the epoxidation of (S)-sulcatol
210978218_响应面优化金丝小枣碱提多糖工艺及其抗氧化活性研究

王娜,刘玉叶,刘美玲,等. 响应面优化金丝小枣碱提多糖工艺及其抗氧化活性研究[J]. 食品工业科技,2023,44(7):163−169. doi:10.13386/j.issn1002-0306.2022060026WANG Na, LIU Yuye, LIU Meiling, et al. Optimization of Alkali Extraction Process of ‘Jinsixiaozao’ Polysaccharide by Response Surface Methodology and Its Antioxidant Activity[J]. Science and Technology of Food Industry, 2023, 44(7): 163−169. (in Chinese with English abstract). doi: 10.13386/j.issn1002-0306.2022060026· 工艺技术 ·响应面优化金丝小枣碱提多糖工艺及其抗氧化活性研究王 娜1,刘玉叶1,刘美玲1,刘孟军1,2,赵智慧1,2, *(1.河北农业大学园艺学院,河北保定 071001;2.河北农业大学枣研究中心,河北保定 071001)摘 要:以金丝小枣为原料,通过单因素考察提取温度、时间、料液比和NaOH 浓度对多糖得率的影响,在此基础上进行Box-Behnken 试验来优化碱提多糖的提取工艺,并对其进行总糖含量、扫描电镜、红外光谱及抗氧化活性测定。
结果表明,碱提多糖最佳工艺为:料液比1:35(g/mL ),提取温度80 ℃,提取时间120 min ,NaOH 浓度为0.2 mol/L ,在此条件下多糖的得率为11.44%,总糖含量为69.39%。
扫描电镜表明其具有不规则块状结构,内部紧实;红外光谱表明枣碱提多糖具有C=O 和C-H 等酸性多糖的特征吸收峰。
HOBT EDCI

Design,Synthesis,and Biological Activity of Boronic Acid-Based Histone Deacetylase Inhibitors Nobuaki Suzuki,†Takayoshi Suzuki,*,†Yosuke Ota,†Tatsuya Nakano,‡Masaaki Kurihara,§Haruhiro Okuda,§Takao Yamori,# Hiroki Tsumoto,†Hidehiko Nakagawa,†and Naoki Miyata*,†,§Graduate School of Pharmaceutical Sciences,Nagoya City Uni V ersity,3-1Tanabe-dori,Mizuho-ku,Nagoya,Aichi467-8603,Japan,Di V ision of Medicinal Safety Science,National Institute of Health Sciences,1-18-1Kamiyoga,Setagaya-ku,Tokyo158-8501,Japan,Di V ision of Organic Chemistry,National Institute of Health Sciences,1-18-1Kamiyoga,Setagaya-ku,Tokyo158-8501,Japan,and Di V ision of Molecular Pharmacology,Cancer Chemotherapy Center,Japanese Foundation for Cancer Research,3-10-6Ariake,Koto-ku,Tokyo135-8550,Japan Recei V ed February2,2009Guided by the proposed catalytic mechanism of histone deacetylases(HDACs),we designed and synthesizeda series of boronic acid-based HDAC inhibitors bearing an R-amino acid moiety.In this series,compounds(S)-18,20,and21showed potent HDAC-inhibitory activity,highlighting the significance of the(S)-aminoacid moiety.In cancer cell growth inhibition assays,compounds(S)-18,20,and21exerted strong activity,and the values of the ratio of the concentration causing50%growth inhibition(GI50)to the concentrationcausing50%enzyme inhibition(IC50),i.e.,GI50/IC50,were low.The potency of these compounds was similarto that of clinically used suberoylanilide hydroxamic acid(SAHA)(2).The results of Western blot analysisindicated that the cancer cell growth-inhibitory activity of compounds(S)-18,20,and21is the result ofHDAC inhibition.A molecular modeling study suggested that the hydrated boronic acid interacts with zincion,Tyr residue,and His residue in the active site of HDACs.Ourfindings indicate that these boronic acidderivatives represent an entry into a new class of HDAC inhibitors.IntroductionThe acetylation status of lysine residues in nucleosomal histones is tightly controlled by two counteracting enzyme families,the histone acetyl transferases and the histone deacety-lases(HDACs a).1The latter family can be divided into two categories:Zn2+-dependent enzymes and NAD+-dependent enzymes.2The Zn2+-dependent HDACs are closely connected with control of gene expression and cell cycle progression.3The inhibition of HDACs causes histone hyperacetylation and leads to transcriptional activation of genes such as p21WAF/CIP1,Gadd 45,FAS,and caspase-3,which are associated with growth arrest and apoptosis in tumor cells.4Indeed,HDAC inhibitors,such as trichostatin A(TSA,1),5suberoylanilide hydroxamic acid (SAHA,also known as vorinostat,2),6PXD-101(3),7and MS-275(4)8(Chart1),have been reported to inhibit cell growth, induce terminal differentiation in tumor cells,and prevent the formation of malignant tumors,and they have been developed as antitumor agents.Among them,SAHA has recently been approved by the FDA for treatment of cutaneous T-cell lymphoma.In addition,there are several lines of evidence indicating that HDAC inhibitors are effective as therapeutic agents for other diseases,including inflammation and neuro-degenerative diseases.9Moreover,it has recently been reported that HDAC inhibitors,such as TSA(1),SAHA(2),and valproic acid,improve the efficiency of induction of pluripotent stem cells without introduction of the oncogene c-Myc,suggesting the importance of HDAC inhibitors in regenerative medicine.10Consequently,there are many ongoing research programs to find more potent and selective HDAC inhibitors.11To date,X-ray crystal structures of human HDAC4,12 