超支化聚合物制备方法的研究进展

基金项目:国家自然科学基金资助项目(N o.20334030,N o.50403024);
作者简介:张传海(1984-),男,中国科学院研究生院硕士研究生,主要从事聚烯烃功能化的研究。
3通讯作者:Email :zhangly @https://www.360docs.net/doc/c4834566.html,
超支化聚合物制备方法的研究进展
张传海a ,李化毅b ,张明革b ,张辽云a 3
(a 中国科学院研究生院,北京 100049;b 中国科学院化学研究所,
北京分子科学国家实验室,高分子科学与材料联合实验室,工程塑料重点实验室,北京 100080) 摘要:超支化聚合物是一类可以通过一步法来合成的具有高度支化结构的体型大分子。经过二十年的研
究,超支化聚合物由于其独特的结构和性能特点以及可实现规模化生产的特点,已经迅速成为一类重要的和具
有广阔应用潜力的高分子材料。本文从单体类型的角度介绍了超支化聚合物的主要制备方法及其发展历程,主要涉及AB x 型,AB 3型,A 2+B 3型以及潜在AB x 型单体(包括开环聚合和偶合单体法)等,同时论述了各制备方法的优点和局限性。
关键词:超支化聚合物;制备方法;缩聚反应;自缩合乙烯基聚合概述
树状支化大分子(Dendritic macrom olecules )是近年来高分子材料领域研究的热点之一[1~6]。根据树状
支化大分子的结构特征,可将其划分为树枝状大分子(Dendrimer )和超支化聚合物(Hyperbranched
polymers )[1]。树状支化大分子由于具有高度支化的结构,因而表现出与线形聚合物不同的性能,例如其分子链不易缠结、溶液和本体的粘度低、分子结构呈球形、以及分子链末端带有大量的官能团等[2]。其中树枝状大分子由于其结构上的完美(无缺陷和高度的对称性等),最先受到学界的关注,但是树枝状大分子无论是通过发散法合成还是通过收敛法合成,都需要经过多步反应和纯化,繁琐的合成过程和高昂的成本大大妨碍了其工业化的应用。另一方面,在很多领域的应用中,完美的树枝状结构并不是必须的条件,例如在改善粘度的时候需要的主要是其高度支化形成的球形结构,作为涂料的交联剂需要的则主要
是其低粘度和大量的末端活性基团[3]。因此,超支化聚合物作为与树枝状大分子结构类似的一类高分子
开始进入人们的视野,并且由于其与树枝状大分子相似的结构和特性以及其合成上远胜于树枝状大分子的优势,因此从一开始就被认为极具大规模工业化的前景。因为超支化聚合物并没有对于完美结构的追求,反应不需要经过多步的合成和纯化,通过一步法即可由单体合成得到所需的聚合物,从而大大降低了合成的成本。此外,大量的官能团的存在使其可以通过改性得到各种不同特性和特殊用途的聚合物,显示了从涂料、粘合剂、流变助剂到超分子化学、纳米科技、生物材料、光电材料、药物运载等诸多领域巨大
的应用价值[4]。
超支化聚合物的历史最早可以追溯到19世纪末,Berzelius 首先报道了一种用酒石酸和甘油合成的具有支化结构的聚酯[5]。1901年,Wats on 报道了邻苯二甲酸酐与甘油合成聚酯的反应,Daws on 、H owell 、K ienle 等对此反应作了进一步的深入研究,结果表明所得到的产物的粘度相对于一般的线形聚合物的粘度要低。但是当时既没有超支化聚合物的概念,也缺乏相关的理论模型,对其进一步的研究一直处于停滞状态。
直到20世纪40年代,Flory 才利用统计力学的方法建立了超支化聚合物的相关理论[7]。他指出AB x (x ≥2)型单体的缩聚反应将生成可溶性的高度支化的聚合物,并预测,由于它们的高度支化结构,这类高分子具有较宽的分子量分布,分子链之间没有缠结,而且不能结晶,相关结果被写进了他的著作《高分子
化学原理》一书中。但是由于这类聚合物缺乏优良的力学性能,不能像普通聚合物那样用于制备现代材料,多年来没有能够引起科学家们的兴趣。
尽管超支化聚合物的出现距今已有一百多年的历史,但是其受到人们广泛关注却是在最近的二十年间。1985年T omalia[8]和Newkome等[9]分别发表了有关树形大分子的最初文章。树形大分子一出现,很快就成为高分子领域的研究热点。最初发表的关于树形大分子的工作主要集中在完美单分散树形大分子的合成上,这些具有精确结构的分子虽然表现出非常有趣的性质,但其合成常常是非常耗时费力的,研究工作应用困难重重。杜邦公司实验部门的研究人员很快认识到这一点,K im和Webster研究将树形大分子作为流变学改性剂和球形多官能团引发剂,开发出一步法合成的支化聚苯。这种聚合物具有多分散性,在线性链段的形成过程中存在缺陷,有高度支化的树枝状结构[10,11]。K im和Webster将其命名超支化聚合物(Hyperbranched P olymer),并于1987年申请了第一项关于超支化聚合物的专利。此后,由于人们对其独特的结构与性能的逐渐重视,使得超支化聚合物的合成和物性研究逐渐成为热点。
2 超支化聚合物制备方法
211 AB x型单体缩聚
(x≥2)型单体通过缩聚反应合成。
超支化聚合物最初的合成方法基于Flory的理论模型,即使用AB
x
其基本要求是:官能团A和B之间可相互反应,但是自身之间不会反应;官能团A和B的反应活性不随反应进行而变化;分子内不会发生环化反应。基于这一理论模型和合成方法,人们合成了大量的超支化聚合物,如聚苯[10,11]、聚醚[12,13]、聚酯[14]、聚酰胺[15]、聚氨酯[16]、聚醚酮[17]、聚醚砜[18]、聚碳酸酯[19]、聚硅氧烷[20]等,这极大地丰富了超支化聚合物的家族种类。
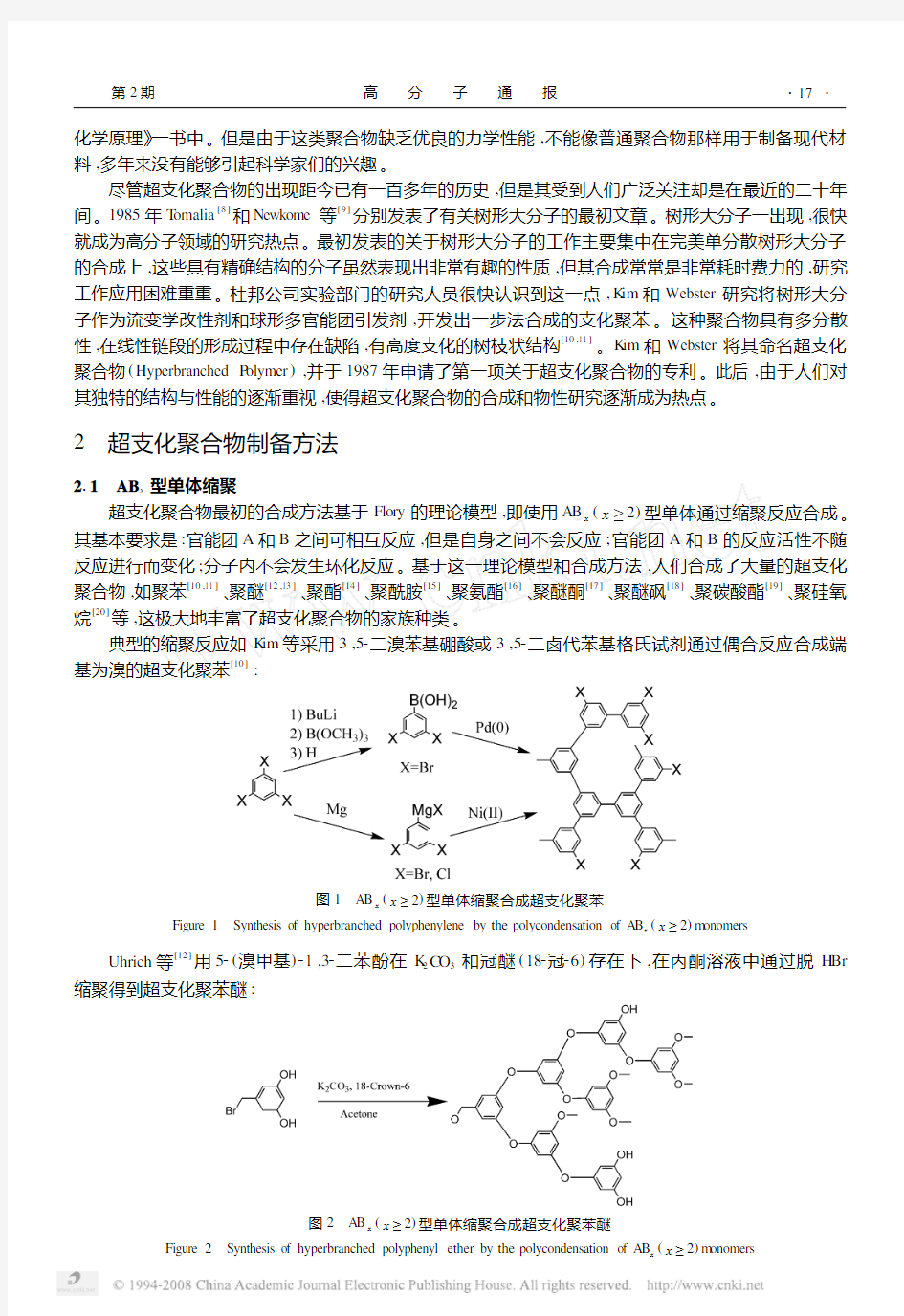
典型的缩聚反应如K im等采用3,52二溴苯基硼酸或3,52二卤代苯基格氏试剂通过偶合反应合成端基为溴的超支化聚苯[10]:
(x≥2)型单体缩聚合成超支化聚苯
图1 AB
x
Figure1 Synthesis of hyperbranched polyphenylene by the polycondensation of AB x(x≥2)m onomers Uhrich等[12]用52(溴甲基)21,32二苯酚在K2C O3和冠醚(182冠26)存在下,在丙酮溶液中通过脱H Br 缩聚得到超支化聚苯醚:
(x≥2)型单体缩聚合成超支化聚苯醚
图2 AB
x
Figure2 Synthesis of hyperbranched polyphenyl ether by the polycondensation of AB x(x≥2)m onomers
在所有合成超支化聚合物的反应中,AB
x型单体是最符合Flory的早期设想的,通过设计合成新型的AB x型单体,可以有效地得到预期功能和用途的超支化聚合物,一直是合成新型超支化聚合物,拓展超支
化聚合物应用领域最有力的工具。或者基于树枝状大分子的研究,利用其相对简单的合成步骤,或者基于线性分子的研究,利用其良好的流变性能,超支化聚合物的种类一直不断地被扩充。
芳杂环线形聚合物因为其出色的热性能以及医学、光学、电学等特性而受到人们关注,但是加工的困
难阻碍了其进一步的推广应用。设计合成新型的AB
2型单体,再由此合成出超支化聚芳杂环可以有效地解决其加工上的问题。Back等[21]合成了三官能团的化合物1,在多聚磷酸溶液中通过缩聚合成了超支化喹喔啉-苯并唑聚合物,该聚合物能够溶解于大部分的极性非质子溶剂和酸性溶剂,进一步的末端修饰可以赋予其多种功能性。
图3 两种合成超支化聚合物的ABx型单体
Figure3 T w o kinds of ABx type m onomers for the synthesis of hyperbranched polymers Li等[22]合成了一种新型的AB2型单体2(BP BA),通过缩聚反应制备了完全的芳香超支化聚合物,超
支化聚苯并咪唑(HP BI)。基本保留了相应线形聚合物的耐高温性能,T
g保持在318~381℃之间,同时超支化结构赋予其良好的加工性能。研究表明末端基团对超支化产物的溶解性、稳定性等有显著的影响。
Che等[23]在众多线形和树枝状偶氮类大分子聚合物的基础上,通过偶氮偶联反应制备了一系列超支化偶氮类聚合物3。末端偶氮类生色团的引入可以明显改变聚合物的热性能、光谱特征和表面浮雕光栅行为。这类超支化偶氮类聚合物在诸如可逆光学数据存储、光控开关、传感器,以及其它光控器件中均有潜在的应用价值。
图4 偶氮类超支化聚合物的合成
Figure4 Synthesis of hyperbranched azo2polymers
AB x型单体合成超支化聚合物的反应并不复杂,结果也相当理想,并且不会产生凝胶,而避免凝胶正是后来发展出的A
+B3型单体合成超支化聚合物面临的主要难题。AB x型单体合成超支化聚合物最大2
的缺陷来自于单体的合成,因为AB
x型单体目前缺乏商业化的产品,因此制备超支化聚合物一般需要先制备出相应的单体,而新型的AB
型单体的制备通常需要经过多步的反应,合成和纯化的过程都较为繁
x
琐,如上例中BP BA的合成中间经历了六个步骤,导致其最终的产率不足10%。单体合成的困难导致了
AB x型单体法进一步的应用受到了很大的限制。
212 开环聚合
另一种合成超支化聚合物的方法是环状单体的开环聚合(又称为Multibranching Ring2opening P olymerization,M BP or M BROP)。作为一种简单易行的合成超支化聚合物的方法,用于开环聚合的单体本身并不含有支化点,支化点产生于增长反应的过程中,因此,也可以被认为是一种潜在AB
x型单体。
1992年,Suzuki等[24]首先报道了以52亚甲基222全氢化21,32 嗪为单体,在25℃,Pd催化下,苯胺引发脱羧开环聚合反应,制得了超支化聚合物。此后这方面的研究逐渐展开,各种环状单体包括氧环丁烷[25]、THF[26]、内酯[27]、环氧乙烷[28,29]等都被用于超支化聚合物的合成。
典型的开环聚合如Frey等[28]利用缩水甘油进行的阴离子聚合(见图5):
