钯等过渡金属催化的卤代芳烃和胺的偶联反应_张贞发

2002年第22卷第10期,685~693
有机化学
Chinese J ournal of Organic Chemistry
Vol.22,2002
No.10,685~693
·综述与进展·
钯等过渡金属催化的卤代芳烃和胺的偶联反应
张贞发 周伟澄
(上海医药工业研究院化学制药事业部 上海200437)
摘要 综述了钯等过渡金属催化的卤代芳烃或芳基磺酸酯和胺的偶联反应以及催化这一新反应的催化剂的发展和应用.用于该反应的配体由P(o-tolyl)
3
发展到BINAP及二烷基芳基膦,其底物由溴代芳烃扩展到经济易得的氯代芳烃及磺酸酯和各种胺.
关键词 芳香胺,钯催化剂,膦配体,胺基化,芳基化
Coupling Reaction of Aryl Halides or Triflates with
Amines Catalyzed by Palladium and Other Transition Metal
ZHANG,Zhen-Fa ZHOU,Wei-Cheng
(Department of Chemistry,Shanghai Ins titute o f Pharmaceutical Industry,Shanghai200437)
A bstract The coupling reaction of aryl halides or triflates with a mines catalyzed by transition metal has rapidly
become a valuable synthetic protoc ol for a variety of applications.The ligands applied in this r eaction develop from the stereo-hindered monodentate phosphine to the chelating ligand and then to the dialkylarylphosphine.
And the substrates cover aryl halides or ar yl triflates and nearly all kinds of a mines.In this review the progress made in the ligands,mechanism and the applications of this reaction was described.
Keywords aromatic amine,palladium catalyst,phosphine ligand,amination,arylation
在天然产物、药物及其它化工产品中芳香胺结构普遍存在.尽管现有的芳香胺的合成方法很多[1].但目前常用的方法都各有其局限性:(1)芳香环的硝化还原法和其他官能团的相容性差.(2)Ullmann 合成法经常需要较高的温度,而且副反应多,通用性差.(3)S N Ar反应则只适用于缺电子芳环.因此,发展新的形成C Ar—N键的方法是很有意义的.
通过过渡金属催化的偶联反应来形成C Ar—C 键在近来已发展成为一种重要的合成方法,例如Kumada,Stille,Suzuki和Negishi等反应[2].相对而言,通过过渡金属催化的偶联反应形成C Ar—杂原子键的发展远不如形成C Ar—C键成熟,但最近几年对C Ar—N键形成的研究发展很快.有逐渐成为构建C Ar—N键的常用合成方法之势.Hartwig和Buchwald 几年前分别联系他们自己的工作做过很好的综述[3~5].最近对这一反应的研究又有许多新的进展,本文以配体的发展为线索联系其实际应用综述该反应近年来的发展.
1 钯催化的胺和卤代芳烃的偶联反应的发现
1983年Kosugi等发现三丁基胺基锡在二价钯络合物催化下可以和溴代芳烃反应生成相应的芳香胺(方程式1)[6].该反应只适用于电中性邻位无位阻的溴代芳烃和脂肪仲胺的锡化物的反应.研究的配体中P(o-tolyl)3是最好的选择.
E-mail:zhangzhenfa@yahoo.co m
R eceived October12,2001;revised January8,2002;accepted February19,2002.
1994年美国的两个研究小组几乎同时报道了这一反应的进一步研究进展
[7]
.他们实现了含有烷
氧基,胺基,烷氧羰基取代的溴代芳烃和仲胺的偶联
(方程式2)[8]
.这些反应的缺点是显而易见的.胺基锡化物的毒性大而且不稳定,底物的适用面窄
.
过渡金属催化的胺和卤代芳烃的偶联反应真正引起人们的注意是在锡试剂被成功替代之后.1995年Hartwig 及B uchwald 研究小组同时发现,将胺和卤代芳烃在碱的存在下直接进行钯催化偶联却能得到更好的结果.伯胺与缺电子和 或邻位有取代的溴代芳烃的偶联也能实现
[9]
.这反应还可以成功地推广
到卤代芳代烃和胺,以至酰胺的分子内偶联[10]
.
2 配体的发展
碱存在下胺和卤代芳烃的催化偶联确立了这一反应的基本要素,但以P (o -tolyl )3络合的钯做催化剂的体系仍具有明显的局限性.这主要表现在许多伯胺、链状仲胺、卤代杂环以及芳基磺酸酯参加的反应.因此以后的研究主要集中于考察不同配体对络合物催化作用的影响.
2.1 最初应用的螯合型配体
1996年Hartwig 和Buchwald 又同时分别报道了两种不同的螯合型配体DPPF 和BI NAP .它们和钯的络合物催化的反应大大地扩展了胺和卤代芳烃偶联的适用范围.螯合型配体的应用是这一反应发展中重要的一步.这在上述综述中已有阐述,本文只涉及最近的发展
.
2.1.1 BINAP 配体
BINAP 目前已成为这一反应中应用得最广泛的
配体,Buchwald 最近对它的适用范围做了详细的探
讨
[11]
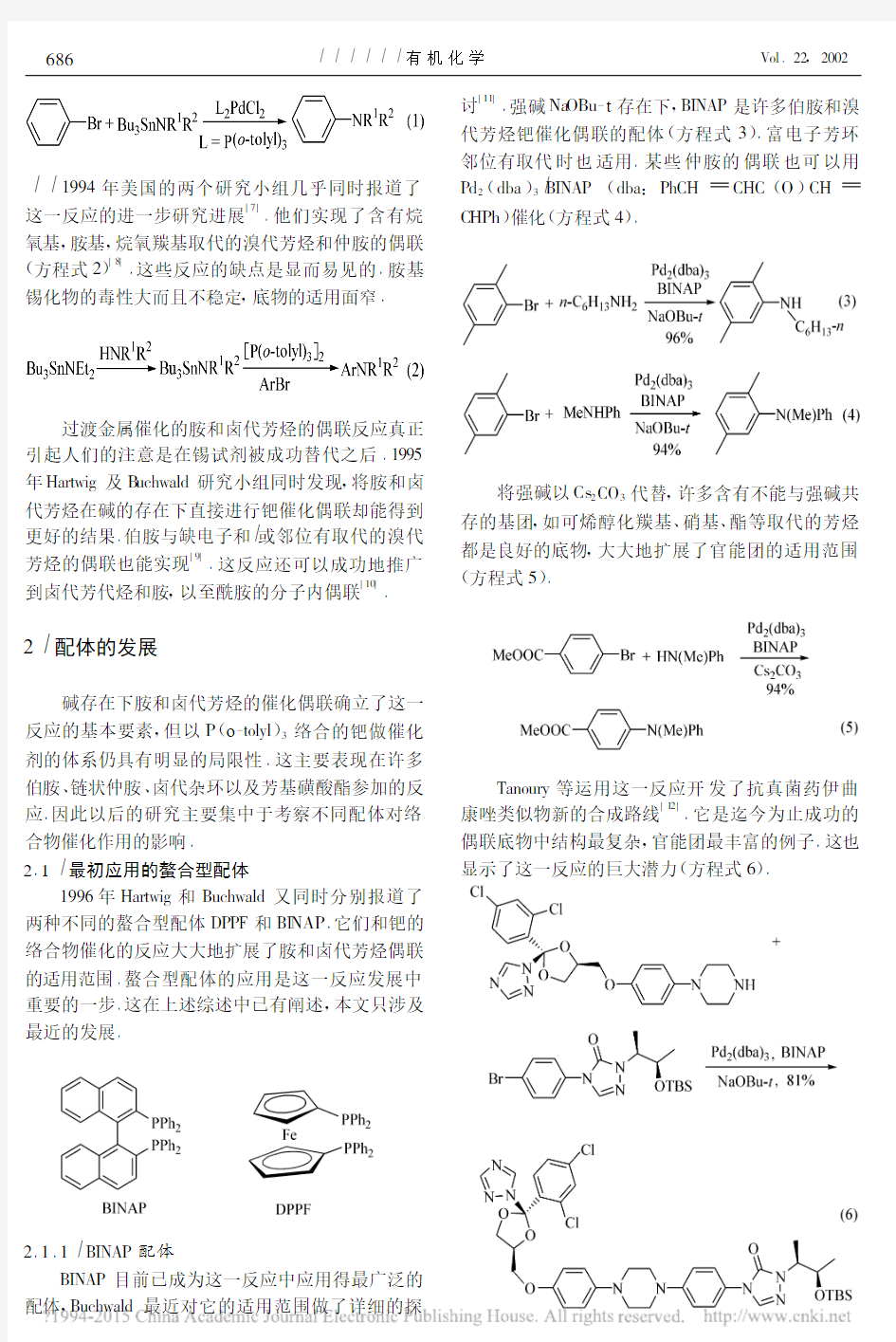
.强碱Na OBu -t 存在下,BINAP 是许多伯胺和溴
代芳烃钯催化偶联的配体(方程式3).富电子芳环
邻位有取代时也适用.某些仲胺的偶联也可以用
Pd 2(dba )3 BINAP (dba :PhCH CHC (O )CH C HPh )催化(方程式4).
将强碱以Cs 2CO 3代替,许多含有不能与强碱共存的基团,如可烯醇化羰基、硝基、酯等取代的芳烃都是良好的底物,大大地扩展了官能团的适用范围(方程式5).