HDAC7,13HDAC8,14and archaebacterial HDAC-like protein (HDLP)15have been published.A number of potent and selective inhibitors have been identified by structure-based drug design using these crystal structures.11Recently,the catalytic mechanism of the deacetylation of acetylated lysine substrates by HDACs was proposed,based on a density functional theory QM/MM study of HDLP,16and this also offers many clues for the design of HDAC inhibitors.Herein we report the synthesis and biological activity of boronic acid-based HDAC inhibitors, which were designed on the basis of the proposed catalytic mechanism of HDACs.ChemistryCompounds5-26prepared for this study are shown in Tables 1-3.The routes used for the synthesis of the compounds are shown in Schemes1-5.Scheme1shows the preparation of R-amino acid derivatives5-10.These compounds were syn-thesized from diethyl pound27was treated with(Boc)2O to yield N-Boc pound 28was allowed to react with5-bromopent-1-ene in the presence of NaOEt in refluxing EtOH to give C-alkylated compound29. Hydrolysis of the ethyl ester of29with an equivalent amount of LiOH gave monocarboxylic acid30,and subsequent decar-boxylation reaction afforded R-amino acid derivative31. Compound31was hydrolyzed with LiOH followed by treatment with an appropriate aromatic amine in the presence of EDCI and HOBt to give amides33-38.The hydroboration reaction of amides33-38with pinacolborane using[Ir(cod)Cl]2catalyst and dppm or dppe ligand gave terminal boronic esters39-44.17 Hydrolysis of the boronic esters39-44using NH4OAc and NaIO4in acetone-water yielded boronic acids5-10.18 Scheme2shows the preparation of boronic acids11-16,in which the R-amino moiety of compounds5-10was removed.*To whom correspondence should be addressed.For T.S.:phone andfax,+81-52-836-3407;e-mail,suzuki@phar.nagoya-cu.ac.jp.For N.M.:phone and fax,+81-52-836-3407;miyata-n@phar.nagoya-cu.ac.jp.†Nagoya City University.‡Division of Medicinal Safety Science,National Institute of HealthSciences.§Division of Organic Chemistry,National Institute of Health Sciences.#Japanese Foundation for Cancer Research.a Abbreviations:HDAC,histone deacetylase;TSA,trichostatin A;SAHA,suberoylanilide hydroxamic acid;HDLP,HDAC-like protein.J.Med.Chem.2009,52,2909–2922290910.1021/jm900125m CCC:$40.75 2009American Chemical SocietyPublished on Web04/14/2009These compounds were synthesized from hept-6-enoic acid 45using the same synthetic approach as described for compounds 5-10.Scheme 3shows the preparation of R -amino acid derivatives 17-24,in which the Boc group of 6was replaced with various carbonyl moieties.These compounds were prepared from 40in three steps:deprotection of the Boc group,condensation with an appropriate aromatic carboxylic acid,and hydrolysis of the boronic esters.Scheme 4shows the preparation of malonic acid derivatives 25and 26,in which the R -aminocarbonyl moiety of compounds 18and 20was replaced with the corresponding carboxamide.These compounds were synthesized from tert -butyl ethyl malonate 66.Treatment of 66with 5-bromopent-1-ene in the presence NaH in THF yielded compound 67.The ethyl ester of 67was hydrolyzed with an equivalent amount of LiOH,followed by condensation with biphenyl-3-ylamine to give amide 69.Hydroboration,deprotection of tert -butyl ester using TFA,condensation with an appropriate aromatic amine,and hydrolysis of the boronic ester gave the malonic acid derivatives 25and 26.Scheme 5shows the preparation of optically active R -amino acid derivatives (S )-and (R )-18,20,21.These compounds were synthesized using the procedure reported by Nishino and co-workers.19Compound (RS )-31was subjected to the action of subtilisin from Bacillus licheniformis in a mixture of DMF and water to yield (S )-32as a white solid.The recovered (R )-31was hydrolyzed with LiOH to afford (R )-32.The synthesis of (S )-and (R )-18,20,21was accomplished from (S )-32or (R )-32using the procedure described for the synthesis of R -amino acid derivatives (shown in Schemes 1and 3).The enantiomeric excess of (S )-and (R )-18,20,21was determined to be >95%by chiral column chromatography.Results and DiscussionDrug Design.In 1999,Finnin et al.reported X-ray crystal structures of HDLP complexed with inhibitors.15The enzyme contains a zinc ion at the bottom of the active site,and the active center consists of a tyrosine,two aspartic acids,and three histidines.In 2006,Corminboeuf et al.carried out density functional theory QM/MM studies on the