图5 缩水甘油的阴离子开环聚合
Figure5 The anionic ring2opening polymerization of glycidol
他们同时利用所合成的缩水甘油超支化聚合物作为亲水性客体的分子胶囊,引起了广泛的关注。环氧类单体开环聚合合成的超支化聚合物因为具有良好的生物相容性或者生物可降解性,而在生物材料等领域有着巨大的潜在应用价值。
Imai等[30]致力于脱水糖的开环聚合,成功地合成了一大类超支化糖类聚合物,扩展了多聚糖的家族。所用的单体包括1,2:5,62二缩水2D2甘露醇,1,62缩水2β2D2吡喃甘露糖,1,62缩水22β2D2吡喃葡萄糖,1,62缩水2β2D2吡喃半乳糖等,所得超支化聚合物表现出高的水溶性和很低的水溶液粘度。此外,他们还利用缩水赤藻糖醇和缩水苏糖醇合成了一类超支化聚丁糖醇,进一步扩充了超支化多聚糖的种类。
Y u等[31]使用一种简单的方法合成了羟基化的1,42二氧杂环己222酮(DON)单体4:62羟甲基21,42二氧杂环222酮(62H DON)。使用62H DON合成的超支化聚合物因为重复单元由甘油和羟基乙酸组成,因此可能具有生物可降解性和生物相容性;其末端含有大量的羟基,可以做进一步的表面修饰。Parzuchowski 等[32]随后报道了52H DON的开环聚合,所得的超支化聚合物含有大量的伯羟基。大量的羟基官能团的存在能够明显改善其亲水性,并可以通过进一步的修饰使得到的超支化聚合物成为一种有前景的药物传输材料。
图6 几种开环聚合的单体
Figure6 Several m onomers for ring2opening polymerization
因为开环聚合单体的特殊性,其合成的聚合物通常具有强的亲水性,对末端官能团的修饰可以得到体型的两亲性聚合物。Mai等[33]以32乙基232羟甲基2氧环丁烷(EH O,6)的超支化聚合物PEH O为核,以线形聚环氧丙烷PPO为臂,设计合成了新型的两亲性超支化多臂醚共聚物。共聚物呈三维结构形态,能
自组装形成大的三维胶束(直径>120nm)。PPO臂与PEH O的摩尔比对胶束的大小,以及产物的T
g 、T
d
等都有影响。所得到的PEH O2star2PPO胶束表面带有大量的羟基,因此在有机功能化、分子纳米胶囊、中尺度空心球制备模板等领域都有着潜在的应用价值。
开环聚合能够制备一些AB
x型单体和AB 3型单体无法制备的超支化聚合物,尤其是制备超支化的
生物可降解材料和生物相容性材料,具有巨大的潜在应用价值。
213 自缩合乙烯基聚合
1995年Frechet等[34]报道了一种新的合成超支化聚合物的方法,称之为自缩合乙烯基聚合(Self2 condensing Vinyl P olymerization,SC VP)。所用单体既非AB x型单体,也非潜在AB x型单体,而是被称为AB 3型的一类新单体。这类单体包含一个乙烯基基团(A)和一个能够引发乙烯基聚合的活性基团B3。活性基团可以为自由基、阳离子或者阴离子。反应中活性基团B3引发单体中含有乙烯基的A单元增长,增长过程中活性位点并不随反应消失,而是迁移形成新的活性位点,并且能够继续引发乙烯基聚合,从而生成超支化聚合物。
基本反应过程如下图所示:
图7 自缩合乙烯基聚合的反应过程示意图
Figure7 Schematic representation for the reaction process of SC VP
SC VP最早的实例来自32(12氯乙基)苯乙烯的阳离子聚合[34]:
图8 32(12氯乙基)苯乙烯的阳离子聚合
Figure8 The cationic polymerization of32(12chloroethyl)styrene
进一步的研究工作将活性聚合引入SC VP中。Hawker等[35]利用稳定自由基聚合(TE MPO)制备了超支化聚苯乙烯。G aynor等[36]将原子转移自由基聚合(ATRP)反应引入SC VP中,使用商业化的产品对氯甲基苯乙烯(C MS)制备了超支化的聚对氯甲基苯乙烯。Frechet等[37]随后对同一体系进行了更详细的研究。反应的历程如图9所示。陈欢等[38]对这一体系制备超支化聚合物过程中的有关问题进行了进一步的探讨。
AB3型单体与AB线形单体的共聚合能够控制产物的支化度[39],并赋予超支化聚合物新的性能,同时利用ATR2SC VP反应活性聚合的特征进行AB3型单体与线形单体的共聚合,可以合成出大量结构可控
图9 对氯甲基苯乙烯ATRP反应合成超支化聚合物
Figure9 The synthesis of hyperbranched polymer by the ATRP of42(chloromethyl)styrene 的、新颖的聚合物。
含氟超支化聚合物最早由AB
x型单体合成[40],其保留了大量线形含氟聚合物出色的性质,如低摩擦
系数和低表面能等,同时获得了更好的加工性。Wang等[41]使用氯三氟乙烯与对氯甲基苯乙烯共聚得到
新的含氟超支化聚合物,其中含氟的单元的含量可以达到50%。Cheng等[42]使用2,3,4,5,62五氟代苯乙烯作为共聚合的线形单元,与对氯甲基苯乙烯(C MS)或对溴甲基苯乙烯(BMS)共聚,合成了一系列共聚物,同时研究了两种AB3型单体以及不同单体配比对共聚物支化度和热性能的影响。
马来酰亚胺是一种强的受电子单体,能与苯乙烯等强供电子单体形成电子转移络合物,并可通过自由基引发形成交替共聚物。计兵等[43]以C MS作为引发剂单体,通过ATRP引发N2苯基马来酰亚胺(NPMI)与C MS进行SC VP共聚合,在单体比例f NP MI=014~017时,得到了分子量可控的超支化交替共聚物。并以此交替共聚物为大分子引发剂,通过ATRP进一步引发甲基丙烯酸甲酯(M M A)聚合,得到了多臂超支化接枝共聚物。
利用双单体或多单体方法进行自缩合乙烯基聚合制备超支化聚合物的研究也在不断地取得进展。Baskaran等[44]利用二乙烯基苯和1,32二异丙烯基苯原位生成AB3型单体的方法成功地制备了超支化聚合物。Isaure等[45]使用乙二醇作为支化单元成功合成了超支化聚甲基丙烯酸甲酯。Ren等[46]使用二乙烯基苯作为支化单元,二乙烯基苯、(12溴乙烯)苯与苯乙烯共聚合成了超支化聚苯乙烯。二乙烯基苯的加入量为苯乙烯的1Π5~1Π30,共聚单体的加入量和催化剂的使用量对产物的支化度,分子量等均有显著影响。
与AB
x
型单体缩聚相比,自缩合乙烯基聚合将超支化聚合物的合成扩展到了乙烯基单体的应用领域,并且赋予超支化聚合物以C—C骨架,从而获得了比杂原子骨架更好的稳定性。另外,SC VP可以制备
分子量更大的超支化聚合物,且AB3型单体的制备和纯化都较AB
x型单体容易。
AB3型单体制备超支化聚合物也面对一些难题。首先,因为链的增长端和引发端的活性不同,导致
聚合物的支化度较低,而不像AB
x型单体可以制备出高支化度的聚合物
(部分体系甚至可以接近100%[47])。此外,SC VP合成超支化聚合物的过程中的副反应容易导致凝胶,这在几种利用ATRP方法合成的例子中尤其明显。凝胶将导致最终产物的产率降低,甚至得不到需要的产物。这两点正是相关研究中致力解决的问题。
214 A2+B3型单体缩聚
如前所述,超支化聚合物相对于树枝状大分子最显著的优势在于其一步法合成的便捷。因此寻找更简便的合成方法一直是超支化聚合物合成中的一个重要内容。AB
x型单体卓越的单体设计能力并不能掩盖其单体合成的复杂,寻找现有商业化的产品直接合成超支化聚合物是最理想的方案。
1999年K akim oto[48]和Frechet[49]的实验小组各自独立地报道了一种新型的合成超支化聚合物的方
法,即使用A
2
+B3型单体的缩聚。一般的,A2+B3型单体缩聚反应会发生凝胶,是制备热固性塑料的经典反应,尽管很早就被用来合成通用的高分子材料,但是直到最近人们才意识到在凝胶点出现之前,体系中呈现的高度支化的结构实际上就是超支化聚合物。
Aharoni[50]在20世纪90年代初首先研究了芳香基二胺(A2)和芳香基三羧酸(B3)的反应,得到了一种网状交联的产物。K akim oto等对其作了进一步的研究,使用三苯基亚磷酸盐和嘧啶作为反应介质,在低
浓度(0121m olΠL,313wt%)下得到了无凝胶的可溶性产物,其中A
2、B3单体按照1∶1的比例投料;减少三苯
基亚磷酸盐的用量,或者提高单体的用量,或者A
2、B3单体按照3∶2的比例投料均会产生凝胶。
Frechet等使用1,2,7,82二环氧基辛烷(A2单体)和1,1,12三羟甲基乙烷(B3单体)成功地合成了超支化聚醚,在反应接近凝胶点的时候将反应瓶从油浴中移出,结束反应,即可得到可溶的超支化聚合物。
相对于合成所需的AB
x 型单体,A
2
+B3型单体更容易找到相应的商业化产品,因为A2型单体大量
的用于线性聚合物的制备,B
3型单体也大量地被当作交联剂使用。A2+B3型单体合成超支化聚合物中的主要问题就在于如何控制反应条件使之不产生凝胶。
Flory提出的A2+B3型单体反应生成交联聚合物的理论模型中,其理想的反应条件是:(1)无分子内环化反应的竞争,链增长中无终止;(2)所有的A基团活性相等,所有的B基团活性也相等;(3)反应只在A、B基团之间进行。
一般而言,针对交联固化生成的条件,避免凝胶的实施方法主要有以下三种方法[51]:
(1)在凝胶点之前结束反应[48,52]。这是最直接、最有效的避免交联固化的途径;
(2)在稀溶液中聚合[53],环化反应能有效避免凝胶[54]。即给出偏离经典交联固化理论模型的反应体系;
(3)在反应体系中缓慢添加单体[55]。
Choi等[56]介绍了一种新的方法,利用两种单体在溶剂中不同的溶解度,同时加入的两个组分一个完全溶解,另一个部分溶解,随着反应的进行,溶解性较差的组分因为反应的消耗而逐步溶解进入反应体系,实现了自动缓慢地添加单体,有效地避免了凝胶化的发生。
由于A
2
+B3型单体的诸多优点,现已成为超支化聚合物合成研究中的热点之一[57,58]。此外,添加另
外的双官能团的单体进入反应体系,即构成A