Tanour y 等运用这一反应开发了抗真菌药伊曲
康唑类似物新的合成路线[12]
.它是迄今为止成功的偶联底物中结构最复杂,官能团最丰富的例子.这也显示了这一反应的巨大潜力(方程式6).
686
有机化学Vol .22,2002
Hong 则因此在仲胺存在下实现了伯胺和仲胺的选择性芳基化(方程式7)
[13,14]
.
BINAP 较P (o -tolyl )3另一个优越性还在于它和钯的络合物对分子间的催化偶联反应的立体化学有
良好的预见性.Marinetti 应用这一特点合成了Azew -tidine 的衍生物,没有消旋产物生成(方程式8)[15]
.
BINAP 和钯的络合物也催化亚胺和卤代芳烃的偶联.因此它也可以作为合成苯胺的一种方法[16,17]
.Bolm 首先用Pd (OAc )2 BINAP 实现磺亚胺和卤代芳烃的偶联
[18,19]
.最近Hamat 利用这一反应发展了[3
+3]成环合成苯并噻嗪的新方法(方程式9)[20]
.
酰胺和卤代芳烃的分子间偶联远不如分子内偶联容易.Madar 最近则在新型抗菌药唑烷酮
的合
成中以Pd 2(dba )3 BI NAP 催化的偶联反应构建了
Dup -721的C Ar —N 键(方程式10)
[21]
.
2.1.2 DPPF 配体
DPPF 也有广泛的适用性,它和钯的络合物催化
苯胺和卤代芳烃的偶联(方程式11)[23]
.亦催化脂肪伯胺及甲基苯胺和缺电子芳烃的偶联(方程式12)
.
最近Song 等将DPPF 和钯的络合物对伯胺偶联良好的催化作用推广到肼的分子内偶联,合成了吲唑(方程式13)[24]
.
DPPF 和钯的络合物运用于唑类和亚胺等sp 2
-N 的偶联也是合适的
[25]
.咪唑、吲哚和咔唑都能和溴
代芳烃偶联.亚胺也是如此,而且速度更快.
2.2 最近发展的新型配体2.2.1 三芳基膦系列
BINAP 和钯的络合物催化苯胺与溴代芳烃的偶
联需要较高的催化剂载量.Buchwald 在1998年发展了对此类偶联效率更高但却相对便宜的螯合配体DPEphos (1)[26]
.1尤其适用于苯胺和溴代芳烃的钯催化偶联,对底物的立体位阻有很好的适应性(方程式14).
687
No .10张贞发等:钯等过渡金属催化的卤代芳烃和胺的偶联反应
Leewen 发现配体Xantphos (2)的应用范围可以
扩展到脂肪伯胺或烷基哌嗪的偶联[27]
.而Buchwald 则利用2的络合物实现了伯胺的分步芳基化(方程式15)[28]
.
之后Buchwald 又发现对腙和卤代芳烃的钯催化偶联2是比BI NAP 更有效的配体[29]
.并据此发展
了新的吲哚合成法(方程式16)
.
最近他又以2和钯的络合物为催化剂顺利实现
酰胺和卤代芳烃的分子间偶联[30]
.这是这一类偶联的首次成功,只对钝化的卤代芳烃及氯代芳烃难于发生.而Artamkina 则运用此方法合成了一系列的双芳基化的脲(方程式17)
[31]
.
2.2.2 二烷基膦类
氯代芳烃经济易得,其偶联反应对实际生产有
重要意义,所以它成为最近研究的重点.目前发现的配体中对这类底物最成功的就是二烷基膦类.Har -twig 发现3,4和钯的络合物都是氯代芳烃和胺偶联的催化剂[33]
,苯胺、脂肪伯胺和仲胺都是很好的底物,这是脂肪伯胺和非活化氯代芳烃偶联的首例(方程式18),溴代和碘代芳烃的偶联条件很温和,以此为催化剂首次实现了芳基苯磺酸酯的偶联(方程式19)
.
在所有的底物胺中以链状胺的偶联最难于有效
进行,Buchwald 发现弱的双齿配位体5a 和钯的络合
物能有效地改善这一现象.最近B ei 发现的Pd -(dba )2 5b 催化氯代芳烃的和某些链状胺的偶联有较好效果(方程式20)
[34]
.
688
有机化学Vol .22,2002
在寻找氯代芳烃钯催化偶联的配体的工作中,
Buchwald 发现6和钯的络合物能催化氯苯和吡咯的偶联但需要加热
[35]
.用7这种单膦弱双齿配位体最
早实现了活化的氯代芳烃的室温偶联[36]
.9,10和
钯的络合物则催化未活化的氯代芳烃的室温偶联.
这些都是卤代芳烃及芳基磺酸酯钯催化偶联很好的配体
[37]
.它们是迄今为止所研究的催化体系中最好
的配体.对几乎所有研究过的底物组合都适用
.
对于氯代芳烃偶联出色的催化作用是这一系列配体的钯络合物最突出的优点:室温下10和钯的络合物催化各种胺和氯代芳烃的偶联(方程式21和
22),加热条件下适用范围更广.含有不能耐受强碱的官能团的氯代芳烃的偶联在用K 3PO 4代替强碱时,通常硝基、酯基、氰基都能得到保留.此时配体以7,9更适合
.
卤代吡啶的偶联不能用P (o -tolyl )3和钯的络合物有效催化.虽可以通过螯合型配体BINAP 而取
得[22]
,但也只适用于溴和碘代吡啶.10和钯的络合物则催化各种氯代吡啶和胺的偶联,配体7,9有时
更有效(方程式23)
.
10和钯的络合物催化芳香胺和溴代芳烃的室温偶联,加热条件下适用范围更广,对于芳胺的偶联,这一系统是迄今为止发现的对这类底物最合适的催化剂之一(方程式24)
.
缺电子芳基磺酸酯的偶联常因为强碱的破坏而收率降低.10和钯的络合物首先实现了芳基磺酸酯的室温偶联.虽然缺电子芳环仍然存在上述缺点,但
可以通过在加热时选择K 3PO 4而扩展其适用范围(方程式25)
.
通常碘代芳烃不是比溴代芳烃更好的反应底
物.最近Buchwald 发现以7,9为配体,碘代芳烃的钯催化偶联明显改观(方程式26)[38]
.
689
No .10张贞发等:钯等过渡金属催化的卤代芳烃和胺的偶联反应
对于唑的芳基化,用DPPF 和钯的络合物催化
对富电子芳烃的反应时间较长,用P (Bu -t )3则存在碳芳基化的副反应.Buchwald 选择这一系列的合适配体发现其应用范围大大扩展
[39]
.对于富电子卤代
芳烃和唑的位阻都有很大的适应性(方程式27)
.
2.2.3 三烷基膦类
Masakaz 最早将P (Bu -t )3应用于单取代芳基哌嗪[40]
,三芳胺[41]
的制备和咪唑的氨基化[42]
.Hartwig
发现它和钯的络合物催化溴代芳烃的室温偶联[43a ]
.氯代芳烃偶联的温度比相应的溴代物高(方程式28)
.
P (Bu -t )3和钯的络合物催化唑类和卤代芳烃的偶联比用DPPF 和钯的络合物条件温和得多.Hartwig 用P (Bu -t )3为配体首先实现了氨基甲酸酯的分子间偶联.最近Hartwig 发现P (Bu -t )3和钯的络合物对于催化小分子的芳胺和溴代芳烃的偶联聚合反应产率高(方程式29).而且几乎不存在碳碳偶联,芳基迁移和还原去卤成芳烃的产物.以此催化剂制得的聚合物分子量大大提高,而且环聚产物几率小,钯
残留低[43b ]
.2.2.4 其他配体
由于这类偶联反应独特的适用范围,寻找经济有效的催化剂越来越成为关注的焦点.最近几年有
许多有意义的尝试.
Guido 等在含水体系中以BI NAS -6(11)为配体的钯催化体系实现活化卤代芳烃和苯胺的偶联
[44]
.
产物分离以后催化剂可以重复使用.Buchwald 最近
以树脂12成功地实现了各种卤代芳烃和各种胺的偶联
[45]
.收率高而且产物分离异常简单,催化剂可
以再循环多次而保持相似的收率(方程式30).
Hartwig 等发现用N -杂环卡宾配体13和Pd (OAc )2配合可以催化氯代芳烃和仲胺的室温偶联[46]
.它是首次实现氯代吡啶和胺室温偶联的配体.也是目前非活化氯代芳烃钯催化偶联的催化体系中效率最高的.最近Li 等发现非常简单的配体亚膦酸R 1
R 2
POH 即能和Pd (dba )2配合催化哌啶与氯苯的偶联.它因其简单经济而且对氯代苯有效的优
点而具有诱人的开发前景[47]
(方程式31).
690
有机化学Vol .22,2002
3 钯催化的卤代芳烃和胺偶联的反应机理
由于有钯催化的形成C A r —C 键的理论基础,这一反应的机理研究一开始就和它的应用联系在一起展开了.Hartwig 对卤代芳烃和胺偶联的机理作了详细的研究
[48]
,并对单齿配位体的反应机理做过很好
的综述,在此不再赘述.最近的发展是对双齿配位体
络合物催化的偶联反应机理的全面了解.3.1 双齿配位体络合物催化的偶联反应机理
Hartwig 最近报道了对于双齿配体钯络合物的催化机理的研究结果.它和单齿配位体的络合物催化机理在很多方面是明显不同的.详细的速率方程支持这样的过程
[49]
:
对Pd (binap )2催化的伯胺的偶联见Scheme 1.