deacetylation reaction catalyzed by HDLP.16The proposed mechanism suggested by the calculation results is depicted in Figure 1a.In this mecha-nism,the carbonyl oxygen of the substrate binds the zinc ion and is located adjacent to a water molecule.The electrophilicity of the carbonyl carbon is increased by coordination to the zinc ion,and so the carbonyl carbon is attacked by the water molecule activated by His 140and His 141(HDAC1number-ing).This nucleophilic attack results in a tetrahedral transition state,which is stabilized by a zinc -oxygen interaction and by hydrogen bonds with Tyr 303and His 140.In the final step,proton transfer from His 141to the nitrogen of the intermediate triggers scission of the carbon -nitrogen bond to afford the acetate and lysine products.In designing selective HDAC inhibitors,we focused on the proposed deacetylation mechanism.On the basis of this mech-anism,we designed R -amino acid derivatives A (Figure 1b)in which the acetamide of the acetylated lysine substrate is replaced by boronic acid.Because the boron atom of the boronic acids A has a vacant p-orbital,it could be attacked by a water molecule in the active site.The generated boronic acid -H 2O “ate”complex could act as a transition state analogue of the deacetylation of acetylated lysine substrates.That is,the hydrated boronic acid would bind the zinc ion and form two hydrogen bonds with Tyr 303and His 140,which would lead to HDAC inhibition.Enzyme Assays.In our exploratory study on boronic acid-based HDAC inhibitors,we initially focused on the R 1group (Table 1and Supporting Information Figure S1).We chose various aromatic rings as the R 1group because we had already discovered that thiolate compounds bearing aromatic groups such as phenyl,3-biphenyl,and 3-quinolinyl potently inhibited HDACs both in enzyme assays and in cellular assays.11BoronicChart 1.Examples of HDACInhibitorsScheme 1aaReagents and conditions:(a)(Boc)2O,Et 3N,THF,room temp,81%;(b)NaOEt,5-bromopent-1-ene,EtOH,reflux,89%;(c)LiOH ·H 2O,EtOH,H 2O,0°C,94%for 32,99%for 30;(d)toluene,reflux,94%;(e)R 1-NH 2,1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI),1-hydroxybenzo-triazole hydrate (HOBt ·H 2O),DMF,room temp,67-95%;(f)[Ir(cod)Cl]2,bis(diphenylphosphino)methane (dppm)or 1,2-bis(diphenylphosphino)ethane (dppe),pinacolborane,CH 2Cl 2,room temp,32-93%;(g)NH 4OAc,NaIO 4,acetone,H 2O,room temp,10-92%.2910Journal of Medicinal Chemistry,2009,Vol.52,No.9Suzuki et al.acids 5-10,in which R 1is various aromatic groups and R 2is -NHBoc,were initially tested with an in vitro assay using HeLa nuclear extract with high HDAC activity (Table 1and Figure S1).Compounds 5-10showed HDAC-inhibitory activity at a concentration of 100µM,while the 3-biphenyl 6,3-quinolinyl 7,and benzthiazole 8compounds were more potent (78-86%inhibition at 100µM).Compounds 6,7,and 8inhibited HDAC activity dose-dependently with IC 50values of 12,16,and 23µM,respectively;these values are much lower than those of the other aromatic analogues 5,9,and 10.We also tested compounds 11-16,which lack the NH-Boc group of com-pounds 5-10,and they were found to be much less potent inhibitors than compounds 5-10(Table 1and Figure S1),suggesting the significance of the R -amino acid moiety.To find more potent HDAC inhibitors,we next focused on the tert -butoxy group of compound 6,the most potent inhibitorScheme 2aaReagents and conditions:(a)R 2-NH 2,EDCI,HOBt,DMF,room temp,72-95%;(b)[Ir(cod)Cl]2,dppm or dppe,pinacolborane,CH 2Cl 2,room temp,13-74%;(c)NH 4OAc,NaIO 4,acetone,H 2O,room temp,35-94%.Scheme 3aaReagents and conditions:(a)HCl,AcOEt,CHCl 3,room temp,100%;(b)R 3-COOH,EDCI,HOBt,DMF,room temp,11-100%;(c)R 3-COCl,Et 3N,CH 2Cl 2,N,N -dimethyl-4-aminopyridine (DMAP),room temp,41%;(d)NH 4OAc,NaIO 4,acetone,H 2O,room temp,19-82%.Scheme 4aaReagents and conditions:(a)NaH,5-bromopent-1-ene,THF,reflux,52%;(b)LiOH ·H 2O,tert -BuOH,H 2O,0°C,100%;(c)biphenyl-3-ylamine,EDCI,HOBt,DMF,room temp,76%;(d)[Ir(cod)Cl]2,dppm,pinacolborane,CH 2Cl 2,room temp,70%;(e)TFA,room temp,100%;(f)(i)oxalyl dichloride,DMF,CH 2Cl 2,0°C;(ii)R 4-NH 2,Et 3N,CH 2Cl 2,0°C,29%for 71,51%for 72;(g)NH 4OAc,NaIO 4,acetone,H 2O,room temp,39%for 26,71%for 25.Boronic Acid-Based Inhibitors Journal of Medicinal Chemistry,2009,Vol.52,No.92911in Table 1.We initially changed the Boc group of compound 6to a Cbz group (compound 17)(Table 2and Supporting Information Figure S2).Compound 17exhibited about 3-fold higher potency than compound 6.We