2
+A2’+B3体系,被认为是控制超支化聚合物结构,尤其是支化度的有效方法。
线形聚芴因为优异的光电性能而受到关注,但是难溶难熔的性质限制了其应用范围。虽然双单体合成的超支化聚芴在一定程度上解决了线形聚芴的难题,但是因为支化度较高,在一般的有机溶剂中的溶解性能依然不理想。Tsai等[59]基于suzuki反应,设计使用了下图7、8、9三单体组成的体系,通过改变各单体的当量比合成了一系列低支化度的超支化聚芴,并且可以溶解于氯仿等常见的有机溶剂。
215 偶合单体法
尽管A
2
+B3型单体合成超支化聚合物克服了AB x型单体商业化原料较少的主要缺陷,使合成的路线得到了很大的简化,但是为了克服凝胶化的产生,反应通常被设计成缓慢的过程,或者在极稀的溶液中进行,或者严格在凝胶点之前结束反应,在一定程度上限制了其大规模工业化的应用前景。
为了解决A
2
+B3型单体面临的困境,另一种新的合成路线被提出,称之为偶合单体法[5](C ouple2 m onomer Methodology,C M M)。根据Flory的理论以及大量通过AB x型单体合成超支化聚合物的实例可以
知道,AB
x型单体在反应的过程中并不产生凝胶。因此在A2+B3型双单体工艺的基础上,综合AB2型单
图10 A
+A2’+B3型单体体系合成超支化聚芴
2
Figure10 The synthesis of hyperbranched poly fluorene from A2+A2’+B3type m onomers
体法合成的优点,通过寻找具有不同反应活性的反应性基团的单体,在反应体系中原位生成AB
2型单体,
再通过AB
2型单体的进一步反应生成超支化聚合物,理论上讲能够有效地避免凝胶的产生。
颜德岳[60]和Froehling[61]的研究小组各自独立地报道了这一合成方法,可简单地标记为AA’+B’B
2型单体反应体系。实验证明,这类单体的反应完全可以在不产生凝胶的情况下完成超支化聚合物的制备。目前通过偶合单体方法成功制得的超支化聚合物已经包括聚芳醚[62]、聚乙氧基硅烷[63]、聚(酯2胺)[64]、聚酰胺2胺[65]、聚胺[66]、聚砜胺[5]、聚(脲2氨酯)[5]、聚(酯2酰胺)[5]等多种类型。
图11 偶合单体法合成超支化聚合物
Figure11 Synthesis hyperbranched polymers by couple2m onomer methodology
反应的关键在于寻找合适的单体对,其中A、A’可以是同一基团,如图12所示,利用偶合单体法合成超支化聚(酯2胺)[64],其中A2型单体10和C B2型单体11于低温下在DMF溶液中发生羧酸和氨基的反
应,生成典型的AB
2型单体,之后除去DMF,升温缩聚,即可制得无交联的超支化聚合物;或者A、A’也可以是反应活性不相同的两个基团,如图13所示,利用迈克尔加成反应生成超支化聚酰胺2胺[65],AA’型单
体13中两个氨基氢的反应活性不同,与B
3型单体12反应后原位生成AB2型的中间产物14;相应的,官能团B、B’也是如此。改变单体的投料比可以影响产物的分子量和末端官能团的比率。
图12 偶合单体法合成超支化聚(酯2胺)
Figure 12 The synthesis of hyperbranched poly (ester 2amide )s by C M M
图13 偶合单体法合成超支化聚酰胺2胺
Figure 13 The synthesis of hyperbranched poly (amido amine )by C M M
3 结语
自1987年至今,超支化聚合物已经经历了二十年的发展,其间不断涌现的新合成方法和大量新材料的合成相互促进,在此基础上出现的大量新型超支化聚合物极大地丰富了高分子材料的家族[67~70]
,而特殊的性能和工业化的良好前景亦使其受到越来越多的关注。超支化聚合物良好的溶解性能、低粘度、低链缠结、多官能团等本体性质不仅使其在传统高分子工业上有巨大的应用前景,更使其在新兴的领域,诸如纳米技术、药物传输、超分子化学、传感器等方面有着诱人的应用前景。
从合成的角度看,AB x 型单体开创了超支化聚合物合成的先河,开环聚合则将其扩展到了环状化合物的领域,SC VP 成功地将乙烯基单体应用到了超支化聚合物的合成中来,发展出了新型的AB 3型单体法。A 2+B 3型单体合成法的开发应用和有效的控制又将超支化聚合物的工业化前景向前推进了一大步。偶合单体法则是综合了先前数种合成方法的优点发展起来的新方法,利用与开环聚合类似的原位生成AB x 单体的方法,结合A 2+B 3型单体和AB x 单体各自的优点,同时在一定程度上克服了各自的缺点,将超支化聚合物的研究推向了一个新台阶。
从聚合机理上看,缩聚反应作为超支化聚合物的合成中最成功的方法,一直是超支化聚合物制备的主要手段;开环聚合和自缩合乙烯基聚合等合成方法则将自由基聚合、阴离子聚合、阳离子聚合等其它传统聚合方法成功地应用于超支化聚合物的合成,不仅扩展了超支化聚合物的合成方法,而且丰富了超支
化聚合物的种类。另一方面,新的聚合实施方法也在不断地用于超支化聚合物的合成[71]。
在现有合成方法下,开发新型单体、制备不同结构和功能的超支化聚合物仍将是超支化聚合物合成中的重点问题。在此之外,对超支化聚合物结构的控制越来越成为超支化聚合物合成中的关键。早期的研究已经表明对其大量的末端官能团的修饰可以有效地控制其诸多性质,如玻璃化转变温度、溶解性、两亲性等;随后的研究工作则大量集中在对其分子量、分子量分布,尤其是支化度等参数的控制上,实验证明这些参数对于超支化聚合物的性能有很大的影响。对结构和性能的控制,结合大量对超支化聚合物潜
在应用领域的开发,使得人们对其大规模应用持有乐观的看法[72,73]。
参考文献:
[1] Hult A,Johanss on M,M almstrom E.Advances in P olym,1999,143:1~341
[2] Jikei M,K akim oto M.Progress in P olym Sci,2001,26:1233~12851
[3] K ainthan R K,Muliawan E B,Hatzikiriakos S G,et al.M acrom olecules,2006,39:7708~77171
[4] Y ates C R,Hayes W.Eur P olym J,2004,40:1257~12811
[5] G ao C,Y an D.Progress in P olym Sci,2004,29:183~2751
[6] 谭惠民,罗运军.超支化聚合物.北京:化学工业出版社,20051
[7] Flory P J,J Am Chem S oc,1952,74:2718~27231
[8] T omalia D A,Baker H,Dewald J,et al.P olym J,1985,17:117~1321
[9] Newkome G R,Y ao Z,Baker G R,et al.J Org Chem,1985,50:2003~20041
[10] K im Y H,W ebster O W.P olymer Preprints,1988,29,(2):310-3111
[11] K im Y H,W ebster O W.J Am Chem S oc,1990,112:4592-45931
[12] Uhrich K E,Hawker C J,Frechet J M J,et al.M acrom olecules,1992,25:4583~45871
[13] K ou H,Asif A,Shi W,et al.P olymers for Advanced T echnologies,2004,15:192~1961
[14] Blencowe A,Davids on L,Hayes W.Eur P olym J,2003,39:1955~19631
[15] Jikei M,K akim oto M.J P olym Sci,Part A:P olym Chem,2004,42:1293~13091
[16] H ong L,Cui Y,W ang X,et al.J P olym Sci,Part A:P olym Chem,2002,40:344~3501
[17] F ossum E,T an L.P olymer,2005,46:9686~96931
[18] F ossum E,H immelberg P.J P olym Sci,Part A:P olym Chemi,2005,43:3178~31871
[19] Bolton D H,W ooley KL.J P olym Sci,Part A:P olym Chem,2002,40:823~8351
[20] S i Q,W ang X,Fan X,et al.J P olym Sci,Part A:P olym Chem,2002,43:1883~18941
[21] Baek J,S imko S R,T an L.M acrom olecules,2006,39:7959~79661
[22] Li Z X,Liu J H,Y ang S Y,et al.J P olym Sci,Part A:P olym Chemi,2006,44,5729~57391
[23] Che P,He Y,W ang X.M acrom olecules,2005,38:8657~86631
[24] Suzuki M,Ii A,Saegusa T.M acrom olecules,1992,25:7071~70721
[25] Y an D,H ou J,Zhu X,et al.M acrom ol Rapid C ommun,2000,21:557~5611
[26] Imai T,Satoh T,K aga H,et al.M acrom olecules,2003,37:3113~31191
[27] Albertss on A,Varma I K.Biomacrom olecules,2003,4:1466~14861
