢钠联合是否影响肿瘤的生长、转移及水通道蛋白的表达 摘要:这项研究目的在于探索乙酰唑胺影响肿瘤的生长、转移以及可能的机制。使用感染了Lewis肺癌的大鼠作为动物模型。相比于联合使用碳酸氢钠是否对于肿瘤的生长转移以及碳酸脱苷酶的活动,使用咪唑影响肺癌的影响。同时使用Western-blot技术和免疫组化技术研究水通道蛋白1在肿瘤组织中的可能发挥的作用。结果显示单独使用乙酰唑胺可以大量降低肺癌的转移和原发肿瘤的生长,并且是一个独立影响发挥作用的因素。乙酰唑胺可以极大的抑制碳酸脱氧苷酶在肿瘤组织中的活动。在附加了碳酸氢钠之后,乙酰唑胺对肿瘤生长、转移的数量、以及原始肿瘤中碳酸酐酶发挥的作用并没有被改变,但是在肺组织中转移的抑制率以及在肺组织碳酸苷酶的活动似乎产生了一个明显的逆转,于单独使用乙酰唑胺相比。明确的作用机制需要在未来被一步一步证明。Wwstern-blot技术和免疫组化技术同时显示水通道蛋白1在肿瘤组织中的表达明显高于正常组织和单独接受过乙酰唑胺治疗的肿瘤组织。乙酰唑胺与碳酸氢钠结合使用并不能明显降低水通道蛋白的表达。结果显示乙酰唑胺的抗肿瘤机制,其中尤其是对抗转移的作用可能一部分包含了抑制碳酸酐酶的活动或者降低了水通道蛋白中水通道蛋白的表达,无论联合碳酸氢钠与否。 简介: 肿瘤转移是肿瘤的一大特点,并且是死亡的直接原因,碳酸托苷酶(CA)是锌结合金属蛋白的一个家族,这一家族可以促进二氧化碳
可逆的水合作用,产生氢离子和碳酸氢根离子,并诱导ph值下降,一些碳酸托苷酶同工酶被显著的发现仅仅在肿瘤细胞中表达,这些CA同工酶通过细胞膜在邻近细胞中通过感应PH值和翻到质子和碳酸氢盐方面产生了一个重大的作用。研究已经显示酸性PH可以增加肿瘤细胞的侵袭性。乙酰唑胺是磺胺类的一种,被当做CA抑制剂。 T等报道CA抑制剂,作为化疗原则的一部分可以增加化疗药物的作用并且有助于延缓肿瘤进展,一些新型CA抑制剂,在试验中被当做抑制肿瘤细胞生长的有效成分,例如白血病、非小细胞肺癌、黑色素瘤、卵巢、肾、前列腺、乳腺细胞系列肿瘤。尽管存在一些CA 抑制因子作用机制。人们依旧认为在直接抑制肿瘤相关CA同工酶后,导致了酸化肿瘤细胞外环境的抑制作用,这一切似乎与之前描述的理论相矛盾。因此,我们假设一定存在通过CA抑制因子抑制肿瘤转移的其他途径。 水通道蛋白,是一种作用于高选择性水通道膜蛋白的大家族,目前研究强调了他们在生物学上的作用,以及人类许多种疾病中,包含快速水通道的疾病,并且已经成为了介入治疗的目标靶向蛋白。AQP1是第一种水特点通道蛋白,广泛存在于红细胞膜、肾输尿管、脉络膜、眼睛、肺、血管内皮、肝管上皮细胞以及其他肿瘤细胞。许多肿瘤显示出极高的血管通透性和高的间隙渗透性,但是肿瘤中的水运输通路仍旧未知。 在之前已经出版的关于AQP1以及CA研究报道中,尤其是CA2和CA4,我们发现他们具有共同的生物学特点。例如,它们均在液体通
水通道蛋白相关疾病阅读材料