also examined the activity of compound 18,in which the benzyloxy group of compound 17was replaced with a simple benzene ring,and the activity of 18(IC 50) 2.0µM)was about 2-fold higher than that of compound 17.By the mimicking of the partial structure of TSA (1),we introduced the dimethylamino group at the 4-position of the phenyl ring of compound 18,but compound 19displayed a 6.5-fold decrease in potency compared to the parent compound 18.Next,we converted the phenyl ring of compound 18to various heteroaryl rings.The 2-indole 23and 2-quinoline 24compounds exhibited HDAC-inhibitory activity less potent than that of the phenyl compound 18,whereas the 3-pyridine 20,5-thiazole 21,and 4-thiazole 22derivatives showed potencies similar to or greater than that of the phenyl compound 18(IC 50values:20)2.0µM,21)1.4µM,22)4.7µM),highlighting the importance of small ring size.To confirm the importance of R -amino acid structure,we examined the activity of malonic acid derivatives 25and 26in which the R -aminocarbonyl moiety of compounds 18and 20is replaced by the corresponding carboxamide (Table 2and Figure S2).Compounds 25and 26showed about 15-and 25-fold decreases in potency compared with the parent compounds 18and 20,respectively.Scheme 5aaReagents and conditions:(a)subtilisin,H 2O/DMF (1:3),37°C,42%for (R )-31,50%for (S )-32;(b)LiOH ·H 2O,EtOH,H 2O,room temp,100%;(c)biphenyl-3-ylamine,EDCI,HOBt,DMF,room temp,56-65%;(d)[Ir(cod)Cl]2,dppm,pinacolborane,CH 2Cl 2,room temp,79-82%;(e)HCl,AcOEt,CHCl 3,room temp,100%;(f)R 5-COOH,EDCI,HOBt,DMF,room temp,64-90%;(g)NH 4OAc,NaIO 4,acetone,H 2O,room temp,4-87%.Figure 1.(a)Proposed mechanism for the deacetylation of acetylated lysine substrates.(b)Proposed mechanism of inhibition of HDACs by boronic acids A .2912Journal of Medicinal Chemistry,2009,Vol.52,No.9Suzuki et al.Next we examined the influence of optical -pounds (R )-18,20,21and (S )-18,20,21were prepared and evaluated for their inhibitory effects on HDAC1,HDAC2,HDAC6,and HDAC8,as well as total HDACs from nuclear extracts.As shown in Table 3and Supporting Information Figure S3,in all cases,(S )-18,20,and 21were at least 10-fold more active than (R )-18,20,and 21,respectively.Since the stereochemistry of (S )-18,20,and 21is the same as that of natural lysine,these results suggest that these boronic acids act in the active site of HDACs.Among (S )-18,20,and 21,(S )-21was found to be the most potent pound (S )-21inhibited all HDACs with IC 50values in the micromolar or submicromolar range.Although (S )-21was less potent than SAHA (2),the isoform inhibition profile of (S )-21was similar to that of 2,which is the only HDAC inhibitor currently used in clinical practice.Cellular Assays.To examine the effectiveness of boronic acid-based HDAC inhibitors as anticancer drugs and tools for biological research,compounds (S )-18,20,and 21,as well as SAHA (2),were tested in a cancer cell growth inhibition assay.For initial screening,we used stomach cancer MKN45cells because HDAC inhibitors have been reported to inhibit the cell growth of MKN45cells.20The results are summarized in Ta-ble 4.SAHA (2)and its derivatives have been reported to show potent HDAC-inhibitory activity in enzyme assays and cancer cell growth inhibition assays.21However,a relatively large shift in potency between enzyme assay and cellular assay (GI 50/IC 50,abbreviated as potency shift in this paper)was observed with those hydroxamate HDAC inhibitors.21Indeed,as shown in Table 4,SAHA (2)exhibited a large potency shift in the cancerTable 1.HDAC Inhibitory Activity of Boronic Acids 5-16aaValues are the mean of at least three experiments.IC 50of SAHA (2)is 0.28µM.Table 2.HDAC Inhibitory Activity of 17-26aaValues are the mean of at least three experiments.Table 3.HDAC Inhibitory Activity of (S )-and (R )-18,20,21aIC 50(µM)class Icompd HDAC (nuclear extract)HDAC1HDAC2HDAC8class II,HDAC620.280.350.25 1.70.028(S )-18 1.6>10b 3.6>100 1.1(R )-1817>100>100>10024(S )-20 1.4 2.80.83230.32(R )-2015>10099>10014(S )-210.92 2.20.53 6.60.11(R )-2124>100>100>1005.6aValues are the mean of at least three experiments.b 38%inhibition at 10µM.Table 4.Growth-Inhibitory Activity on MKN45Cells and GI 50/IC 50Values of SAHA (2)and (S )-18,20,21aGI 50/IC 50compd GI 50(µM)HDAC HDAC1HDAC227.3262129(S )-189.3 5.8<0.93 2.6(S )-20 5.5 3.9 2.0 6.6(S )-213.53.81.66.6aValues are the mean of two separate experiments.Boronic Acid-Based InhibitorsJournal of Medicinal Chemistry,2009,Vol.52,No.92913cell