[28] Sunder A,Hanselmann R,Frey H,et al.M acrom olecules,1999,32:4240~42461
[29] 朱宝库,赵永红,孔力,等,高分子通报,2007,4:23~28
[30] Imai T,Nawa Y,K itajy o Y,et al.M acrom olecules,2005,38:1648~16541
[31] Y u X,Feng J,Zhuo R.M acrom olecules,2005,38:6244~62471
[32] Parzuchowski P G,G rabowska M,T ryznowski M,et al.M acrom olecules,2006,39:7181~71861
[33] M ai Y,Zhou Y,Y an D.M acrom olecules,2005,38:8679~86861
[34] Frechet J M J,Henmi M,G its ov I,et al.Science,1995,269:1080~10831
[35] Hawker C J,Frechet J M J,G ubbs R B,et al.J Am Chem S oc,1995,117:10763~107641
[36] G aynor S G,Edelman S,M atyjaszewski K.M acrom olecules,1996,29:1079~10811
[37] W eimer M W,Frechet J M J,G its ov I.J P olym Sci,Part A:P olym Chemi,1998,36:955~9701
[38] 陈欢,王国建.高分子材料与工程,2005,21(4):59~621
[39] S im on P F W,Muller A H E.M acrom olecular Theory and S imulations,2000,9:621~6271
[40] Pitois C,W iresmann D,Lindgren M,et al.Advanced M aterials,2001,13:1483~14871
[41] W ang W,Y an D,Bratton D,et al.Advanced M aterials,2003,15:1348~13521
[42] Cheng C,W ooley KL,K hoshdel E.J P olym Sci,Part A:P olym Chem,2005,43:4754~47701
[43] 计兵,刘翠华,杨金田,等.化学学报,2006,64(6):556~5621
[44] Baskaran D.P olymer,2003,44:2213~22201
[45] Isaure F,C ormack P A G,G raham S,et al.Chem C ommun,2004,9:1138~11391
[46] Ren Q,G ong F,Liu C,et al.Eur P olym J,2006,42:2573~25801
[47] Fu Y,Vandendriessche A,Dehaen W,et al.M acrom olecules,2006,39:5183~51861
[48] Jikei M,Chon S H,K akim oto M,et al.M acrom olecules,1999,32:2061~20641
[49] Emrick T,Chang H T,Frechet J M J.M acrom olecules,1999,32:6380~63821
[50] Aharoni S M.M acrom olecules,1991,24:235~2391
[51] Unal S,Long T E.M acrom olecules,2006,39:2788~27931
[52] W ang D,Liu Y,H ong C,et al.J P olym Sci,Part A:P olym Chem,2005,43:5127~51371
[53] Unal S,Lin Q,M ourey T H,et al.M acrom olesules,2005,38,3246~32541
[54] K richeldorf H R,Vakhtangishvili L,Fritsch D.J P olym Sci,Part A:P olym Chem,2002,40:2967~29781
[55] Lin Q,Long T E.M acrom olesules,2003,36:9809~98161
[56] Choi J,T an L,Baek J.M acrom olesules,2006,39:9057~90631
[57] 车鹏超,和亚宁,王晓工.高分子学报,2007,1:21~251
[58] 王海侨,王换方,卢红斌,等.高等学校化学学报,2007,2:388~3901
[59] Tsai L,Chen Y.M acrom olecules,2007,40:2984~29921
[60] Y an D,G ao C.M acrom olecules,2000,33:7693~76991
[61] Froehling P,Brackman J.M acrom olecular Sym posia,2000,151:581~5891
[62] K im YJ,K akim oto M,K im S Y.M acrom olecules,2006,39:7190~71921
[63] Zhu X,Jaumann M,Peter K,et al.M acrom olecules,2006,39:1701~17081
[64] Li X,Lu X,Lin Y,et al.M acrom olecules,2006,39:7889~78991
[65] W ang D,Zheng Z,H ong C,et al.J P olym Sci,Part A:P olym Chem,2006,44:6226~62421
[66] Liu J H,Ren C,Y ang Z,et al.J P olym Sci,Part A:P olym Chem,2007,45:699~7081
[67] 易昌凤,李全涛,徐祖顺.化学进展,2007,19(2Π3):356~3611
[68] 侣庆法,范晓东,王生杰,等.高分子通报,2006,6:44~561
[69] 李爱香,鲁在君,谭业邦,等.高分子通报,2005,2:6~131
[70] 赵雅青,董炎明,李 ,等.化学进展,2005,17(5):868~8751
[71] Blencowe A,Caiulo N,C osstick K,et al.M acrom olecules,2007,40:939~9491
[72] V oit B.C om ptes Rendus Chimie,2003,6:821~8321
[73] V oit B.J P olym Sci,Part A:P olym Chem,2005,43:2679~26991
Development of I nvestigation on the Synthesis of H yperbranched Polymers
ZH ANG Chuan2hai a,LI Hua2yi b,ZH ANG Ming2ge b,ZH ANGLiao2yun a3
(a Graduate Univer sity o f Chinese Academy o f Sciences,Beijing100049,China
b Beijing National Laboratory for Molecular Science(BNLMS),Joint Laboratory o f Polymer Science and Materials,
K ey Laboratory o f Engineering Plastics,Institute o f Chemistry,Chinese Academy o f Sciences,Beijing100080,China)
Abstract:Hyperbranched polymers are one type of dendritic macrom olecules which can be synthesized by one2 step polymerization process.Because of the special structures and properties,as well as the possibility of large2scale production,twenty years of research on hyperbranched polymers have made it become one of the m ost im portant kinds of polymer materials rapidly,which have broad application potentials.This paper reviews the main synthetic methods of hyperbranched polymers and the developing history from the aspects of m onomers,which inv olving ABx m onomers, AB3m onomers,A2+B3m onomers,and the latent AB x m onomers(including ring2opening polymerization and couple2 m onomer methodology),etc.The excellence and the limiting of each method are als o introduced.