对于Pd (binap )2催化的仲胺的偶联.中间体的稳
定性不如相应的伯胺.主要的副反应来自于芳基迁移,所以Pd (binap )2催化仲胺的偶联总的说来比伯胺的偶联产率低而且催化剂载量大.
对于Pd (dppf )2催化的苯胺的偶联见Scheme 2
决定催化效率的步骤是氧化加成一步.对于Pd -(dp -pf )2催化的苯胺的偶联中间体虽然存在芳基迁移,但更明显的是氨基交换.这也解释了二芳基化副反应的原因.
3.2 副反应及其影响因素
卤代芳烃和胺偶联的主要副反应是卤代芳烃的还原脱卤形成芳烃.通常认为是通过中间体的β-摄氢然后还原消除形成的.但对于双齿配位体,标记试验表明,其芳烃的来源不只β-摄氢一种.
对单齿配位体立体位阻越大还原消除产物比例越大.Hartwig 系统地阐述了双齿配位体的立体因素、电子因素和夹角对络合物催化性能的影响.中等位阻和小夹角的配体具有较高的偶联产物比例.对于底物而言,胺的立体位阻会增加芳烃副产物的比例,卤代芳烃的位阻则相反
[50]
.
Scheme
1
Scheme 2
691
No .10张贞发等:钯等过渡金属催化的卤代芳烃和胺的偶联反应
4 发展中的其它过渡金属催化剂
Buchwald
[51]
首先报道了Ni 催化的氯代芳烃和胺的偶联.Fort [52]
最近以Ni (bpy )2催化实现了氯代
芳烃和脂肪仲胺的偶联.多氯代芳烃可以完全胺基
化
[53]
,哌嗪可以不经保护而实现选择性单芳基化或
双芳基化(方程式32)[54]
.最近Lipschutz
[55]
发现的催
化剂Ni C 也可以在DPPF 的配合下催化氯代芳烃的
偶联(方程式33)
.
Gujadhur 发现的Cu (PPh 3)3Br 能催化碘代芳烃和苯胺的偶联
[56]
.这些方法由于底物和催化剂相当
经济,对于工业化的研究有重要意义.马大为等则研究了Pd (PPh 3)4 CuI 对溴代芳烃和氨基酸的偶联,以易得的试剂实现了较好的收率
[57]
.
5 结论
综上所述,我们可以发现钯催化的卤代芳烃和胺的偶联反应最近已得到迅速的发展.其配位体由最初的P (o -tolyl )3发展到双齿配位体使该反应的适用范围大大扩展.烷基磷配位体的发展则使这一反应的条件更温和而且适于氯代芳烃等经济的底物.包括Ni ,Cu 等金属催化的偶联反应的发现也将使这一反应走向生产成为可能.
References
1 M arch ,J .A dvanced Organic Chemis try ,4th ed .,Weily ,New York ,1992,p .641.
2 Suzuki ,A .In M etal Catalyzed Cr oss -Coupling Reactions ,Eds .:Diederich ,
F .;Stang ,
P .J .,Wieley -VCH ,
Weiheim ,1998,Chapter 2.
3 Hart wig ,J .F .A ngew .Chem .,Int .Ed .Engl .1998,37,2046.
4 (a )Wolfe ,J .P .;Wagaw ,S .;Marcous ,J .F .;Buchwald ,
S .L .Acc .Chem .Res .1998,31,805.
(b )Belfield ,A .J .;Brown ,G .R .;Fobister ,A .J .Tetra -
h edr on 1999,55,11399.
(c )Hartwig ,J .F .A cc .Chem .Res .1998,31,852.5 Yang ,B .H .;B uchwald ,S .L .J .O rganomet .Chem .1999,576,125.
6 Kosugi ,M .;Kameyama ,M .;Migita ,T .Chem .Lett .1983,927.
7 Paul ,F .;Patt ,J .;Hartwig ,J .F .J .A m .Chem .Soc .1994,116,5969.
8 Guram ,A .S .;Buch wald ,S .L .J .Am .Chem .Soc .1994,116,7901.
9 Guram ,A .S .;Rennels ,R .A .;Buchwald ,S .L .Angew .
Chem .,Int .Ed .Engl .1995,34,1348.
10 Wolfe ,J .P .;Rennels ,R .A .;B uchwald ,S .L .Tetrahe -dron 1996,52,7525.
11 Wolfe ,J .P .;Buch wald ,S .L .J .Org .Chem .2000,65,
1144.
12 Tanoury ,G .J .;Senanayake ,C .H .;Hett ,R .;Kuhn ,A .
M .;Kessler ,D .W .;Wald ,S .A .Tetr ahedr on Lett .1998,39,6845.
13 Lopez -Rodriguez ,M .L .;Ben hamu ,B .;Ayala ,D .;
Rominguera ,J .L .;Mucia ,M .;Ramos ,J .A .;Viso ,A .Tetrahedron 2000,56,3245.
14 Hong ,Y .;Senanay ake ,C .H .;Xiang ,T .;Vanden -bossche ,C .P .;Tanoury ,G .J .;Bakale ,R .P .;Wald ,S .A .Tetrahedron Lett .1998,39,3121.
15 Marinetti ,A .;Hubert ,P .;Genet ,J .P .Eur .J .O rg .
Chem .2000,1815.
16 Wolfe ,J .P .;Ahman ,J .;Sadighi ,J .P .;Singer ,R .A .;
Buchwald ,S .L .Tetr ah edr on Lett .1997,38,6367.17 Purohit ,V .;Basu ,A .K .Org .Lett .2000,2,1871.18 Bolm ,C .;Hildebrand ,J .P .Tetrahedron Lett .1998,39,
5731.
19 Bolm ,C .;Hildebrand ,J .P .J .Org .Chem .2000,65,
169.
20 Harmata ,M .;Pavri ,N .A ngew .Chem .,Int .Ed .1999,
38,2419.
21 Madar ,D .J .;Kopecka ,H .;Pireh ,D .;Pease ,J .;Pli -ushchev ,M .;Sciotti ,R .J .;Wiedeman ,P .E .;Djuric ,S .W .Tetrahedron Lett .2001,42,3681.
22 Brown ,J .A .Tetrahedro n Lett .2000,41,1623.23 Driver ,M .S .;Hartwig ,J .F .J .A m .Chem .So c .1996,
118,7217.
24 Song ,J .J .;Yee ,N .K .Org .Lett .2000,2,519.25 Mann ,G .;Hartwig ,J .F .;Driver ,M .S .;Fernandez -Ri -vas ,C .J .A m .Chem .Soc .1998,120,827.
26 Sadighi ,J .P .;Harris ,M .C .;Buch wald ,S .L .Tetrahe -dron Lett .1998,39,5327.
692
有机化学
Vol .22,2002
27 Guari,Y.;Van Es,D.S.;Reek,J.N.H.;Kamer,P.
C.J.;Van Leeu wen,P.W.N.M.Tetrahedro n Lett.
1999,40,3789.
28 Harris,M.C.;Geis,O.;Buchwald,https://www.360docs.net/doc/e26095001.html,.
Chem.1999,64,6019.
29 Wagaw,S.;Yang,B.H.;Buch wald,S.L.J.Am.
Chem.Soc.1999,121,10251.
30 Yin,J.;Buchwald,https://www.360docs.net/doc/e26095001.html,.Lett.2000,2,1101.
31 Artamkina,G.A.;Sergeev,A.G.;Beletskaya,I.P.Tet-rahedro n Lett.2001,42,4381.
32 Sridharan,V.Annu.Rep.Prog.Chem.,Sect.B.:Org.
Chem.2000,96,85.
33 Hamann,B.C.;Hartwig,J.F.J.A m.Chem.Soc.
1998,120,7369.
34 Bei,X.;Guram,A.S.;Turner,H.W.;Weiberg,W.H.
Tetr ahedr on Lett.1999,39,1237.
35 Old,D.W.;Wolfe,J.P.;Buchwald,S.L.J.Am.
Chem.Soc.1998,120,9722.
36 Wolfe,J.P.;Buch wald,S.L.Ange w.Chem.,Int.Ed.
1999,38,2413.
37 Wolfe,J.P.;Tomori,H.;Sadighi,J.P.;Yin,J.;Buch-wald,S.L.J.O rg.Ch em.2000,65,1158.
38 Ali,M.H.;Buchwald,https://www.360docs.net/doc/e26095001.html,.Chem.2001,66, 2560.
39 Old,D.W.;Harris,M.C.;Buchwald,https://www.360docs.net/doc/e26095001.html,.Lett.
2000,2,1403.
40 Nishiy ama,M.;Yamamoto,T.;Koie,Y.Tetrahedron Lett.
1998,39,617.
41 Yamamoto,T.;Nishiyama,M.;Koie,Y.Tetrahedron Lett.
1998,39,2367.
42 Watanabe,M.;Nishiyama,M.;Yamamoto,T.;Koie,Y.
Tetr ahedr on Lett.2000,41,481.43 (a)Hartwig,J.F.;Kawatsura,M.;Hauck,S.I.;
Shaughnessy,K.H.;Alcazar-Roman,L.M.J.O rg.
Chem.1999,64,5575.
(b)Goodson,F.E.;Hauck,S.I.;Hartwig,J.F.J.
Am.Chem.Soc.1999,121,7527.