growth inhibition assay using MKN45cells.22The reason for the large potency shift of SAHA (2)is unclear,but it seems reasonable to assume that it is at least partly due to poor membrane permeability resulting from the highly polar character of hydroxamic ing Advanced Chemistry Development (ACD/Laboratories)software,version 8.14for Solaris,the log D (pH 7)values of methylhydroxamic acid (CH 3CONHOH)and methylboronic acid (CH 3B(OH)2)were calculated to be -1.59and 1.0,respectively.The calculation results suggest that boronic acid is more lipophilic than hydroxamic acid under physiological conditions,and boronic acid derivatives might permeate through the cell membrane more efficiently than hydroxamates;there-fore,they might not show such large potency shifts as hydroxamates like SAHA (2).We then evaluated the growth-inhibitory activity of (S )-18,20,and 21on MKN45cells.As we expected,(S )-18,20,and 21inhibited the growth of MKN45cells and showed smaller potency shift values than SAHA (2).Among (S )-18,20,and 21,compound (S )-21showed the highest activity,being more potent than SAHA (2).Next,we evaluated growth inhibition by SAHA (2)and compound (S )-21against nine human cancer cell lines (Table 5).Compound (S )-21exerted potent growth inhibition against various human cancer cells,with GI 50values ranging from 0.73to 9.1µM,and these inhibitory activities were similar to those of SAHA (2)(average GI 50of (S )-21is 4.0µM,that of SAHA is 6.0µM).In addition,compound (R )-21did not show strong activity against these cancer cell lines (Table S1and Figure S4in Supporting Information)with GI 50s values ranging from 13to 21µM,suggesting the responsibility of HDAC inhibition for cancer cell growth repression.Next,we performed Western blot analysis to examine the HDAC-inhibitory effects of boronic acid derivatives in cells.Since nuclear HDACs such as HDAC 1and HDAC2catalyze the deacetylation of histones,3d the acetylation level of histone H4in human colon cancer HCT116cells was analyzed after treatment of the cells with compounds (S )-18,20,and 21.As can be seen in Figure 2,the level of acetylated histone H4was elevated dose-dependently.The level of acetylated R -tubulin was also investigated because (S )-18,20,and 21displayed potent inhibition of HDAC6,which is reported to catalyze the deacetylation of R -tubulin.23As expected,compounds (S )-18,20,and 21caused a dose-dependent increase in acetylation of R -tubulin.These results suggest that the cancer cell growth inhibition by boronic acids (S )-18,20,and 21was the result of HDAC inhibition.Molecular Modeling.Since the results of the enzyme assays (Table 3)suggested that boronic acids act within the active center of HDACs,we studied the binding mode of hydrated (S )-21within this site.We calculated the low energy conforma-tion of the hydrated (S )-21complex docked in a model based on the crystal structure of HDAC8(PDB code 1T67)using Macromodel 9.6software (Figure 3).An inspection of the complex shows that one of the oxygen atoms of the hydrated boronic acid can coordinate to the zinc ion (distance between oxygen and zinc,2.04Å)(Figure 3,left).In addition,the hydrogen of the OH group of Tyr 306is located 1.95and 2.20Åfrom the two oxygen atoms of the hydrated boronic acid,Table 5.Growth Inhibition of Various Cancer Cells Using SAHA (2)and (S )-21aGI 50(µM)cell2(S )-21HBC-5breast cancer17 6.5SNB-78central nervous system 169.1HCT116colon cancer 0.580.73DMS114lung cancer 2.9 2.4LOX-IMVI melanoma 1.3 1.3SK-OV-3ovarian cancer 2.5 2.2RXF-631L renal cancer 2.0 3.2MKN45stomach cancer 7.3 3.5PC-3prostate cancer 4.6 6.7mean6.04.0aValues arethe mean of two separate experiments.Figure 2.Western blot detection of acetylated R -tubulin and histone H4levels in HCT116cells after 8h treatment with (S )-18,20,21and reference compound 2.Figure 3.View of the conformation of (S )-21(ball-and-stick)docked in the HDAC8catalytic core.Residues within 5Åfrom the zinc ion are displayed in the tube graphic (left),and the surface of the enzyme is displayed in gray (right).2914Journal of Medicinal Chemistry,2009,Vol.52,No.9Suzuki et al.which suggests that Tyr306can form two hydrogen bonds with the hydrated boronic acid.Moreover,a short distance(2.22Å) between one of the hydrogens of the hydrated boronic acid and the nitrogen of His142was also observed.This indicates that hydrated boronic acids inhibit HDACs by interacting with the zinc ion,Tyr residue,and His residue in the active