K ey w ords:Hyperbranched polymers;Synthetic method;C ondensation polymerization;Self condensation vinyl polymerization
超支化聚合物阻垢剂
一种新型超支化聚合物阻垢剂 摘要 在超支化聚乙烯亚胺中添加阴离子乙烯基单体,乙烯基磷酸、乙烯基磺酸、丙烯酸、马来酸和丙烯三羟酸来制备一系列聚合物,并对其作为防止碳酸钙和硫酸钡沉积的阻垢剂的性能进行研究。使用高压管阻塞设备对其在1200磅和100℃条件下进行动态力学测试,发现这些新型阻垢剂可以抑制碳酸盐和硫酸盐结垢,其中丙烯酸类共聚物对碳酸盐垢效果最好,膦酸基类共聚物对硫酸盐垢效果最好。 此前还没有关于超支化聚乙烯亚胺在海水中生物降解数据的报告。用 OECD306测试技术对分子量为300和1200的聚合物进行测试,得到了在28天时对海水的生物降解率分别是10%和19%,马来酸或丙烯酸功能化的分子量为1200的超支化聚乙烯亚胺表现出了很高的生物降解率,在28天内可以达到34%,到60天可以升高到60%。这反映了细菌对烯烃基羧酸盐组分的攻击和消化比对胺基聚合物骨干更容易。 关键词:垢,晶体生长,石油,阻垢剂,聚合物 1、前言 结垢通常定义为无机盐在水溶液中的沉积。在上游石油天然气工业中,水垢最常见的组分是碳酸钙和硫酸锶/硫酸钡(Sallis 等,1995;Frenier、Ziauddin,2008;Kelland,2009;mjad, 2010)。结垢是石油天然气工业中的一个主要问题,垢对油井和管道的阻碍和堵塞会导致生产中显著的延迟和损失。多种带有功能组分的水溶性分子或水溶性高分子化学药剂被用作阻垢剂来防止结垢,其中最常见的功能组分就是膦酸盐、羧酸盐和磺酸盐。高分子和低分子膦酸盐都是有效的阻垢剂,但有效的油田阻垢剂只有带有多个羧酸或磺酸基团的高分子。 氨基膦酸盐是最常见的非高分子类膦酸基油田用阻垢剂,图1所示是两个例子,包括最常见的氨基膦酸盐类油田用阻垢剂二乙烯三胺五甲叉膦酸(DTPMPA)(Stewart 、Walker,2003;Tomson等, 2003; Sorbie、Laing, 2004)。高分子膦酸盐也是熟知的阻垢剂但是由于环保特性差在北海地区并不使用,这主要
树形、超支化聚合物的研究进展
树形、超支化聚合物的研究进展 董璐斌 (天水师范学院化学系,甘肃天水,741000)摘要:随着社会的高度发展,对原材料的性能提出了越来越高的要求,也推动了新型高分子化合物和新材料的发展。树形、超支化聚合物由于其独特的分子结构和物理化学性质使之在众多领域有着广泛的用途。故本文对树形、超支化聚合物的应用研究进展进行综述。 关键词:树枝状聚合物;超支化聚合物;应用;进展 树形聚合物和超支化聚合物为高度支化的聚合物,性质的独特性,引起了众多领域科学家的广泛关注,在此主要介绍树形聚合物在超分子化学、生物医学、光化学与电化学、催化剂等领域的研究进展;超支化聚合物在热固性树脂增韧剂、染色助剂、缓释剂、超支化液晶、涂料及聚合物薄膜方面的应用研究进展。 一、树形聚合物的应用研究进展 1、超分子化学 由于树形聚合物的结构、尺寸、表面和内部的官能团种类及数目等分子参数都可以精确控制,使得其非常适合作为超分子体系的构筑单元和研究超分子体系的模型,因此,从树形聚合物的出现开始就在超分子领域引起了极大的兴趣。 Cardulls等合成了一种两亲的C60树枝状聚合物,并在空气-水界面上形成了单分子层的L2B膜。C60树枝状聚合物共轭体系是由富勒烯二酸合成的。这种膜有可能应用于光学技术或生物传感器领
域。 Crooks等用在金箔表面重复沉淀的方法,通过第四代的聚酰胺2胺树形聚合物(PAMAM)与马来酸酐-甲基乙烯基醚共聚物自组装成渗透选择性膜,该膜对外部刺激、pH值变化具有响应性。此膜作超分子“门”的功能是pH的函数:在低pH值时阴离子容易穿透而阳离子被排除在外;在高pH值时,结果相反。 2、生物和医学 树形聚合物的大小、内部空腔和表面管道决定了它可以作为蛋白质、酶和病毒理想的合成载体,再加上它们很容易进行官能化作用,树形聚合物在很多与生物和医学相关的领域都得到了应用。这些领域包括药物载体、基因载体、DNA生物传感器、硼中子俘获治疗试剂、核磁共振造影剂、免疫制剂等。 Roy和Zanini等在糖型树形聚合物方面进行了部分研究工作。他们合成的L2赖氨酸树形聚合物能有效的抑止红血球的凝聚。这一点已通过流感A病毒试验证实。 硼中子俘获治疗(BNCT)是一种最新治疗癌症的方法。在这种疗法中,低能中子与10B核子进行的核裂变反应所产生的能量及细胞毒素用来破坏恶性细胞。PAMAM树形聚合物(G2,G4)首先连接到异氰酸根络硼烷,再被接到单克隆抗体上,这样就具有了通过免疫结合来选择靶向肿瘤的能力。 树形聚合物在医学上的另一个重要应用是用作核磁共振造影剂(MRI)。它与螯合剂相连可对靶器官进行成像,以检查脑或器官血池
超支化聚合物的研究进展
超支化聚合物的研究进展 李璇 化学与环境学院 1105班 111030210 摘要超支化聚合物由于具有高度支化三维球状结构以及众多的端基的独特结构特征,与传统的线型高分子在性能上有很大差异,因而引起科学家们高度关注。本文通过对其结构、合成及应用的介绍,旨在加深人们对该领域的了解,从而促进该领域的快速发展。 关键词树枝状分子;超支化聚合物;结构特征; Progress of Hyper-branched Polymers Li Xuan (College of Chemical and Environment Class 1105 No.111030210) Abstract Hyper-branched polymers due to the unique characteristics of the highly branched three-dimensional spherical structure and a large number of end group structure, has the very big difference performance with the traditional linear polymers, which attracted the attention of scientists. This paper describes the structure, synthesis and application of hyper-branched polymers, in order to deepen the understanding of the people in this field, thus contributing to the rapid developments in the field. Key Words Dendrimer,Hyper-branched polymer,Structural characteristic 在过去的很长一段时间,聚合物化学家们发现了一种由一系列支化单元组成的树状支化大分子--新的“树状分子”,它可分为树枝状大分子和超支化聚合物两大类。树状大分子的合成为了控制分子的尺寸和形状,通常需要多步反应。而超支化高分子因其分子结构而得名,它是一种经一步法合成得到的高度支化的聚 型单体分子间的缩聚合物[1]。早在1952年,Flory[2]就首先在理论上论述通过AB x 制备高度支化大分子超支化聚合物的可能性。但是,对于这种非结晶、无链缠绕的超支化聚合物,当时并未引起足够重视。直到90年代初,Kim等[3]制备了超支化的聚苯之后,人们才开始对它产生兴趣。 1 超支化聚合物简介 1.1 超支化聚合物支化度 超支化聚合物有三种不同的重复单元,即树状单元、线型单元和由未反应的B官能团所决定的的末端单元。1991年,Fr chet 把支化度作为描述超支化聚合物结构的一个因素, 如式1 所示: 支化度(DB)=(D+T)/(D+T+L)(1) 在这里,D 代表树状单元数, T 代表末端单元数,L 代表线型单元数。 Frey 基于反应过程, 将式1 修改成如式2 所示: (2) 这里,N 是分子数。因为式(2)中的N 可被忽略, 所以式(1)和(2)给出的DB 几乎相同。
超支化材料的应用
超支化聚苯的性能及应用 超支化聚合物可以简单描述为具有高度支化结构的聚合物,他既与支化聚合物不同,也与树形分子有别。换句话说,其支化度大于支化聚合物,而小于树形分子。超支化聚合物的名称并没有像树形分子那么复杂,在文献中,见到报道的只有高支化聚合物和超支化聚合物,目前文献中已普遍采用后一名称。 超支化聚合物的制备比较简单通常不需要应用多步的保护与脱保护过程,而是采用一步法(无控制增长)和准一步法(逐步控制增长)。最常见的反应是缩聚反应,,也使用活性聚合、开环聚合、离子聚合等方式 超支化聚合物的特殊结构决定了他有与普通线形高分子不同的特殊性质。影响超支化聚合物性能的结构因素很多,包括支化结构、重复单元、端基官能团以及核结构等等,其中支化结构比化学结构对性能的影响更大。超支化聚合物的最终性能主要由支化重复单元的结构和端官能团性质决定,只有当这两个因素确定以后,才可以考虑支化度的影响,这个影响还可能会被分子量分布的影响所掩盖。 与分子量相近的线形高分子相比,超支化聚合物的溶解性有很大的提高,例如超支化聚苯和芳香族聚酰胺可溶解在有机溶剂中,而对应的线形聚合物则由于主链的刚性,在有机溶剂中几乎是不能溶解的。超支化聚合物的分子尺寸小,有大量的短支链存在,以及分子链本身及分子之间无链的缠绕使得分子间相互作用力小,因而粘度较低。 超支化聚合物的玻璃化温度不等同于一般线形高聚物的玻璃化温度,因在其结构中含有大量的端基,使其链段的运动受支化点和端基的影响更为明显,在玻璃化转变机理上也有所不同。 超支化聚合物的热稳定性同其化学结构密切相关通常芳香族的比脂肪族的更为稳定,与类似的线形聚合物相比,超支化聚合物的结构更为之谜,热膨胀系数和压缩系数较小。 超支化聚合物的表面有大量的官能团存在,端基官能度非常大,一般为12、16、32,如果保留反应活性基团,则反应活性非常高。超支化聚合物的端基被部分或全部全部活化后,可以通过进一步的反应得到类似热固性聚合物的交联网络,从而使之具有良好的力学性能。 另外,超支化聚合物具有结晶性,在胶束环境中显现出典型的胶束性质,具有良好的流动性;利用超支化聚合物的结构特点,通过适当的物理或化学手段,可以赋予超支化聚合物其他一些特殊性能,如光物理及光化学性能,吸附及解吸附性能等。
超支化聚合物增韧环氧树脂的研究进展
超支化聚合物增韧环氧树脂的研究进展 朱 超 林丽娟 (徐州建筑职业技术学院土木工程系,徐州 221008) 摘要 介绍超支化聚合物的结构及特点,着重综述了超支化聚合物增韧改性环氧树脂的研究进展,指出了超支化聚合物在环氧树脂改性方面的发展方向。 关键词 环氧树脂 超支化聚合物 增韧 改性 环氧树脂(EP)作为一种热固性树脂因具有良好的电性能、化学稳定性、粘接性、加工性等特点而被广泛应用于机械、电气电子、航天航空等领域。但纯环氧树脂的最大弱点是固化后质脆、耐冲击性较差和容易开裂,因而难以满足工程技术的要求,使其应用受到一定的限制。为了解决这些问题,需要对环氧树脂进行增韧改性,其方法包括增塑剂增韧、低分子量增韧剂增韧、热塑性树脂增韧、互穿网络聚合物(IPN )增韧、热致性液晶聚合物(TLCP)增韧、橡胶类弹性体增韧及纳米粒子增韧等[1,2]。这些增韧手段都能使环氧树脂的韧性得到较大的提高,但同时却降低了材料的耐热性、硬度、模量和电性能。而近几年出现了一种新的共混改性环氧树脂的方法,即采用超支化聚合物(HBPs)改性环氧树脂。由于超支化聚合物具有独特的性能,可以在保证提高环氧树脂韧性的同时不降低固化物的模量、耐热性等性能,故引起了人们广泛的关注。1 超支化聚合物 超支化聚合物是近10多年才出现的一种新型高分子材料,它是一种以低分子为生长点,通过逐步控制重复反应而得到的一系列分子质量不断增长的结构类似的化合物。常见的3种聚合物的结构如图1 所示。 图1 三种聚合物的结构 用超支化聚合物改性环氧树脂,初始时由超支化聚合物与环氧树脂共混形成均相体系,固化时发生相分离,由于超支化聚合物分子外层可以按要求组装官能团,这样可有效地调控环氧树脂固化物的结构和相态,为实现其改性提供了很大的空间。 2 超支化聚合物改性环氧树脂的研究进展 对于热固性的环氧树脂,其加工性能对应用有着非常重要的影响。加工时通常希望体系具有较低的粘度,使得其在固化后期能发生相分离以达到增韧的目的。但是传统增韧改性剂的分子量较高,这种高分子量往往意味着高粘度,这对加工来说是不利的。超支化聚合物具有独特的结构和良 好的相容性、低粘度等特性,所以可用作环氧树脂的改性剂。 超支化聚合物应用于增韧改性环氧树脂还具有下列优点[3]:(1)超支化聚合物的球状三维结构能降低环氧固化物的收缩率;(2)超支化聚合物的活性端基能直接参与固化反应形成立体网状结构,众多的末端官能团能加快固化速度;(3)超支化聚合物的尺寸和球状结构杜绝了在其它传统的增韧体系中所观察到的有害的粒子过滤效应,起到内增韧的作用。 2.1 超支化聚合物改性环氧树脂的固化行为 由于环氧树脂的固化行为直接影响到环氧树脂材料的制备和最终性能,所以针对超支化聚合物改性环氧树脂固化行为,人们做了大量的研究工作。 2000年韩国的Joon H ak O h 等[4]研究了超支化聚合物与环氧树脂的固化行为。他们采用差示扫描量热(D SC)仪和傅里叶变换红外光谱(FT-IR )仪等分析手段发现环氧树脂/超支化聚合物体系的固化温度比环氧树脂/线性聚合物体系的固化温度高,但环氧树脂/超支化聚合物体系的固化反应活化能较低。当超支化聚合物末端的羟基转变为苯甲酸基团时,固化反应的诱导期变得较长,并且反应热降低,整个反应级数为1.5。随着固化反应的进行,环氧基团的峰特性呈下降趋势,同时H 连接到C O 键上的峰值增加,并随着超支化聚合物含量的增加,H 与C O 连接的峰值不断增强。 日本的M.Okazak 等[5]用超支化聚酰胺多胺与有机硅接枝制得超支化聚合物。结果表明,超支化聚酰胺多胺的端 胺基促进了凝胶化反应。采用接枝有机硅为固化剂,在170e 、48h 条件下超支化聚酰胺多胺固化环氧树脂的凝胶级数达到77%,凝胶程度随其端胺基含量的增加而增加。 2003年D.R a t na 等[6]选用二乙基甲苯-2,6-二胺(DET-DA )为固化剂,使用环氧化超支化聚合物增韧双酚A 型环氧树脂。结果表明,环氧化超支化聚合物的加入对体系的固化速率没有影响。100e 时超支化聚合物与双酚A 型环氧树脂很容易混溶,固化时则发生相分离。随着固化温度的升高,分散相的超支化聚合物的含量也增加。超支化聚合物的加入使得固化物的韧性得到显著提高。 收稿日期:2006-10-12