44 Wullner,G.;Jansch,H.;Kannenberg,S.;Schubert,F.;
Boche,G.Chem.Co mmu n.1998,1509.
45 Parrish,C.A.;Buchwald,S.L.J.Or g.Chem.2001, 66,3820.
46 Stauffer,S.R.;Lee,S.;Stambuli,J.P.;Hauck,S.I.;
Hartwig,https://www.360docs.net/doc/e26095001.html,.Lett.2000,2,1423.
47 Li,G.Y.Angew.Chem.,Int.Ed.2001,40,1513.
48 Hart wig,J.F.Acc.Chem.Res.1998,31,852.
49 Alacazar-Roman,L.M.;Hart wig,J.F.;Rheingold,A.
L.;Liable-Sands,L.M.;Guzei,I.A.J.A m.Chem.
Soc.2000,122,4618.
50 Roy,A.H.;Hartwig,J.F.J.A m.Chem.Soc.2001, 123,1232.
51 Hamann,B.C.;Hart wig,J.F.J.A m.Chem.Soc.
1998,120,7369.
52 Brenner,E.;Schneuder,R.;Fort,Y.Tetrahedron1999, 55,12829.
53 Desmarets,C.;Schneuder,R.;Fort,Y.Tetrahedron Lett.
2000,41,2875.
54 Brenner,E.;Schneuder,R.;Fort,Y.Tetrahedron Lett.
2000,41,2881.
55 Lipschutz,B.H.;Veda,H.Angew.Chem.,Int.Ed.
2000,39,4492.
56 Gujadhur,R.;Venkataraman,D.;Kintigh,J.T.Tetrahe-dron Lett.2001,42,4791.
57 Ma,D.;Jiang,J.Tetrahedron:As ymmetry1998,9,1137.
(Y0110123 QIN,X.Q.)
693
No.10张贞发等:钯等过渡金属催化的卤代芳烃和胺的偶联反应
卤代芳烃的毒性
卤代芳烃的毒性 第九章卤代芳烃的毒性 第一节卤代有机化合物 一、卤代作用 在陆生和淡水水生生物中很少见到以共价结合的卤素作为必要的组分而存在,但甲状腺素中有共价键 结合的碘。 卤离子易于同碳原子结合,尤其是不饱和碳原子,可改变分子的特性。: (1) 增加分子量、比重、熔点和沸点,降低蒸汽压 (2) 增加混合物的稳定性。C-X比C-H结合更强。 二、卤代有机化合物的环境行为 联苯类: ClCl ClCl ClCl 联苯3,3’,4,4’,5,5’-六氯联苯 呋喃: ClClO ClClOO 呋喃二苯并呋喃 2,3,7,8-四氯代二苯并呋喃(2,3,7,8-TCDF) 二噁英: OOOClCl ClClOOO
对-二噁英二苯并二噁英 2,3,7,8-四氯代二苯并二噁英(2,3,7,8-TCDD)有机卤化物水溶性很差。卤化程度越高,水溶性越差。但可与其他非极性物质混溶,即有机卤化物是 亲脂的。易于转移到脂肪组织中储存而很少参与生物转化和排泄过程。 持久性和亲脂性是生物浓缩的主要决定因素,而水溶性和挥发性是在环境中迁移的决定因素。 在水生和海洋环境中,一些低氯代或对位氯代的同系物可溶于水或挥发到大气中,而高氯代的同系物 则附在颗粒物上,并沉降。 PCBs在环境中的迁移转化行为 1、在大气中的转移 PCBs污染最初是在赤道至中纬度地区,然而目前在北极和其他遥远地区都发现了PCB,这其中大气 传输的作用不可忽视。大气:高挥发性的卤代物迅速扩散,低或中等挥发性卤代物被短距离输送或存留 在其他物质中相当时间。 在大气中的损失途径主要有两条: (1)直接光解和与OH、NO3等自由基以及O3作用。尤其是OH自由基,估计全世界每年约有0.6%的 PCBs由于OH基反应而消失。 (2)雨水冲洗和干湿沉降。实现从大气向水体或土壤的转移。 2、在土壤中的迁移 土壤中PCBs主要来源于颗粒沉降,有少量来源于污泥作肥料,填埋场的渗漏以及在农药配方中使用
钯催化交叉偶联反应
钯催化交叉偶联反应 钯催化交叉偶联反应是一类用于碳碳键形成的重要化学反应,在有机合成中应用十分广泛。 简介: 为制造复杂的有机材料,需要通过化学反应将碳原子集合在一起。但是碳原子在有机分子中与相邻原子之间的化学键往往非常稳定,不易与其他分子发生化学反应。以往的方法虽然能令碳原子更加活跃,但是,过于活跃的碳原子却又会产生大量副产物,而用钯作为催化剂则可以解决这个问题。钯原子就像“媒人”一样,把不同的碳原子吸引到自己身边,使碳原子之间的距离变得很近,容易结合——也就是“偶联”。这样的反应不需要把碳原子激活到很活跃的程度,副产物比较少,因此更加精确而高效。赫克、根岸英一和铃木章通过实验发现,碳原子会和钯原子连接在一起,进行一系列化学反应。这一技术让化学家们能够精确有效地制出他们需要的复杂化合物。 发展阶段: 一、大约100年前,法国化学家维克多·格林尼亚发现,将一个镁原子同一个碳原子偶联在一起,会将额外的电子推向这个碳原子,使得它能够更容易同另外一个碳原子连接在一起。不过,科学家们发现,这样的方法在创造简单的分子时起到了效果,但是在对更为复杂的分子进行合成时,却在试管里发现了很多并不需要的副产品。 二、早在上世纪60年代,赫克就为钯催化交叉偶联反应奠定了基础,1968年,他报告了新的化学反应——赫克反应,该反应使用钯作为主要的催化剂来让碳原子连接在一起。 三、1977年,根岸英一对其成果进行了精练,他使用一种有机氯化物作为催化剂;两年后,铃木章发现使用有机硼化合物的效果会更好。应用: 如今,“钯催化交叉偶联反应”被应用于许多物质的合成研究和工业化生产。例如合成抗癌药物紫杉醇和抗炎症药物萘普生,以及有机分子中一个体格特别巨大的成员——水螅毒素。科学家还尝试用这些方法改造一种抗生素——万古霉素的分子,用来灭有超强抗药性的细菌。此外,利用这些方法合成的一些有机材料能够发光,可用于制造只有几毫米厚、像塑料薄膜一样的显示器。科学界一些人士表示,依托“钯催化交
第九章卤代芳烃的毒性
第九章卤代芳烃的毒性 第一节卤代有机化合物 一、卤代作用 在陆生和淡水水生生物中很少见到以共价结合的卤素作为必要的组分而存在,但甲状腺素中有共价键结合的碘。 卤离子易于同碳原子结合,尤其是不饱和碳原子,可改变分子的特性。: (1)增加分子量、比重、熔点和沸点,降低蒸汽压 (2)增加混合物的稳定性。C-X比C-H结合更强。 二、卤代有机化合物的环境行为 联苯类: Cl Cl Cl Cl Cl Cl 联苯3,3’,4,4’,5,5’-六氯联苯 呋喃: O O O Cl Cl Cl Cl 呋喃二苯并呋喃2,3,7,8-四氯代二苯并呋喃(2,3,7,8-TCDF) 二噁英: O O O O O O Cl Cl Cl Cl 对-二噁英二苯并二噁英2,3,7,8-四氯代二苯并二噁英(2,3,7,8-TCDD)有机卤化物水溶性很差。卤化程度越高,水溶性越差。但可与其他非极性物质混溶,即有机卤化物是亲脂的。易于转移到脂肪组织中储存而很少参与生物转化和排泄过程。 持久性和亲脂性是生物浓缩的主要决定因素,而水溶性和挥发性是在环境中迁移的决定因素。 在水生和海洋环境中,一些低氯代或对位氯代的同系物可溶于水或挥发到大气中,而高氯代的同系物则附在颗粒物上,并沉降。 PCBs在环境中的迁移转化行为 1、在大气中的转移 PCBs污染最初是在赤道至中纬度地区,然而目前在北极和其他遥远地区都发现了PCB,这其中大气传输的作用不可忽视。大气:高挥发性的卤代物迅速扩散,低或中等挥发性卤代物被短距离输送或存留在其他物质中相当时间。 在大气中的损失途径主要有两条: (1)直接光解和与OH、NO3等自由基以及O3作用。尤其是OH自由基,估计全世界每年约有0.6%的PCBs由于OH基反应而消失。
钯催化的交叉偶联反应——2010年诺贝尔化学奖获奖工作介绍