center.On the surface of HDAC8,the biphenyl ring is located in the hydrophobic region formed by the benzene rings of Tyr100 and Phe152and the alkyl chain of Lys33,and the thiazole ring lies in the hydrophobic area formed by the benzene ring of Tyr100and the methylene groups of Asp101,Gly151,and Phe208(Figure3,right).It is also suggested that two hydrogen bonds can be formed between the two NH groups and the carboxylate anion of Asp101(the distances are1.72and1.74Å).The observed interactions between(S)-21and HDAC8on the surface of the enzyme suggest the importance of the amino acid structure and the(S)-configuration of the inhibitor. ConclusionOn the basis of the proposed catalytic mechanism for the deacetylation of acetylated lysine substrates by HDACs,we designed a series of boronic acids bearing an R-amino acid moiety as candidate mechanism-based HDAC inhibitors.Among the synthesized compounds,compounds(S)-18,20,and21 displayed potent HDAC-inhibitory activities,suggesting that these boronic acids act in the active site of pounds (S)-18,20,and21also showed cancer cell growth-inhibitory activities as potent as SAHA(2).Intracellular HDAC inhibition by compounds(S)-18,20,and21was confirmed by Western blot analysis of acetylated histone H4and acetylated R-tubulin.A molecular modeling study of the HDAC8/(S)-21complex suggested that the hydrated boronic acid interacts with zinc ion, Tyr residue,and His residue in the active site of HDACs. Thus,we have identified a novel lead structure from which it should be possible to develop more potent HDAC inhibitors. Although boronic acids have been used as inhibitors for various hydrolytic enzymes such as serine protease,24arginase,25and proteasome,26this is thefirst report of HDAC inhibitors containing a boronic acid moiety.The results obtained in this study suggest that boronic acid-based inhibitors of HDACs have considerable potential for the development of novel therapeutic agents and tools for biological research.Experimental SectionChemistry.Melting points were determined using a Yanagimoto micro melting point apparatus or a Bu¨chi545melting point apparatus and were left uncorrected.Proton nuclear magnetic resonance spectra(1H NMR)and carbon nuclear magnetic resonance spectra(13C NMR)were recorded with a JEOL JNM-LA500,JEOL JNM-A500,or Bruker Avance600spectrometer in solvents as indicated.Chemical shifts(δ)are reported in parts per million relative to the internal standard tetramethylsilane.Elemental analysis was performed with a Yanaco CHN CORDER NT-5analyzer,and all values were within(0.4%of the calculated values,which indicates>95%purity of the tested compounds.Fast atom bombardment(FAB)mass spectra were recorded on a JEOL JMS-SX102A mass spectrometer.In positive-mode MS analysis using 3-nitrobenzyl alcohol(NBA)as a matrix,boronic acids are esterified with NBA and the molecular weight detected corresponds to(M+2NBA-2H2O+H)+or(M+2NBA-2H2O)+.In negative-mode analysis using glycerol(Gly)as a matrix,the molecular weight detected corresponds to(M+Gly-2H2O-H)-.HPLC was performed with a Shimazu instrument equipped with a CHIRALPAK IA column(4.6mm×250mm,Daicel Chemical Industries),and the samples eluted at1mL/min with ethanol and n-hexane.Reagents and solvents were purchased from Aldrich,Tokyo Kasei Kogyo,Wako Pure Chemical Industries,and Kanto Kagaku and used without purification.Flash column chromatog-raphy was performed using silica gel60(particle size0.046-0.063 mm)supplied by Merck.6-tert-Butoxycarbonylamino-7-oxo-7-phenylaminoheptylbo-ronic Acid(5).Step1:Preparation of Diethyl2-tert-Butoxy-carbonylaminomalonate(28).To a suspension of diethyl ami-nomalonate(27;10.0g,47.2mmol)and Et3N(32.9mL,236mmol) in THF(20mL)was added a solution of(Boc)2O(20.6g,94.4 mmol)in THF(10mL),and the solution was stirred overnight at room temperature.The reaction mixture was poured into water and extracted with AcOEt.The AcOEt layer was washed with water and brine and dried over Na2SO4.Filtration and concentration in vacuo and purification by silica gelflash column chromatography (AcOEt/n-hexane)1/5)gave10.5g(81%)of28as a colorless oil:1H NMR(CDCl3,500MHz,δ,ppm)5.56(1H,d,J)6.7Hz), 4.95(1H,d,J)7.6Hz),4.27(4H,m),1.45(9H,s),1.30(6H,t, J)7.3Hz).Step2:Preparation of Diethyl2-tert-Butoxycarbonylamino-2-(penten-4-yl)malonate(29).To a solution of28(10.2g,37.2 mmol)obtained above in EtOH(15mL)was added NaOEt(2.78 g,40.9mmol)under Ar gas at0°C.The reaction mixture was stirred at0°C