超支化聚合物
超支化大分子的最新应用进展 超支化大分子独特的构筑使其合成与应用在世界范围内受到人们越来越多的关注。笔者对最近以来国内外超支化大分子的最新应用进行了简要的综述, 对今后超支化大分子的应用前景进行了展望和预测。 最近几年以来, 由于超支化大分子独特的构筑, 使得超支化大分子的合成与应用在世界范围内受到人们越来越多的关注。与线性大分子相比较, 超支化大分子具有内部多孔的三维结构, 表面富集大量的端基, 使超支化大分子具有较佳的反应活性。其独特的分子内部的纳米微孔可以螯合离子, 吸附小分子, 或者作为小分子反应的催化活性点; 由于具有高度支化的结构, 超支化聚合物难以结晶, 也无链缠绕, 因而溶解性、相容性大大提高; 与相同分子量的线性分子相比, 超支化分子结构紧凑( 较低的均方回转半径和流体力学半 径) , 熔融态粘度较低; 并且分子外围的大量末端基团可以通过端基改性以获得所需的性能。此外超支化大分子的合成采用一锅法, 合成方法简单, 无需繁琐耗时的纯化与分离过程, 大大降低了成本. 因此超支化聚合物独特的结构和简单的合成方法使其在许多领域中均有着广泛的应用,现将最近以来国内外超支化大分子的主要应用领域作一简要的总结与展望。 1 超支化大分子嵌段共聚物 在水溶液中具有自组织功能的两亲性嵌段共聚物由于其在生
物工程、信息材料和药物传输等领域的潜在应用前景而备受人们关注, 被人们称作 architectural copolymer!聚乙烯醇共聚物组成的胶束由于具有良好的生物相容性和溶解性而在药物载体运输( 药物缓释) 和基因转移方面具有潜在应用价值。与传统的由表面活性剂组成的低分子胶束相比较, 由大分子组成的胶束具有较低的临界胶束浓度( CMC) 和稳定性, 通过调节不同结构嵌段比例可以使某种嵌段富集于胶束的内部或外部。但是, 大分子两亲嵌段共聚物的扰曲性产生的链缠结和较宽的相对分子质量分布限制了其应用。采用内部具有高度支化结构的单分子胶束可以避免以上问题, 通过对超支化大分子表面的改性可以捕捉不同的分子, 因此此种结构的单分子胶束可以作为纳米反应器。超支化大分子..线型分子的嵌段共聚物具有两亲性自组织功能, 可以形成胶束通过对超支化大分子表面的改性可作为分子载体吸纳不同有机分子, 起纳米胶囊作用。例如富含羟基端基的极性聚酯超支化大分子可通过表面改性形成非极性烷基长链, 形成极性内核和非极性外壳的两亲性胶束结构, 可使非极性的聚烯烃吸纳极性染料等有机物质而不产生明显相分离, 通过对核/ 臂长度和结构的恰当选择可合成出具有两亲性结构的嵌段共聚物。Iyer等采用亲水性线性分子聚环氧乙烷与憎水性超支化聚( 胺/ 酰胺) 共聚合成了一种两段式嵌段共聚物, 其玻璃态转变温度Tg 取决于超支化聚合物的末端官能团的性质和数量。加入的线性聚环氧乙烷链段明显改变了嵌段聚合物的性质: 短链聚环氧乙烷共聚物分子的粘度行为和线性分子类似, 而长链聚环氧乙烷共聚物分子则通过自组装形成了单分子胶束, 这种胶束
超支化聚合物的活性聚合方法
超支化聚合物的活性聚合方法 1 前言 超支化聚合物是一类具有三维椭球状立体结构的高度支化的大分子聚合物[1],分子之间无缠结, 大量的端基暴露在最外层, 因此超支化聚合物表现出高溶解度、低粘度、化学反应活性高等特殊性能, 对其端基进行改性可得到不同特性和各种功能性的聚合物,如共混改性剂、涂料、纳米杂化材料、药物缓释、光电材料、粘合剂以及可降解聚合物等[2-4]。因此, 超支化聚合物一出现就受到了大批研究者的关注与青睐, 成为高分子科学中的热门课题之一[5-8]。超支化聚合物的飞速发展,不但增加了超支化聚合物的制备方法, 也丰富了超支化聚合物的种类[9 ]。科学家们也在不断开发和应用新型的超支化聚合物[10]。 2 超支化聚合的活性/可控自由基聚合方法 传统的自由基聚合由于其反应条件温和、形式多样化(本体、悬浮、溶液、乳液),易于制备,是合成高分子材料的主要方法。而它慢引发、快增长、易转移、链终止等反应特点使得产物的分子量和结构难以控制、分子量分布宽,还易出现支化交联等现象,严重影响了高分子材料的某些方面的性能。直至上世纪七十年代,科学家发现了碘转移自由基聚合[11](ITP),使氟烯烃的自由基聚合得以控制。经过科学家几十年的不懈努力,活性/可控自由基聚合(Control/Living Radical Polymerization,CRP)成为制备分子结构明确、分子量可控及分子量分布窄的聚合物的主要方法,已引起了学术界和工业界的极大兴趣。当前制备超支化聚合物的活性/可控自由基聚合包括原子转移自由基聚合[12-14](ATRP)、可逆加成—断裂链转移聚合[15,16] (RAFT),且他们都可以与点击化学(Click Chemistry)相结合。这些活性/可控自由基都是使增长自由基浓度降低,但链增长反应仍可进行,双基偶合和歧化反应显著减少,从而达到控制反应的目的,从而便利高效地合成各种具有预定结构的聚合物,比如嵌段、梳型、接枝、星型、超支化和环形等。 2.1 原子转移自由基聚合(ATRP) 原子转移自由基以有机卤化物为引发剂,过渡金属络合物作为卤原子载体即催化剂,在“活性种”与“休眠种”之间建立可逆的动态平衡.有效地抑制了自由基双基终止,实现多种单体的活性聚合和可控自由基聚合,最终实现对反应的控制。 Gaynor等[17]最先报道了利用ATRP制备超支化聚合物的研究成果。他们选择分子结构中含有苄基氯和聚合双键的对氯甲基苯乙烯(CMS)作为单体原料,在CuCI/2,2'-联二Ⅱtt啶(bpy)的催化体系中进行ATRP,最终得到了端基含有大量氯原子的超支化聚合物。Weimer等[18]发现只有使用大量催化剂才能制的超支化聚合物。陈云辉等[19]以CuBr/bpy作为催化剂,通过a—溴代苯乙烷引发二苯甲烷双马来酰亚胺的ATRP,可由双烯化合物原位生成自引发单体合成超支化聚合物。 原子转移自由基聚合(ATRP)利用控制自由基来控制分子结构和分子量,制备分子量分布较窄的聚合物,相对分子质量可以控制在103~105,Mw/Mn介于1.05-1.5之间。通过ATRP得到的聚合物,末端带卤素,可被其他亲核基团所取代,用来制备末端功能化的聚合物。迄今为
超支化聚合物
超支化分子(hyperbranched molecular)是最近十几年发展起来的, 在聚合物科学领域引起人们广泛兴趣的一种具有特殊大分子结构的聚合物。早在1952年, Flory就提出了可以由多官能团单体制备高度支化的聚合物。但在过去的几十年中, 高度支化的聚合物并没有引起人们的注意。直到20世纪80年代中期, 杜邦公司的瓦Kim等人有目的地合成了一种超支化聚合物, 并申请了第一项关于这方面的专利, 而且于1988年在美国洛杉矶召开的全美化学会议上公布了这一成果。在早期, 主要是对树枝形聚合物的研究。第一代树枝形聚合物图是通过缩聚反应得到的, 需严格控制反应过程使其结构具有极好的对称性、分子的体积和形状。但是, 因其结构比较规整和完善, 就需要在合成的每一步, 核心分子末端的活性基团必须反应完全, 且每一步的产物需经过彻底的纯化, 因此得到的产物产率很低, 这就大大限制了树枝形大分子的工业化生产。超支化聚合物的结构不要求很完美, 具有一定的相对分子质量分布, 并且与树枝形聚合物相似, 一般可采用一步聚合的方法来合成, 所以易于工业化生产。这两类聚合物在结构上都高度支化, 而且都带有大量官能性的端基, 与线性同系物相比都具有较高的溶解性和较低的粘度, 因此现在一般将这两类聚合物通称为树枝状聚合物。超支化聚合物与线性聚合物在结构上也有很大的差别。线性聚合物中线性部分占大多数, 支化点很少, 分子链容易缠结, 体系的粘度随着相对分子质量的增大而迅速增加。而超支化聚合物中主要是支化部分, 支化点较多, 支化部分至少呈的几率增长。分子具有类似球形的紧凑结构, 流体力学回转半径小, 分子链缠结少, 所以相对分子质量的增加对粘度影响较小而且分子中带有许多官能性端基, 对其进行修饰可以改善其在各类溶剂中的溶解性, 或得到功能材料。摘抄自“超支化聚合物合成及其端基改性”,寇玉霞等,武汉化工学院化工与制药学院,上海涂料第42卷第2期2004.4
超支化聚合物应用研究进展
超支化聚合物研究进展 摘要:本综述的目的是叙述和讨论近年来国内外有关超支化聚合物(HBP)的概述、制备方法、羟基改性引入功能基团以及应用研究进展,并对今后HBP的应用前景进行了展望。方法是以数据库资源为主,查询万方、维普、以及各大外文数据库中有关超支化聚合物研究进展的资料。结果选取其中有代表性的文献进行参考后做出的总结与讨论。本文介绍了超支化聚合物的结构和性能特征,综述了超支化聚合物的制备方法,如缩聚反应、加成反应等,介绍了羟基改性引入功能基团、功能型元素的用途,并对其应用研究进行了说明和分析。Abstract: The purpose of this review is described and discussed the hyperbranched polymer(HBP)'s research in recent years. Method is based on database resources, mainly inquires the ten thousand party, VIP, and other big foreign language database about the hyperbranched polymer. The results is came from making reference to summarize and discuss after selecting representative literature. This paper introduces the hyperbranched polymer structure and performance characteristics,summarized the hyperbranched polymer preparation methods, such as polycondensation reaction,addition reaction.And introduces the hydroxyl modified into functional groups and analysis its application in research. 关键词:超支化聚合物端羟基制备方法应用前景 Keyword:The hyperbranched polymer Hydroxyl Preparation methods Application prospect 正文: 一.超支化聚合物的概述 1.1 结构特征 超支化聚合物(Hyperbranched Polymer)(简称HBP)可以简单描述为具有高度支化结构的聚合物。它既与支化聚合物不同也与树形分子有别。超支化高分子因其分子结构而得名,其结构和树枝状大分子非常相似,树枝状大分子分子结构中只含有末端单元和支化单元,而超支化聚合物不仅含有末端单元、支化单元还有线形单元。如图1所示.