2011年第 31卷 有 机 化 学 V ol. 31, 2011 * E-ma i l: nxwang@ma i l.i https://www.360docs.net/doc/e26095001.html, Received December 9, 2010; revised and accepted March 10, 2011. ·学术动态· 钯催化的交叉偶联反应——2010年诺贝尔化学奖获奖工作介绍 王乃兴 (中国科学院理化技术研究所 北京 100190) 摘要 钯催化的交叉偶联反应是非常实用的合成新方法. 文章给出了Heck 反应、Negishi 反应和Suzuki 反应的概念, 对其反应机理作了详细的说明, 并对其在复杂化合物和天然产物全合成中的应用作了评价. 关键词 钯催化; Heck 反应; Negishi 反应; Suzuki 反应 Palladium-Catalyzed Cross-Coupling Reactions — Introduction of Nobel Prize in Chemistry in 2010 Wang, Naixing (Technical Institute of Physics and Chemistry , Chinese Academy of Sciences , Beijing 100190) Abstract Palladium-catalyzed cross-coupling reactions provide chemists with a more precise and efficient new methodologies. The concepts of the Heck reaction and Negishi reaction as well as Suzuki reaction are given, the reaction mechanisms are proposed, and applications of these reactions in the total synthesis of natural products are commented. Keywords palladium-catalyzed; Heck reaction; Negishi reaction; Suzuki reaction 2009年10月6日, 瑞典皇家科学院宣布, 美国科学家Richard F. Heck(理查德 赫克)、日本科学家Ei-ichi Negishi(根岸英一)和Akira Suzuki(铃木章)共同获得今年的诺贝尔化学奖. 美国教授Richard F. Heck, 1931年出生于美国的斯普林菲尔德, 1954年在美国加利福尼亚大学洛杉矶分校获得博士学位. 随后他进入瑞士苏黎世联邦工学院从事博士后研究, 后在美国特拉华大学任教, 于1989年退休. Richard F. Heck 现为特拉华大学名誉教授. Ei-ichi Negishi 教授是日本人, 1935年出生于中国长春, 1958年从东京大学毕业后进入帝人公司, 1963年在美国宾夕法尼亚大学获得博士学位, 现任美国普渡大学教授. Akira Suzuki 也是日本人, 1930年出生于日本北海道鹉川町, 1959年在北海道大学获得博士学位, 随后留校工作了一段时间. 1963年到1965年, Akira Suzuki 在美国普渡大学从事了两年的博士后研究工作. Akira Suzuki 于1973年任北海道大学工学系教授, 现在是北 海道大学名誉教授. 钯催化的交叉偶联反应是一种可靠而又实用的工具, 对有机合成具有长久和深远的影响力, 该反应得到了合成化学工作者的普遍应用. 笔者于2004年在《有机反应——多氮化物的反应及有关理论问题(第二版)》的第4.13节中列举了5个较新的人名反应[1], 其中有Heck 反应、Negishi 反应和Suzuki 反应. 对其定义分别为: Heck 反应是钯催化下, 不饱和有机卤化物或三氟磺酸酯与烯烃进行的偶联反应. Negishi 反应是钯催化下的不饱和有机锌试剂和芳基或乙烯基卤化物等进行偶联的反应. Suzuki 反应是钯催化下不饱和有机硼试剂和芳基或乙烯基卤化物等进行偶联的反应. 这是钯催化的交叉偶联反应的基本概念. 最初的Suzuki 反应还需要在无氧无水的条件下来进行, 后来发展的一些反应条件已经无需无氧无水操作了. 这几种钯催化的交叉偶联反应机理不尽相同, 对机
新型二恶英类卤代芳烃的研究
#综述# 收稿日期:2006-11-28 基金项目:新乡医学院高学历人才科研启动基金资助 作者简介:杨志军(1972-),男,河南省南乐县人,博士,讲师,主要从事有毒有机污染物的分离分析研究。 新型二口 恶英类卤代芳烃研究进展 杨志军1,梁鑫淼 2 (1.新乡医学院化学教研室,河南 新乡 453003;2.中国科学院大连化学物理研究所1803组,辽宁 大连 116023) 摘要: 许多卤代芳烃化合物具有类似于二口恶英类化合物的结构和性质,都属于持久性有机污染物,其毒性对环境和人类健康存在潜在的危害。作者对部分新型二口恶英类卤代芳烃化合物的性质、环境行为、分离分析方法等进行了详细评述。 关键词: 二口恶英类卤代芳烃;化学表征;毒性 中图分类号:X 708 文献标识码:A 文章编号:1004-7239(2007)01-0099-04 Advance i n ne w types of di o xi n -li k e arom atic halides YANG Zh-i jun 1,L I A NG X in -m i a o 2 (1.D e p ar t m ent of Che m istry,X i nx iang M edical Co llege ,X inx iang 453003,China;2.G roup 1803,D a lian Institute of Che m ical and Phy sics ,Chinese A cad e m y of Sciences ,D alian 116023,China) Abstrac t : The structures and prope rties o fm any po lyha l ogena ted arom atic hydrocarbons are s i m il ar w ith d i ox i n -like co m-pounds ,wh i ch have potential threatens to env iron m ent and hu m an hea lth as persistent organ ic poll utants because of t he ir tox ic-i ty .In th i s paper ,the properties ,env iron m ental behav i o rs ,separa ti on and analysism ethods of som e ne w types o f d i ox i n -li ke aro -m atic ha li des are rev i ew ed i n deta i.l K ey word s : d i ox i n -li ke aro m a tic ha li des ;che m i ca l character i zati on ;t ox ic it y 由于多氯代二苯并二口 恶英/二苯并呋喃(po ly -chlorinated d i b enzo -p -diox ins/d i b enzo f u rans ,PCDD / Fs)、多氯联苯(polych l o rinated biphenyls ,PCB s)已成为公认的典型持久性有机污染物(persistent or -ganic po ll u tants ,POPs),因此,近年来有关PCDD /Fs 、PCBs 的相关研究相对比较多。进入人体内的二 口 恶英大多蓄积在肝脏和脂肪组织中, 因而危害极大。 二口 恶英类环境污染物不仅具有致癌性,且具有免疫和生殖毒性,作为内分泌干扰物可以造成雄性生物雌性化。如果长期低剂量暴露就可以使得其在人体内蓄积,从而可能造成严重的身体损害。即使在很微量的情况下,长期摄取二口 恶英类化合物时也可引 起癌症等顽症。此外,二 口 恶英对人体还会引起皮肤 痤疮、头痛、失聪、忧郁、失眠和新生儿畸形等症,并可能引起诸如染色体损伤、心力衰竭和内分泌失调 等。在其非致癌效应方面,则有神经毒性、免疫抑制、内分泌干扰破坏与生殖毒性等。但是,在环境中除PCDD /Fs 、PCBs 外,还存在种类繁多的未知POPs ,如许多卤代芳烃类化合物,它们都具有类似于二口 恶英类化合物的结构和性质,对环境和人类健 康存在潜在的毒性危害。鉴于其未知性、复杂性和 表征手段的限制,目前此方面的研究还十分有限。其中,特别是一部分结构和性质与二口 恶英类似的卤代芳烃类化合物,其分离分析、对环境和健康的危害 及风险正逐渐成为环境科学、分析化学研究的热点 问题[1] 。 1 部分二口恶英类卤代芳烃 1.1 硫取代的杂环二口 恶英 如果将PCDD /Fs 环上的O 原子换成S 原子,那么得到的化合物分别是多 氯代噻蒽(polychlorinated thianthrenes ,PCTA s)和多氯代二苯并噻吩(po lychlori n ated d i b enzoth i o phenes ,PCDTs),此外还有O 、S 混合的多氯代吩口 恶噻(po l y -chlori n ated phenoxathiins ,PCPAs),它们的结构和性 质与PCDD /Fs 非常相似[2-3]。W iedm ann 等[4] 在城市高速公路隧道的灰尘样品中用HRGC /HR M S 检出了PCDT s ,证明PCDTs 的产生可能与机动车燃料焚烧过程有关,但是没有检出PCTA s 和PCPA s 。这并不能说明PCTA s 和PCPAs 就不存在,而可能与其在样品中含量太小或表征方法手段的限制有关。S i n kkonen 等[2] 研究证明,PCDTs 存在于烟道气、焚烧飞灰、造纸废水、底泥等样品中,从而证实焚烧过程是环境中PCDTs 的主要来源之一,而利用高分辨气相色谱/高分辨质谱(HRGC /HRM S)分析的