for7min.After that,to the solution was added 5-bromopent-1-ene(8.32g,55.8mmol),and the mixture was refluxed for10h.It was then poured into water and extracted with AcOEt.The AcOEt layer was washed with water and brine and dried over Na2SO4.Filtration and concentration in vacuo and purification by silica gelflash column chromatography(AcOEt/n-hexane)1/6)gave11.3g(89%)of29as a colorless oil:1H NMR (CDCl3,500MHz,δ,ppm)5.94(1H,broad s),5.76(1H,ddt,J) 17,10,6.4Hz),5.00(1H,dd,J)15,1.8Hz),4.95(1H,d,J)10 Hz),4.26-4.18(4H,m),2.28(2H,m),2.07(2H,q,J)7.3Hz), 1.43(9H,s),1.30-1.19(8H,m).Step3:Preparation of2-tert-Butoxycarbonylamino-2-(ethoxy-carbonyl)hept-6-enoic Acid(30).To a solution of29(11.3g,33.0 mmol)obtained above in EtOH/H2O(20mL/10mL)was added LiOH·H2O(1.52g,36.3mmol),and the solution was stirred overnight at0°C.The reaction mixture was neutralized with10% citric acid and extracted with AcOEt.The AcOEt layer was washed with brine and dried over Na2SO4.Filtration and concentration in vacuo gave10.3g(99%)of30as a yellow oil:1H NMR(CDCl3, 500MHz,δ,ppm)5.80-5.71(2H,m),5.01(1H,dd,J)17,1.5 Hz),4.97(1H,dd,J)10,1.8Hz),4.31-4.18(2H,m),2.22(2H, m),2.07(2H,m),1.44(9H,s),1.39-1.22(5H,m).Step4:Preparation of Ethyl2-tert-Butoxycarbonylamino-hept-6-enoate(31).A solution of30(10.3g,32.6mmol)obtained above in toluene(40mL)was refluxed for9h.The reaction mixture was concentrated and purified byflash column chromatography (AcOEt/n-hexane)1/6)to give8.51g(94%)of31as a yellow oil:1H NMR(CDCl3,500MHz,δ,ppm)5.77(1H,ddt,J)17, 10,6.7Hz),5.04-4.94(3H,m),4.28(1H,m),4.19(2H,m),2.07 (2H,m),1.81(1H,m,),1.63(1H,m),1.53-1.37(11H,m),1.28 (3H,t,J)7.3Hz).Step5:Preparation of2-tert-Butoxycarbonylaminohept-6-enoic Acid(32).To a solution of31(8.34g,30.8mmol)obtained above in EtOH/H2O(20mL/10mL)was added LiOH·H2O(1.25 g,29.8mmol),and the solution was stirred overnight at0°C.The reaction mixture was neutralized with10%citric acid and extracted with AcOEt.The AcOEt layer was washed with brine and dried over Na2SO4.Filtration and concentration in vacuo gave7.01g (94%)of32as a white solid:1H NMR(CDCl3,500MHz,δ,ppm) 5.78(1H,ddt,J)17,10,6.7Hz),5.06-4.94(3H,m),4.32(1H, m),2.09(2H,m),1.87(1H,m),1.68(1H,m),1.57-1.37(11H, m).Step6:Preparation of tert-Butyl1-(Phenylaminocarbonyl) hex-5-en-1-ylcarbamate(33).To a solution of32(1.00g,4.11 mmol)and aniline(560µL,6.17mmol)in DMF(15mL)were added1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochlo-ride(EDCI;1.18g,6.17mmol)and1-hydroxy-1H-benzotriazole monohydrate(HOBt·H2O;834mg,6.17mmol),and the mixture was stirred overnight at room temperature.The reaction mixtureBoronic Acid-Based Inhibitors Journal of Medicinal Chemistry,2009,Vol.52,No.92915。
银柴胡多糖超声辅助提取工艺优化及抗氧化活性分析

王红,彭励,宋乐,等. 银柴胡多糖超声辅助提取工艺优化及抗氧化活性分析[J]. 食品工业科技,2024,45(1):185−191. doi:10.13386/j.issn1002-0306.2023030025WANG Hong, PENG Li, SONG Le, et al. Optimization of Ultrasonic-assisted Extraction Process and Analysis of Antioxidant Activity of Polysaccharide from Stellariae Radix[J]. Science and Technology of Food Industry, 2024, 45(1): 185−191. (in Chinese with English abstract). doi: 10.13386/j.issn1002-0306.2023030025· 工艺技术 ·银柴胡多糖超声辅助提取工艺优化及抗氧化活性分析王 红1,彭 励1,2, *,宋 乐1,冯 璐1,李振凯1,李彦青1,高 跳1(1.宁夏大学生命科学学院,宁夏银川 750021;2.宁夏天然药物工程技术研究中心,宁夏银川 750021)摘 要:目的:为了提高银柴胡多糖得率,对银柴胡多糖提取工艺参数进行优化,并评价其体外抗氧化活性。
方法:采用超声辅助提取银柴胡多糖,在单因素实验基础上结合响应面法(Box–Behnken Response Surface )对提取工艺参数进行优化,并采用Sevag 法除蛋白得银柴胡粗多糖,进一步对其抗氧化活性进行分析。
结果:优化后银柴胡多糖最佳提取工艺参数为超声温度50 ℃、时间3.20 h 、提取次数2 次,在此条件下多糖得率最高,为28.24%±0.10%,多糖含量为59.13%;体外抗氧化测定结果显示,银柴胡粗多糖清除DPPH 自由基、OH 自由基、ABTS +自由基的IC 50分别是5.47、2.40和1.44 mg/mL ,表明其具有一定的抗氧化能力。
The Chemistry of Green Synthesis and Catalysis

The Chemistry of Green Synthesis andCatalysisIntroductionThe modern world is facing many environmental problems caused by human activities. Environmental pollution, climate change, and depletion of natural resources are the major concerns. To address these issues, scientists are exploring ways to develop sustainable technologies and practices. The field of green chemistry aims to reduce the environmental impact of chemical processes by minimizing waste, using renewable resources, and reducing toxic substances. Green synthesis and catalysis are two important subfields of green chemistry, which have great potential for sustainable development.What is Green Synthesis?Green synthesis refers to the development of chemical reactions that are environmentally benign and sustainable. It involves using renewable resources, reducing the use