超支化聚氨酯热熔胶的合成及性能
超支化聚氨酯热熔胶的合成及性能 曾少敏 刘 丹 陈爱芳 姚 畅 郭小丽 徐祖顺 3 (湖北大学材料科学与工程学院 武汉430062)摘 要 采用AA ′+b B 2法制备了可以直接作为热熔胶使用的超支化聚氨酯(HP U )。以甲苯22,42二异氰酸酯(T D I )与聚碳酸酯二醇(PCDL )为原料合成两端为异氰酸根(NCO )封端的低聚物(A 2),然后在0℃下加入二乙醇胺(DE OA )得AB 2型中间物,进一步高温聚合得支化点间含有长链段的超支化聚氨酯。采用红外光谱(FTI R )、核磁共振(13C NMR )、GPC 对超支化聚氨酯的结构进行了表征。结果表明,所得到的产物具有超支化 结构,在55℃下反应20h 后,支化度可达到0175,重均分子量M w =710×103。对产物进行了热失重和粘接性 能测试。结果表明,超支化聚氨酯的热分解温度为200℃。产物的粘接剪切强度随着链段长度的增加先增大后减小,最大可达到615M Pa 。 关键词 超支化聚氨酯,合成,热熔胶 中图分类号:O631.5 文献标识码:A 文章编号:100020518(2009)022******* 2008201208收稿,2008206226修回 湖北省杰出人才基金资助项目(2004ABB003) 通讯联系人:徐祖顺,男,教授,博士生导师;E 2mail:zushunxu@hubu .edu .cn;研究方向:乳液聚合、分散聚合、微波聚合等 超支化聚合物具有类似于树枝状聚合物的结构和性能,具有大量的空腔、支化点和近似球形的结 构,且其制备步骤简单,无需纯化,近来受到了广泛的关注[1~4]。Fr échet 等[5]和Kumar 等[6]曾分别用光 气法和叠氮化合物法制备了超支化聚氨酯。Davis 等[7]利用高选择性化学反应制备了可溶于水的超支 化聚氨酯。Long 等[8]用A 2+B 3法合成了力学性能较好的高度支化聚氨酯,颜德岳等[9,10]、B ruchmann 等[11,12]报道了利用商业化单体制备超支化聚合物方法,即耦合单体法(C MM )或AA ′+bB 2法。超支化聚合物分子链通常呈刚性,无链缠结,力学性能差,仅作为添加剂或对其外部官能团改性后使用,很少有 直接用作材料的报道[1]。本文利用AA ′+bB 2法首先用异氰酸基封端的低聚物(A 2)与二乙醇胺(bB 2) 在一定条件下反应得到1个异氰酸根和2个羟基的AB 2中间体,经进一步聚合形成超支化聚氨酯。由于产物支化点之间具有较长的链段,外部有大量羟基官能团,因此产物具有一定的力学性能,并可以直接用作热熔胶。 1 实验部分 1.1 试剂和仪器 甲苯22,42二异氰酸酯(T D I ),丁酮,均为分析纯试剂;聚碳酸酯二醇(PCDL,日本旭化成公司),平均相对分子量为1000,分析纯,90~100℃真空干燥2h;二乙醇胺(DEOA )、二月桂酸二丁基锡(DBT L ),均为化学纯试剂;4A 型分子筛浸泡2d 后使用;N 2甲基吡咯烷酮(NMP ),分析纯试剂,使用前先用4A 型分子筛干燥,再减压蒸馏。Spectru m one 型傅立叶变换红外光谱仪(美国Perkin 2Ele mer 公司),产物溶于四氢呋喃后涂于K B r 晶片形成薄膜;I N VOA 2600型核磁共振仪(美国Varian 公司),T MS 为内标,DMS O 2d6为溶剂;AG 210K NA 型电子万能材料试验机(日本岛津公司),参照G B 712421986,以金属铝为基材,拉伸速率10mm /m in;Agilent 1100型高效液相色谱质谱联用仪(美国安捷伦公司),四氢呋喃为淋洗剂,流速110mL /m in,温度30℃,聚苯乙烯为标样。 1.2 聚合物的合成 称取10100g (0101mol )PCDL 于四口烧瓶中,装好温度计、冷凝管(外接干燥器)和N 2气导入管,通入N 2气10m in,排除体系中的O 2,然后加入3148g (0102mol )T D I 和31160g 丁酮(体系质量分数为30%),在搅拌和N 2气保护下于75~80℃回流。采用二正丁胺化学滴定法测定溶液中异氰酸根(NCO ) 第26卷第2期 应用化学Vol .26No .22009年2月 CH I N ESE JOURNAL OF APP L I E D CHE M I ST RY Feb .2009
超支化聚合物的合成及应用
基金项目:武汉市科技攻关基金; 作者简介:易昌凤(1964-),女,副教授,现主要从事乳液聚合、分散聚合、功能高分子等领域的研究工作。 3通讯联系人。 超支化聚合物的合成及应用 易昌凤,陈爱芳,徐祖顺3 (湖北大学化学与材料科学学院,湖北武汉 430062) 摘要:综述了超支化聚合物合成方法的最新研究进展,并对其应用进行了描述,旨在加深人们对该领域的 了解,从而加速该领域的发展。 关键词:超支化聚合物;合成;应用 超支化聚合物因其分子结构而得名,它是一种经一步法合成得到的高度支化的聚合物[1] 。早在1952 年,Flory [2]就首先在理论上论述通过AB X 型单体分子间的缩聚制备超支化聚合物的可能性。但是,对于这种非结晶、无链缠绕的超支化聚合物,当时并未引起足够的重视。直到1987年,K im [3]申请了制备超支 化聚合物的专利,并于1988年在洛杉机美国化学会上公布了这一成果[4]之后,人们才开始对它产生兴 趣。迄今为止,超支化聚合物的研究已经经历了十多年的历程,本文对超支化聚合物的合成及应用的研究进展做一论述。1 超支化聚合物的合成 目前,超支化聚合物的合成方法除了研究的比较成熟的一步缩聚法外,近年来又发展了一些新的合成方法。下面就文献中报道的一些超支化聚合物的合成方法进行介绍。 111 缩聚反应 缩聚反应是合成超支化聚合物最常用的方法,也是最经典、研究得最成熟的方法,主要是采用AB X 型单体通过逐步增长的方式合成的。一般采用最多的是AB 2型单体,有时为了控制支化度,得到结构更复杂的聚合物,可以采用AB 4、AB 6、AB 8型的单体。目前已用此法合成出了各种类型的超支化聚合物,如聚酯类、聚醚类、聚酰胺类、聚醚2酮类、聚硅氧烷类、聚氨酯类 、聚碳酸酯类等。Y ang 等[5] 以3,52二(42氨基苯氧基)苯甲酸为AB 2型单体,在235℃下进行缩聚合成了超支化芳香聚酰胺(见图1)。产物的重均分子量M W 及分子量多分散指数分别为74600和216。图1 超支化芳香聚酰胺的形成 Figure 1 The formation of the hyperbranched polyamides
超支化聚合物涂料
超支化聚合物涂料 苏慈生(天津理工大学,300191) 摘要:介绍了超支化聚合物的发展、特性,合成的简捷性及在涂料中的应用前景。 关键词:超支化聚合物;超支化聚酯;超支化聚酯酰胺;涂料;发展 超支化聚合物是树状大分子同系物,是从一个中心核分子出发,由支化单体(ABx) 逐级扩散伸展开来的结构,或者是由中心核、数层支化单元和外围基团通过化学键连接而成的。早在1952 年Flary 就首先在理论上提出由ABx 型单体(x ≥2 ,A 、B 为反应基团) 分子间缩聚,制备高度支化聚合物的可能,同时还就其特性作了一些预测。直到20 世纪80 年代才相继合成出此类聚合物,并深入地对其合成、性质及应用进行了研究。至今主要品种有超支化聚酯、酰胺、醚、芳烃、有机硅等,有些已经商品化,如超支化聚酯Boltron20 , Boltron 30 ,Boltron 40 , Perstorp Speciality Chemicals AB 。超支化聚合物的特性是其分子结构规整,分子体积、形状和末端官能可在分子水平上设计与控制,因此成为高分子科学中的热门课题之一,也引起了涂料界的关注。树状大分子、超支化聚合物和传统的线型聚合物的分子结构模型如图1 所示。 图1 树枝状大分子、超支化聚合物、线型聚合物的分子结构模型 1 超支化聚合物的特性概述 树枝状大分子和超支化聚合物均可由ABx 单体合成,二者既有相同之处,也有区别。前者分子具有高度规整的分支结构,分子中无缺陷,呈园球形,后者的分子规整性较前者差,呈椭球形。二者分子的表面均密布着大量有反应活性的末端官能团。其次,前者是分步合成的,在进行下一步合成之前需分离提纯, 其所合成的高度规整分子结构,可作为模型分子供理论研究,后者是由一釜法合成的,制备较简便、经济、易于工业化。再有一点是超支化聚合物的相对分子质量分布较树状大分子宽,具有多分散性。该不足之处可以采用多官能度的核分子,在降低核分子浓度, 以及采取缓慢滴加单体的条件下,是可以改进的。试验证明这是减少分散性和增加分支度的有效方法。经研究发现超支化聚合物与树状大分子在结构和性能上的相似性,加之其在工业上的易合成性,使得超支化聚合物可以满足实际应用的需要。由AB2 单体合成的超支化聚合物分子结构见图2 。
超支化聚合物研究进展_超支化聚合物的合成.