四.水通道蛋白相关疾病 当水通道蛋白的调节出现紊乱的时候,则可能引起多种疾病。 (一)肾脏水通道蛋白和相关疾病 研究表明,水通道蛋白基因突变将引起尿崩症(diabetesinsipidus,DI)。尿崩症广义上讲是指多饮、低比重尿和低渗尿为特征的一组综合征。目前报道的多数遗传性肾性尿崩症病例是以X连锁方式遗传的,由编码V2 受体的基因突变引起,另外的病例则是由于编码AQP2基因的突变引起,以常染色体显性或隐性方式遗传[11]。 (二)肺部水通道蛋白和相关疾病 肺水通道蛋白的异常与肺疾病的关系已有诸多实验报道。AQP可能参与肺水肿的发病机制。在各种肺损伤中,存在着大量的水的异常跨膜转运及在肺组织中的异常聚集等情况,这些情况均可能与水通道蛋白有关。在小鼠病毒性肺炎模型中,发现AQP1和AQP5在鼠肺中的表达降低,这说明肺水在肺间质中聚集的重要原因就是水通道蛋白的减少,导致水不能及时排出而出现水肿。 哮喘发作时,水分子运动在气道阻塞中起重要作用,特别在冷哮喘或运动哮喘时, 上皮黏膜下血管(含AQP1) 、气管及支气管(含AQP3 和AQP4) 的肿胀是形成气道阻塞的重要原因[1]。从而说明了水通道蛋白和哮喘的发生也有密切关系。 (三)水通道蛋白及癌症 水通道蛋白在肿瘤组织的表达及其与肿瘤细胞转移的关系可能将会是今后研究的热门。 多年研究表明,为满足快速增殖、分裂和侵袭转移的需要,肿瘤细胞内一系列酶的活性和表达会发生改变,细胞基本结构成分如蛋白质、脂类和核酸的合成加强。癌细胞的所有生命活动都离不开水的微环境和参与,癌细胞比正常细胞更需要水分子的快速跨膜转运。
目前的研究表明,部分AQPs在肿瘤组织中表达明显增高或降低。在脑胶质瘤中水通道蛋白的表达明显增多,脑胶质瘤多伴有脑水肿的发生。经证实,AQP 1和AQP4在脑胶质瘤中的表达明显高于正常组织,且在星型细胞的表达量与恶性程度有直接关系[8]。 AQPs同时还可能促进肿瘤血管增生,增强肿瘤血管渗透性,在肿瘤的生长和扩散、侵袭和转移中有重要作用。多数肿瘤有很高的组织间隙液体压力,其新生血管对血浆蛋白及其他循环体系中的高分子物质具有很高的通透性。目前认为这种不正常的通透性是因为许多肿瘤细胞分泌的血管渗透性因子(vascular pe rmeability factor,VPF)和血管内皮生长因子(vascular endothelial growth factor,VEGF)。 目前已知AQP1表达于红细胞膜上,毛细血管内皮细胞,人大、小动脉以及动脉粥样硬化斑的血管平滑肌细胞上。AQP1基因敲除小鼠表明AQP1提供了一定数量的水通路的排出,引起水通过毛细血管内皮细胞的渗透性升高。AQP1通路代表了微血管壁整个通过细胞间的10%~45%的液体渗透性,因此认为AQP1可能与血管渗透性有很大关系。 同时还发现应用AQP抑制剂可以部分抑制肿瘤侵袭和转移。肿瘤侵袭和转移是一个高度选择性的过程,其依赖于肿瘤特性和它们周围独特的微环境之间的复杂反应。据报道恶性肿瘤细胞外环境的酸化可以提高肿瘤的侵袭性,在正常组织,酸性产物可以被碳酸酐酶催化,碳酸酐酶在肿瘤生长和转移中有重要作用,某些碳酸酐酶在特定的肿瘤上有过量表达。大部分肿瘤的高血管渗透性和高组织间隙渗透压可能是由于肿瘤微环境的酸化,以及广泛分布于肿瘤上的水通道蛋白的活性引起。AQP1是唯一表达于肾脏近曲小管上皮细胞的水通道,对尿液的浓缩和稀释过程起重要作用。碳酸酐酶抑制剂是一类作用于肾脏近曲小管的利尿剂,如乙酰唑胺,其作用位点与AQP1的组织分布一致,体外实验表明可以抑制肿瘤细胞的侵袭能力。荷瘤小鼠癌组织中AQP1的蛋白水平显著高于正常组织,用乙酰唑胺治疗后,AQP1表达明显降低,显著抑制肿瘤转移。因此认为乙酰唑胺抑制肿瘤转移的作用是因为下调了AQP1的表达。以上理论为我们提供了一种全新的思路,或许可以开发应用水通道蛋白抑制剂来治疗肿瘤。[8]