钯催化交叉偶联反应
钯催化的交叉偶联反应 一、偶联反应综述 1.交叉偶联反应 偶联反应,从广义上讲,就是由两个有机分子进行某种化学反应而生成一个新有机分子的过程。狭义的偶联反应是涉及有机金属催化剂的碳-碳键生成的反应,根据类型的不同,又可分为自身偶联反应和交叉偶联。交叉偶联反应是一个有机分子与另一有机分子发生的不对称偶联反应。 2.碳碳键形成的重要性 新碳-碳键的形成在有机化学中是极其重要的。人们了解了天然有机物质的结构和性能,并根据有机物质的结构,通过碳原子组装成链,建立有机分子,最终实现天然有机物质的人工合成。目前为止,人类已经利用有机合成化学手段创造出几千万种物质,且越来越多的有机物质已经广泛应用到制药、建材、食品、纺织等人类生活领域,我们的生活也几乎离不开有机物了。合成药物、塑料等有机物质时,需要用小的有机分子将碳原子连接在一起构建新的复杂大分子,因而有机合成中高效的连接碳-碳键的方法是有机合成化学中的重要工具。从以往该领域诺贝尔化学奖的授予情况也可以看出合成新碳-碳键的重要性:1912年维克多·格林尼亚因发明格林尼亚试剂——有机镁试剂获奖,1950年迪尔斯和阿尔德因发明双烯反应迪尔斯-阿尔德反应获奖,1979年维蒂希与布朗因发明维蒂希反应共同获奖,2005年伊夫·肖万、罗伯特·格拉布、理查德·施罗克因在有机化学的烯烃复分解反应研究方面作了突出贡献获奖。 3.有机合成中的钯催化交叉偶联反应 随着时代发展,合成有机化学的研究愈加深入,20世纪后半期,科学家们发现了大量通过过渡金属催化来创造新有机分子的反应,促使有机合成化学快速发展。
特别是赫克、根岸英一和铃木章发现的钯催化交叉偶联反应,为化学家们提供了一个更为精确有效的工具。三位科学家发现的钯催化交叉偶联反应中都使用了金属钯作为反应的催化剂,当碳原子与钯原子连在一起时,钯原子唤醒了“懒惰”的碳原子但又不至于使它太活泼,于是形成温和的碳-钯键,在反应过程中,钯原子又可以把别的碳原子吸引过来,形成另一个金属-碳键,此时两个碳原子都连接在钯原子上,它们的距离足够接近而发生反应,生成新的碳-碳单键。以下两个反应式代表了典型的两类钯催化交叉偶联反应。 上述两个反应的催化剂都是零价的金属钯,都使用卤代烃RX(或卤代烃的类似物)作为亲电偶联试剂。区别在于两个反应所选用的亲核偶联试剂,在反应(1)中,选用的是烯烃,反应(2)中则是一种有机金属化合物R〃M(M为Zn,B,Al 或Sn)。我们所熟知的赫克反应属于反应(1)这一类的交叉偶联反应,根岸反应和铃木反应属于反应(2)这一类。由于反应底物不同,三个反应的应用范围和适用途径也各不相同。 4.“钯催化的交叉偶联反应”内容及反应原理 (1)Heck反应 Heck反应以有机钯配合物为催化剂得到具有立体专一性的芳香代烯烃(图1)。反应物主要是卤代芳烃(碘、溴)与含有吸电子基团的烯烃。该反应的催化剂通常用Pd(0),Pd(II)或含Pd的配合物(常用醋酸钯和三苯基膦)。卤代烃首先与A 发生氧化加成反应,C-X键的断裂与Pd-C和Pd-X键的形成是同步进行的。氧化加成反应是偶联反应中最常见的决速步骤,经过氧化加成化合物A生成中间体B,B再经过配体解离,得到化合物RPdLX。RPdLX先与烯烃配位,然后再经烯烃插入,配
钯催化反应及其机理
钯催化反应及其机理研究 摘要:目前过渡金属催化的有机反应研究一直是一个比较热的话题,其中由于钯催化的反应活性和稳定性等原因,使其在有机反应中得到了广泛的使用,被全球广泛关注。本文主要列举了钯催化的交叉偶联反应的机理,及与偶联反应相关的钯催化的碳氢键活化反应、钯催化的脂肪醇的芳基化反应等的机理。 关键词:过渡金属催化偶联反应钯催化机理 1.引言 进入二十一世纪以后,钯催化的偶联反应已经建立了比较完整的理论体系,研究的侧重点也和以前有所不同化学键的断裂和形成是有机化学的核心问题之一。在众多化学键的断裂和形成方式中,过渡金属催化的有机反应有着独特的优势:这类反应通常具有温和的反应条件,产率很高并有很好的选择性(包含立体、化学、区域选择性)。很多常规方法根本无法实现的化学反应,采用了过渡金属催化后可以很容易地得到实现。在众多过渡金属中,金属钯是目前研究得最深入的一个。自上世纪七十年代以来,随着Kumada,Heck,Suzuki,Negishi [1]等偶联反应的陆续发现,钯催化的有机反应发展十分迅速,时至今日,钯催化的偶联反应作为形成碳-碳、碳-杂键最简洁有效的方法之一,已经得到了广泛应用。 2.钯催化各反应机理的研究 2.1.钯催化的交叉偶联反应 自上世纪七十年代以来,随着Kumada,Heck,Suzuki,Negishi 等偶联反应的陆续发现[1],钯催化的有机反应发展十分迅速,时至今日,钯催化的偶联反应作为形成碳-碳、碳-杂键最简洁有效的方法之一,已经得到了广泛应用[2]。交叉偶联,就是两个不同的有机分子通过反应连在了一起(英文中交叉偶联为crosscoupling,同种分子偶联为homo coupling)。 2.1.1Heck反应 Heck 反应是不饱和卤代烃和烯烃在强碱和钯催化下生成取代烯烃的反应,是一类形成与不饱和双键相连的新C—C 键的重要反应[3]。反应物主要为卤代芳烃(碘、溴)与含
一、项目名称发现致癌性卤代芳烃自由基产生新机制
一、项目名称 发现致癌性卤代芳烃自由基产生新机制 二、提名者及提名意见 提名专家:江桂斌 项目完成人朱本占研究员长期从事自由基化学生物学与环境污染物自由基毒性机理方面的研究,取得了具有独创性和系统性的研究成果,特别是突破了经典羟基自由基产生理论(Fenton反应),发现了一类不依赖于过渡金属离子的卤代醌介导产生羟基自由基,化学发光和重排反应的新型分子机制,首次检测并鉴定了一类新型的以碳为中心的醌自由基,发现卤代醌介导产生的多种活性自由基中间体能诱导产生DNA等生物大分子的氧化损伤,提出五氯酚及其它持久性卤代芳烃污染物致癌的自由基新机制。该项目研究成果丰富和发展了经典羟基自由基产生理论,在有机污染物自由基产生机制和毒性效应研究方面有重要突破,形成了比较系统的理论与方法体系,在国际权威综合性学术期刊《美国科学院院刊》PNAS连续发表五篇相关论文,有关成果被写入经典教科书《Free Radicals in Biology and Medicine》, 多次被Science等期刊作专栏或亮点/封面介绍,入选中科院“十二五”重大科技成果及标志性进展。朱本占研究员应邀担任美国化学会著名毒理学期刊Chem. Res. Toxicol.等多个学术期刊的编委或特约/专刊编辑,并多次在重要国际学术会议如Gordon Conference上做大会/特邀报告;曾获中科院“百人计划”、国家基金委“杰青”和“创新群体”基金支持,终期评估三项皆为“优秀”;荣获中科院“杰出科技成就奖” 和徐元植顺磁共振发展奖励基金“杰出贡献奖”。 提名该项目为国家自然科学奖二等奖。 三、项目简介 自由基,特别是羟基自由基(?OH)的形成机制,在化学、生物医学及环境科学与技术等多项前沿研究领域中均十分重要。本项目属于环境化学与毒理学、物理有机化学等多学科交叉领域。项目完成人长期从事自由基化学与卤代芳香环境污染物分子毒性机理研究,取得以下具有独创性和系统性的研究成果: 1. 突破经典?OH产生理论,发现了一类不同于经典Fenton反应的卤代醌介导的?OH产生新机制,提出五氯酚等卤代芳烃致癌的自由基新机理;首次检测/鉴定了一类新型的碳中心醌自由基,为先前提出的卤代醌诱导的氢过氧化物分解的亲核取代/均裂分解机制提供了直接实验证据;采用新型自由基捕获剂,首次分离纯化了醌碳加合物的自由基形式,建立了ESR自旋捕获与HPLC/MS联用的未知自由基检测鉴定新方法;发现卤代醌介导产生的多种活性自由基能导致DNA损伤。?OH产生新体系和未知自由基鉴定新方法被国内外多个课题组采用,引发并引领了系列后续研究。
钯催化的交叉偶联反应——2010年诺贝尔化学奖简介