超支化聚合物研究进展( 超支化聚合物的合成 赵辉 1,2 ,罗运军 1* ,宋海香 1 (1 北京理工大学材料科学与工程学院,北京100081;2 开封大学,河南开封475004 摘要:综述了超支化聚合物合成方法的最新研究进展。关键词:超支化聚合物;合成;结构;性质 中图分类号:T Q316 64 文献标识码:A 文章编号:1002-7432(200405-0031-04 1 引言 早在20世纪50年代Flory [1]就提出了超支化大分子的概念,首先在理论上描述了AB x 型单体分子间无控缩聚制备超支化大分子的可能性,并与线型高分子和交联高分子进行了比较。Flory 指出由于具有超支化结构,这类高分子将具有很宽的分子质量分布,并且无缠绕、不结晶。因此,这类超支化聚合物材料的力学强度不高,所以当时并未引起足够的重视。1987年Kim [2]申请了制备超支化大分子的专利,1988年在洛杉矶美国化学会上公布了这一成果[3],1990年发表了关于超支化聚苯的论文并创造了!超支化?(hyperbranched这一名词,并逐渐成为聚合物化学中的1个重要的分支。
超支化聚合物独特的魅力在于其具有大量的高度支化的三维球状结构的端基,分子之间无缠绕和高溶解性、低粘度、高的化学反应活性等性质。由于各种优异的性质和简单的制备方法,超支化大分子在许多领域里都显示出其诱人的应用前景。特别是在作为添加剂改善工程塑料及其他热固性聚合物的韧性等性质的应用[1~5] ,越来越受到人们的重 视。 从第1次有意识地成功合成超支化聚合物至今已有十多年,并且已经取得了重大进展,使之成为合成化学中的1个新的热点而广受关注。本文重点综述超支化聚合物合成。2 超支化聚合物合成 目前,超支化大分子的合成方法除研究得比较成熟的一步缩聚法外,近年来又发展了一些新的合 成方法。下面就文献中报道过的一些超支化聚合物的合成方法进行简单的介绍。 2 1 逐步聚合 通常,超支化聚合物的合成可分为无控制增长!一步法?和逐步控制增长!准一步法?。2 1 1 !一步法? !一步法?指由AB x 型单体不加控制一步反应。是合成超支化聚合物最常用的也是研究得较成熟的方法。其优点是合成方法简单,一般无需逐步分离提纯,且聚合物仍可保持树形大分子的许多结构特征和性质,其缺点是常得到多分散性的聚合物,分子质量无法控制。目前已用该方法合成出一系列超支化大分子,如聚醚酮类、聚醚类、聚氨酯类、聚酰胺类、聚碳酸酯、聚酯类、聚硅烷类等等。 Shu [7]等以5-苯氧基间苯二酸为AB 2型单体,以五氧化二磷和甲磺酸为缩合剂,采用一步法合成了带有羧酸端基的超支化聚(醚-酮,通过亲电芳香取代反应形成芳香
超支化聚合物
超支化聚合物的性质和应用 摘要:超支化大分子独特的构筑使其合成与应用在世界范围内受到越来越多的关注。本文对超支化大分子的结构、性质及其应用进行了简要的综述。 关键字:超支化聚合物;性质;应用 20世纪80年代末,Tomalia 等人发表了有关树形分子的第一篇文章。与线形聚合物相比,具有精确支化结构的单分散树形分子表现出许多独特的性质。例如:在分子量足够高时,发现它们具有球形结构、分子的外层具有大量的端基、分子内存在空腔、粘度随分子量的增加出现极大值和具有分子胶束的性质等[1]。因此,树形分子一出现,很快就成为高分子领域的研究热点。Kim [2]等则将工作集中在将树形分子作为流变学改性剂和球形多官能度引发剂,从而使得他们集中开发了一种合成支化聚苯的一步法。这种聚合物是多分散性的,在线形链段的形成过程中存在缺陷,但它们是高度支化的树枝形分子,Kim 等将其命名为超支化聚合物(Hyperbranched Polymer ,简称HBP)。从Kim 等第一次有意识地成功合成超支化聚合物至今已近二十年,并且已经取得了突破性进展,使之成为合成化学中的一个新的增长热点 而广受关注[3,4]。 1 超支化聚合物的结构及性能特点 超支化聚合物一般由ABx 型(x≥2,A ,B 为反应性基团)单体制备,对其反应过程中生成的中间产物通常不作仔细纯化,聚合条件也不如树枝状分子严格,与完美结构的树形大分子相比,它们具有较高的缺陷率,并且具有很宽的分子质量分布。如果在体系中加入“核分子”,可形成具有类似球形的三维立体结构的 超支化聚合物[5]。图1-11所示为超支化聚合物、树枝状聚合物和线型聚合物的分 子结构示意图。 超支化聚合物 树枝状聚合物 线型聚合物 图1-1超支化聚合物、树枝状聚合物、线型聚合物的分子结构示意图 由图1-1可以看出,相对于普通的线型高分子及树枝状高分子,超支化聚合物具有独特的结构,超支化聚合物的分子中只含1个未反应的A 基团,而含多个未反应的B 基团。超支化聚合物与树枝状聚合物一样,单个分子的形状是球形的,但是树枝状聚合物的分子具有完美的分枝结构,整个分子中无缺陷,因此,树枝状聚合物的单分子是圆球形,而超支化聚合物的分子中有缺陷,整个分子并不完全对称,所以,超支化聚合物的单分子形状是椭球形,但这两种结构的分子表面均密布着大量具有反应活性的末端官能团。传统的线型聚合物在无外力作用下总是自发地呈蜷曲形态,当与线型高分子具有相同的端基数目时,超支化聚合物
超支化聚合物制备方法的研究进展_张传海
基金项目:国家自然科学基金资助项目(No.20334030,No.50403024); 作者简介:张传海(1984-),男,中国科学院研究生院硕士研究生,主要从事聚烯烃功能化的研究。 *通讯作者:Email:zhangly@https://www.360docs.net/doc/c4834566.html, 超支化聚合物制备方法的研究进展 张传海a ,李化毅b ,张明革b ,张辽云 a*(a 中国科学院研究生院,北京 100049;b 中国科学院化学研究所, 北京分子科学国家实验室,高分子科学与材料联合实验室,工程塑料重点实验室,北京 100080) 摘要:超支化聚合物是一类可以通过一步法来合成的具有高度支化结构的体型大分子。经过二十年的研 究,超支化聚合物由于其独特的结构和性能特点以及可实现规模化生产的特点,已经迅速成为一类重要的和具 有广阔应用潜力的高分子材料。本文从单体类型的角度介绍了超支化聚合物的主要制备方法及其发展历程, 主要涉及AB x 型,AB*型,A 2+B 3型以及潜在AB x 型单体(包括开环聚合和偶合单体法)等,同时论述了各制备 方法的优点和局限性。 关键词:超支化聚合物;制备方法;缩聚反应;自缩合乙烯基聚合 概述 树状支化大分子(Dendritic macromolecules)是近年来高分子材料领域研究的热点之一 [1~6]。根据树状支化大分子的结构特征,可将其划分为树枝状大分子(Dendrimer )和超支化聚合物(Hyperbranched polymers)[1]。树状支化大分子由于具有高度支化的结构,因而表现出与线形聚合物不同的性能,例如其 分子链不易缠结、溶液和本体的粘度低、分子结构呈球形、以及分子链末端带有大量的官能团等[2]。其中 树枝状大分子由于其结构上的完美(无缺陷和高度的对称性等),最先受到学界的关注,但是树枝状大分子无论是通过发散法合成还是通过收敛法合成,都需要经过多步反应和纯化,繁琐的合成过程和高昂的成本大大妨碍了其工业化的应用。另一方面,在很多领域的应用中,完美的树枝状结构并不是必须的条件,例如在改善粘度的时候需要的主要是其高度支化形成的球形结构,作为涂料的交联剂需要的则主要是其低粘度和大量的末端活性基团[3]。因此,超支化聚合物作为与树枝状大分子结构类似的一类高分子开始进入人们的视野,并且由于其与树枝状大分子相似的结构和特性以及其合成上远胜于树枝状大分子的优势,因此从一开始就被认为极具大规模工业化的前景。因为超支化聚合物并没有对于完美结构的追求,反应不需要经过多步的合成和纯化,通过一步法即可由单体合成得到所需的聚合物,从而大大降低了合成的成本。此外,大量的官能团的存在使其可以通过改性得到各种不同特性和特殊用途的聚合物,显示了从涂料、粘合剂、流变助剂到超分子化学、纳米科技、生物材料、光电材料、药物运载等诸多领域巨大的应用价值[4]。 超支化聚合物的历史最早可以追溯到19世纪末,Berzelius 首先报道了一种用酒石酸和甘油合成的具有支化结构的聚酯[5]。1901年,Watson 报道了邻苯二甲酸酐与甘油合成聚酯的反应,Dawson 、Howell 、Kienle 等对此反应作了进一步的深入研究,结果表明所得到的产物的粘度相对于一般的线形聚合物的粘度要低。但是当时既没有超支化聚合物的概念,也缺乏相关的理论模型,对其进一步的研究一直处于停滞状态。 直到20世纪40年代,Flory 才利用统计力学的方法建立了超支化聚合物的相关理论[7]。他指出AB x (x 2)型单体的缩聚反应将生成可溶性的高度支化的聚合物,并预测,由于它们的高度支化结构,这类高分子具有较宽的分子量分布,分子链之间没有缠结,而且不能结晶,相关结果被写进了他的著作 高分子