doi:10.3969/j.issn0253-9608.2010.06.005 钯催化的交叉偶联反应 ———2010年诺贝尔化学奖简介 肖唐鑫① 刘 立② 强琚莉③ 王乐勇④ ①②博士研究生,③博士,④教授,南京大学化学化工学院,南京210093 关键词 钯催化 偶联反应 诺贝尔化学奖 2010年10月6日,瑞典皇家科学院宣布将2010年诺贝尔化学奖授予美国科学家Richar d F.Heck,日本科学家Ei-ichi Ne g ishi和A kira Suzuki。这三名科学家是因为在有机合成领域中钯催化交叉偶联反应方面的卓越研究而获奖。它为化学家提供了一款精致的工具来合成复杂的有机分子。这一成果广泛应用于制药、电子工业和先进材料等领域。笔者对钯催化交叉偶联反应领域作了粗浅的介绍,以期起到抛砖引玉之作用。 2010年的诺贝尔化学奖揭晓后,很多专业人士对此 并不感到惊讶,认为这次的评选结果实乃众望所归。确实如此,三位科学家都已近耄耋之年,他们所做的贡献早已造福全球,按理早应摘取这个桂冠了。当瑞典皇家科学院在2010年10月6日宣布将诺贝尔化学奖颁发给美国科学家Richard F.Heck和日本科学家Ei-ichi Negishi,Akira Suzuki时,Heck所说的一句话———这是个圆满的结局———道出了所有人的心声。目前,钯催化的交叉偶联反应在全球的科研、医药生产、电子工业和先进材料等领域都有广泛应用。以在此领域有卓越贡献的科学家名字命名的有机反应对于从事化学的人来说是耳熟能详的,如Heck反应、Negishi反应、Suzuki反应、Stille反应、Kumada反应、Sonogashira反应以及Hiyama反应等等。 众所周知,有机合成化学以其强大的生命力制造出了几千万种新的物质,并且这个数目仍在迅速的膨胀,而有机合成化学的基础核心是新型、高效有机合成方法学的研究和发展。我们从21世纪这10年来三次与有机合成方法学相关的诺贝尔化学奖授予情况可以看出这一领域的重要性:2001年W.S.Knowles,R.Noyori 和K.B.Sharpless因在发展催化不对称合成研究方面获奖;2005年Y.Chauvin,R.H.Grubbs和R.R. Schrock因在发展烯烃复分解反应所作出的贡献而获奖;最后就是2010年的钯催化交叉偶联反应的获奖。下面对钯催化交叉偶联反应的早期研究、反应机理以及发展应用等做一个粗浅的介绍,以期达到抛砖引玉之作用。1早期研究 有机合成化学制造出的这几千万种新的物质绝大多数都是以碳原子为主来构建的。为了制备结构更复杂、功能更强大的新型材料,就要想办法通过各种化学反应将碳原子连接在一起。然而碳原子本身是十分稳定的,在化学反应中并不活泼,所以就得想办法来激活碳原子,让它更容易参与反应并与其他碳原子连接起来,逐步形成更高层次的碳基骨架。1912年,法国人Grignard因发明有机镁试剂(格氏试剂)而荣获诺贝尔化学奖,可以说是碳基活化史上的第一个里程碑。随着时代的发展,人们对碳基的研究愈加深入。在研究的前期,要么无法活化碳基,化合物难于参加反应;要么使碳原子过于活跃,虽然能有效地制造出很多简单的有机物,但要是合成复杂分子却有大量的副产物生成。正如大家所知,在有机合成操作中提纯是一项繁琐的工作。Heck,Negishi和Suzuki等人通过实验发现,当碳原子和钯原子连接在一起,会形成一种“温和”的碳钯键,在这里钯既活跃了碳基,又使其不至于过于活泼,然后又可以把别的碳原子吸引过来,这样使得两个碳原子距离拉近,容易成键而偶联起来。在这里钯原子就相当于“媒人”的作用,只需使用催化剂就行。所以“钯催化交叉偶联反应”就是一款精致的工具,让化学家得以像艺术家一样来雕刻和拼接类似积木的模块(小的基团),构筑令人叹为观止的艺术品(有机复杂分子)。与此同时还避免了过多不必要副产物的生成。 Heck1931年出生于美国麻省斯普林菲尔德(Spri- · 332·Chinese J ournal o f N ature V ol.32N o.6 Brief Introduction of No bel Prize
芳烃的命名与结构
第十章 芳烃 第一节 芳烃的命名与结构 一、苯衍生物的命名与苯的结构 1.苯衍生物的命名 a.常见基团的命名优先顺序 常见基团的命名优先顺序:COOH>SO 3H>COOR>CONH 2>CN>CHO>CO>OH>NH 2>C ≡C>C=C>C 6H 5(苯环)>R (烷基)>X ,NO 2(这两种官能团在命名时总是作为取代基)。 当分子中存在多种官能团时物质的类名由命名优先的官能团决定而其它官能团只看作取代基。 b.确定母体名称 苯的同系物、卤代物和硝基化合物以苯作为母体名称。对结构复杂或侧链带有官能团的化合物将侧链当母体而苯环作为取代基。 当苯环上有命名优先的官能团时将苯环与优先官能团一起作为母体其它作为取代基。 c.编号 当苯环上有两个基团时可用词头—“邻”、“间”和“对”表示两个基团在环上的位置但也可用编号数来表示,当苯环上有多个基团时基团在环上的位置只能用编号数来表示,编号遵循官能团最小位次(如果有的话)规则和(优先级最低的)取代基最小位次规则。 d.从左至右按先取代基后母体的顺序书写名称。 e.当存在多种取代基时取代基按优先级增大的顺序从左至右排列,即取代基按CIP 次序规则依次写出。例如, OH CH 3 OH CO 2H 这两个化合物可以分别命名为:邻羟基苯甲酸和2-甲基-5-氯苯酚。 2.苯的结构 苯是一个平面分子,六个碳原子以SP 2杂化轨道相互重叠形成六个碳碳σ键组成一个平面正六边形,每一个碳原子未参与杂化的P 轨道相互平行重叠组成闭合的离域大π键(一个六中心六电子π键)。如果采用共振论,苯的结构可以写成如下两个Kekule 式的杂化体: 由此苯具有大的共振稳定性,根据燃烧热或氢化热数据得到苯的共振能是150kJ/mol 。 可以用如下的非Kekule 式表示苯的结构,其中圆圈表示离域π电子: 但也常常用一个Kekule 式: 或 作为苯杂化体的简写,但必须牢记π键不是孤立的而是离域的。 二、萘衍生物的命名与萘的结构 1.萘衍生物的命名
钯催化的偶联反应
AgNO3/KF作用下的Pd催化2-溴噻吩S原子邻位上的C-H键选择性偶联反 应 摘要: 溴噻吩的衍生物与芳基碘在加入了钯的硝酸银/氟化钾催化剂的催化下发生C—H键的偶联反应,而C—Br键未发生变化。这些含有C —Br键的偶联产物在钯的进一步催化下使溴噻吩和芳基碘的C—C键相连接从而得到理想的产量。 引言: 狭义上的偶联反应是涉及由基金属催化剂的C-C键生成的反应,根据类型不同,可分为交叉偶联反应和自身偶联反应。交叉偶联反应是一个有机分子与另一有机分子发生的不对称偶联反应。例如:烯丙基锂与2-氯辛烷可以发生交叉偶联反应生成4-甲基-1-癸烯。格利雅试剂、有机铝、有机锌、有机锡、有机铜、有机铅、有机汞等多种有机金属化合物也都可以与卤化烷等烃基化试剂发生交叉偶联反应,生成相应的不对称烃,是合成不对称烃,特别是单烷基芳烃和含有三级碳原子的链烃的有效方法。交叉偶联反应的范围很广,像芳烃重氮盐与苯酚或N,N-二甲基苯胺的偶联反应,也属于交叉偶联反应。
正文: 芳香族化合物与有机卤化物的C-H键取代反应和那些含金属试剂与相同的有机卤化物的偶联反应相比,在有机合成中更有前景。【1】相比之下,C-H键上的直接反应将有利于含有不同种类的官能团的衍生物的合成,并且,反应也会加强合成中原子的效应。我们注意到噻吩衍生物的偶联反应是发生在C-H键上,从而形成了联噻吩。在添加了AgF后,反应效率得到了提高。【2】当噻吩与2-溴噻吩反应生成正联溴噻吩时,仍然是C-H键发生偶联,而C-Br键未发生变化。我们的注意力集中到溴噻吩衍生物C-H键的交叉耦合上,来介绍噻吩环上的取代基。【3】溴噻吩上的C-H键偶联,如果可以通过C-Br键的反应而进一步改变偶联产物,那么C-H键和C-Br键的偶联反应的相互结合将得到一种新的合成取代噻吩的方法。这将把人们的注意力都吸引到设计更先进的有机金属材料来揭示液晶、光发射和有机半导体的特点。【4】在此,我们报告一个新的催化剂系统—AgNO3/KF,它有助于提高钯催化下溴噻吩衍生物C-H键的取代反应发的效率。 2-溴噻吩与对甲氧基碘苯的反应在添加了钯催化剂的含AgF的条件下进行。在60℃的条件下搅拌5小时后,将得到占总产物50%的正偶联产物。类似的反应如用AgNO3/KF来替代AgF,将会得到占总产物42%的上述产物。值得注意的是尽管最后的终产物是适度的,我们并没有发现C-Br键上有反应发生,而是在溴噻吩的与S原子相邻的C-H 键上有反应发生。噻吩衍生物的芳基化反应是在钯和体积较大的磷化氢配合基的混合物催化下在150℃利用Cs2CO3作为碱的反应条件下
芳烃总结
芳香烃 芳香族碳氢化合物简称芳香烃或芳烃,它们基本上都是含有稳定结构苯环,或者苯环稠和环的不饱和烃,有的并没有香味,这类化合物实际上比较稳定。芳烃按其结构可分为3类:单环芳烃、多环芳烃、非苯芳烃。通常所说的芳烃是指分子中含有苯环结构的芳烃,而不含苯环结构的芳烃,称为非苯芳烃。单环:含有一个苯环;多环:含有两个或两个以上独立苯环;非苯芳烃:除苯以外的符合4n+2规则的烃。 物理性质:1苯基本的同系物多为液体,不溶于水,易溶于有机溶剂。单环芳烃的相对密度小于1,但是比佟碳原子数的脂肪烃和脂环烃大,一般在0.8~0.9。有特殊气味,蒸汽有毒,由于含碳量较多燃烧时往往带有黑烟。沸点随相对分子质量的增大而增大,苯的同系物中平均每增加一个CH2沸点升高25℃。含有同碳原子数的苯各种异构体沸点相似。熔点除与相对分子质量有关外,还与其结构有关,分子对称性好,熔点高,如对位结构。 化学性质:一:加成反应 1加氢反应(还原反应):当 分子中,具有侧链的双键时,再 换和条件下,侧链选择加氢,苯环不选择加氢。但是在激烈条件下苯环也可以发生加氢反应。 这是工业上制备环己烷的方法。 Birch还原:碱金属(NaKLi)在液氨与乙醇的混合液中,与芳香化合物反应,苯环可被还原为1,4环己二烯类化合物,叫伯奇还原,例如苯可以被还原为1,4环己二烯。机理为: 首先是钠和液氨作用生成溶剂化电子,此时体系为一蓝色溶液。然后,苯环得到一个电子仍是环状共轭体系,但有一个单电子处在反键轨道上,从乙醇中夺取一个质子生成再取得一个溶剂化电子转变成是一个强碱,可以再从乙醇中夺取一个质子生成1,4一环己二烯。苯的同系物也能发生Birch还原,一取代烃基苯经Birch还原生成1-烃基一1,4一环己二烯。 例如左图。若取代基上有与苯环共扼的双键Birch还原首先在共 轭双键处发生。 不与苯环共扼的双键不能发生Birch还 原。Birch还原反应与苯环的催化氢化不 同,它可使芳环部分还原生成环己二烯类 化合物,因此Birch还原有它的独到之处, 在合成上十分有用。萘同样可以进行Birch还原。蔡发生Birch还原时,可以得到1,4一二氢化蔡和1,4,5,8一四氢化 蔡。蒽和菲的加氢反应也容 易发生在9,10位。 2.加氯反应:紫外光照射下,苯与氯在40℃即可发生加成反应,生成六氯环己烷。 无论加氢还是加氯,反应都不容易停在加一分子或者两分子的阶段,因为加氢或者加氯的中间产物,比苯更容易发生加成反应。蒽和菲的取代反应也容易发生在9,10位。 二.氧化反应:1.笨的氧化苯在高温下与高锰酸钾或者铬酸等强氧化剂同煮,也不会被氧化。只有在五氧化二钒的情况下,苯在高温被氧化成,顺丁烯二酸酐。烷基取代的苯可以被氧化,但是苯环不变,只是和苯环相连的烷基被氧化成羧基,而且不管侧链多长,只要是和 苯环相连的碳上有H,侧链均可呗氧化成羧基,如果两个
卤代芳烃胺基化反应研究进展
卤代芳烃胺基化反应研究进展 摘要:钯催化卤代芳烃胺基化反应是形成CAr-N 的重要方法。以卤代芳烃为线索, 对钯催化偶联胺化反应的研究进展和胺化反应从合成化合物到合成高分子的过渡进行了综述, 介绍了本课题组运用胺化反应合成高性能聚合物聚亚胺酮和聚亚胺醚酮的相关研究。 关键词:卤代芳烃; 钯催化胺化反应; 聚亚胺酮 CAr-N 键普遍存在于生物活性物及药物中,芳胺类化合物广泛用作药物、染料、杀虫剂, 因此含有CAr-N 化合物的合成引起了研究者的兴趣。经典的合成方法有硝化还原法、Ulmann 合成法以及SNAr 合成法。但是这些方法通用性差, 合成步骤多, 化学选择不确定, 需要苛刻的反应条件。所以, 研究者们采用过渡金属催化形成CAr-N 键, 其中钯的效果较好。在过去几年里,因为选择性和官能团兼容性的提高, 钯催化卤代芳烃的胺化已广泛应用于合成芳胺, 并且逐渐发展成一个普遍、可靠和实用的方法[1-4] 。20 世纪初期钯催化的胺化反应实现了从合成化合物到合成高分子的过渡, 2005 年本课题组运用该反应缩聚合成了高相对分子质量的聚亚胺酮, 并实现了聚亚胺酮的功能化[5-12] 。 1 氯代芳烃胺化反应合成化合物 相对于溴代芳烃和碘代芳烃, 氯代芳烃活性低, 但它比相应的溴代芳烃更经济易得。在过去几十年里, 钯催化偶联引起研究者的广泛兴趣, 氯代芳烃的胺化反应的研究取得较好的成绩。目前发现的氯代芳烃胺化反应的催化体系多数使用富电性和大体积的烷基膦以及N 杂环碳烯作为配体。主要配体如下: Beller 等首次实现了氯代芳烃的偶联[ 13] 。Reddy 根据C Cl 的特性, 用富电子和大体积的膦配体( PCy 3或Pi Pr3 ) 使C Cl 断裂, 此配体适用于仲胺和氯代芳烃的偶联( 式1) [ 14] 。配体A、B 不仅能高效地催化仲胺和氯代芳烃的偶联, 也适用于伯烷基胺与邻位有取代的氯代芳烃的反应。脂肪伯胺的胺化活性低, 很难与非活化的氯代芳烃反应。H amann 和Hartw ig 利用大体积配体C 和Pd 的络合物首次成功地催化了两者的反 应( 式2) [ 15] 。Bei 等发现D/ Pd2 ( dba) 3 催化偶联环、链、伯、仲胺与缺电子、富电子氯代芳烃都能得到理想的芳胺化合物( 式3) [ 16]
有机合成中钯催化下的交叉偶联反应
有机合成中钯催化下的交叉偶联反应 -2010年诺贝尔化学奖简介 陈明华 ( 兴义师范学院化学生物系,贵州兴义 562400) 摘要:介绍了2010年诺贝尔化学奖的科学背景,即“有机合成中钯催化下的交叉偶联反应”的产生、发展和应用,体现了有机化学已经发展成为一门艺术形式,在这个形式下,科学家们在试管里创造性的产生出不可思议的化学物质的过程。 关键词:钯催化剂;交叉偶联反应;赫克反应;铃木反应;根岸反应 Palladium-Catalyzed Cross Couplings in Organic Synthesis CHEN Ming-Hua (Department of Chemistry and Biological, Xingyi Normal College, Xingyi, Guizhou 562400) Abstract: This paper introduces scientific background of the Nobel Prize in Chemistry for 2010, it’s palladium-catalyzed cross couplings in organic synthesis.And this fack had been presents that “Organic chemistry has developed into an art form where scientists produce marvelous chemical creations in their test tubes”. Key words: palladium catalyst; cross-coupling reaction; heck reaction; suzuki reaction; negishi reaction 2010年10月6日,瑞典皇家科学院决定授予美国特拉华大学(University of Delaware) 理查德-赫克(Richard F. Heck), 普渡大学(Purdue University)根岸荣一(Ei-ichi Negishi)和日本北海道大学(Hokkaido University)的铃木彰(Akira Suzuki)三位教授2010年的诺贝尔化学奖,以表彰他们在“有机合成中钯催化下的交叉偶联反应”作出的贡献[1]。 碳元素是构成生命的主要物质,这些物质是以C-C键(单键或双键)为基础,形成各种形式的碳胳化合物。人们要想制备新药物、新材料、生物分子和了解生命的过程,最先的方法就是合成一系列碳胳化合物。可见,以简单的有机原料为基础,通过化学反应合成更复杂的化合物(增长碳链),是十分重要的,这种重要性体现在过去历年授予的诺贝尔化学奖:格利雅试剂(Grignard reagent,) 维狄反应(Wittig reaction, 1979年),烯烃的转位反应(Olefin metathesis, 2005年)[1]。 作者简介:陈明华(1966,3-),男,兴义师范学院化学生物系教师,理学硕士,高级实验师、教授。主要研究方向:有机合成。
钯催化下的交叉偶联反应
钯催化下的交叉偶联反应 摘要:钯催化交叉偶联反应是一类用于碳碳键形成的重要化学反应,在有机合成中应用十分广泛。本文综合概述了钯催化交叉偶联反应的原理和发展,简单介绍了它的应用领域。 关键词:钯催化剂;交叉偶联反应;赫克反应;铃木反应;根岸反应 引言 2010年10月6日,瑞典皇家科学院决定授予美国特拉华大学理查德-赫克(Richard F. Heck), 普渡大学根岸荣一(Ei-ichi Negishi)和日本北海道大学的铃木彰(Akira Suzuki)三位教授2010年的诺贝尔化学奖,以表彰他们在“有机合成中钯催化下的交叉偶联反应”作出的贡献。 碳是构筑有机物和生命的基本材料,通过在“碳骨架”上嫁接其他功能性“化学模块”,有机物和生命体方才表现出千变万化的特质。化学家,特别是有机化学家们要做的,就是以人工手段,将各种物质分子以碳化合物“裁剪”、“缝合”,创造出自然界所不存在的新物质。但是要“裁剪”碳并不简单。碳原子非常稳定,它们之间要联接起来,必须要找到一种让碳原子活跃起来的方法。2010年度的三位获奖化学家,就是利用钯来作为催化剂。钯催化剂的作用,先是相当于一把剪刀,剪断碳化学键,然后再如针线一般,把新的功能性基团“缝制”到剪开的碳骨架上。 1、早期研究 碳元素是构成生命的主要物质,这些物质是以C-C键(单键或双键)为基础,形成各种形式的碳胳化合物。人们要想制备新药物、新材料、生物分子和了解生命的过程,最先的方法就是合成一系列碳胳化合物。可见,以简单的有机原料为基础,通过化学反应合成更复杂的化合物(增长碳链),是十分重要的,这种重要性体现在过去历年授予的诺贝尔化学奖:格利雅试剂(Grignard reagent,) 维狄反应(Wittig reaction, 1979年),烯烃的转位反应(Olefin metathesis, 2005年)[1]。 有机合成化学制造出的这几千万种新的物质绝大多数都是以碳原子为主来构建的。为了制备结构更复杂、功能更强大的新型材料, 就要想办法通过各种化学反应将碳原子连接在一起。然而碳原子本身是十分稳定的, 在化学反应中并不活泼, 所以就得想办法来激活碳原子, 让它更容易参与反应并与其他碳原子连接起来, 逐步形成更高层次的碳基骨架。1912 年, 法国人Grignard 因发明有机镁试剂( 格氏试剂) 而荣获诺贝尔化学奖, 可以说是碳基活化史上的第一个里程碑。随着时代的发展, 人们对碳基的研究愈加深入。在研究的前期, 要么