of hazardous chemicals, and minimizing waste. Green synthesis is important because traditional chemical processes are often resource-intensive, produce large amounts of waste, and use toxic solvents, which can have adverse effects on human health and the environment.Green synthesis can be achieved by several methods, including using bio-based feedstocks, replacing hazardous solvents with benign ones, using microwave or ultrasound-assisted reactions, and using catalysts. Catalysts are an important tool in green synthesis, as they often allow the reaction to proceed more efficiently and with fewer environmental impacts.What is Catalysis?Catalysis is the process of accelerating a chemical reaction by adding a substance known as a catalyst. A catalyst works by lowering the activation energy required for the reaction to occur. It does not affect the thermodynamics of the reaction and is notconsumed in the reaction. Therefore, catalysts can be used repeatedly to speed up reactions without being depleted.Catalysis plays a crucial role in many industrial processes, including the production of fuels, polymers, and pharmaceuticals. However, traditional catalytic processes often use high temperatures and pressures, require toxic solvents, and produce hazardous waste. This is where green catalysis comes in.What is Green Catalysis?Green catalysis is a branch of green chemistry that focuses on developing sustainable catalysts and catalytic processes. Its goal is to reduce the environmental impact of catalytic reactions by using renewable resources, minimizing waste and toxicity, and improving efficiency.Green catalysis has many benefits over traditional catalysis. For example, it can reduce energy consumption and therefore lower greenhouse gas emissions. It can also use non-toxic and renewable resources, which can reduce the environmental impact of the reaction. In addition, green catalysis often uses less hazardous solvents, which improves the safety of the reaction for workers and reduces the risk of contaminated waste.Green Synthesis and Catalysis in ActionThere are many examples of green synthesis and catalysis being used in industrial processes. For example, the production of biodiesel is a green synthesis process that uses vegetable oil or animal fat as a renewable feedstock. The reaction is catalyzed by sodium hydroxide or potassium hydroxide, which are both inexpensive and non-toxic catalysts.Another example is the production of pharmaceuticals using enzyme catalysis. Enzymes are biocatalysts that work under mild conditions of temperature and pressure and produce little or no waste. Their use in the production of pharmaceuticals can reduce the environmental impact of the process and improve the safety for workers.ConclusionGreen synthesis and catalysis are two important subfields of green chemistry that have great potential for sustainable development. By using renewable resources, minimizing waste and toxicity, and improving efficiency, these processes can reduce the environmental impact of chemical reactions. Green synthesis and catalysis are already being used in many industrial processes, and their use is likely to increase in the future as the demand for sustainable technologies and practices grows.。
中国弯颈霉多糖抗氧化和抑制氧化应激活性研究

白明健,周颖,程昊,等. 中国弯颈霉多糖抗氧化和抑制氧化应激活性研究[J]. 食品工业科技,2024,45(2):333−341. doi:10.13386/j.issn1002-0306.2023030199BAI Mingjian, ZHOU Ying, CHENG Hao, et al. Antioxidant and Oxidative Stress Inhibitory Activities of Tolypocladium sinense Polysaccharide[J]. Science and Technology of Food Industry, 2024, 45(2): 333−341. (in Chinese with English abstract). doi:10.13386/j.issn1002-0306.2023030199· 营养与保健 ·中国弯颈霉多糖抗氧化和抑制氧化应激活性研究白明健,周 颖,程 昊,边 聪,李名正,李 林*,张春晶*(齐齐哈尔医学院医学技术学院,黑龙江齐齐哈尔 161006)O −2摘 要:目的:探究中国弯颈霉菌丝体多糖(Tolypocladium sinense polysaccharide ,TSP )的体外抗氧化活性和抑制过氧化氢诱导小鼠胰岛MIN6细胞氧化应激导致的细胞凋亡。
方法:采用热水浸提法提取中国弯颈霉菌丝体多糖,随后测定其超氧阴离子自由基(superoxide anion ,·)、羟自由基(hydroxy radical ,·OH )、对1,1-二苯基苦基苯肼自由基(P-1,1-diphenylpicryl phenylhydrazine radical ,DPPH·)清除能力;采用200 μmol/L 过氧化氢(hydrogen peroxide ,H 2O 2)诱导小鼠胰岛MIN6细胞氧化应激,给予高剂量和低剂量TSP (0.625、0.156 mg/mL )进行保护,MTT 法测定MIN6细胞生存率;倒置显微镜观察细胞形态;采用试剂盒测定培养基中乳酸脱氢酶(lactate dehydrogenase ,LDH )水平,细胞内超氧化物歧化酶(superoxide dismutase ,SOD )活性和丙二醛(malondialdehyde ,MDA )含量;流式细胞术检测细胞凋亡;Western Blot 检测核因子E2相关因子2(nuclear factor E2-related factor2,Nrf-2)和磷酸化c-Jun 氨基末端激酶(phosphorylated c-jun N-terminal kinase ,pJNK )相对表达量。
不同酶消化法提取猪原代肝细胞的效果比较

532024.4·试验研究0 引言猪圆环病毒(PCV )是Circoviridae 科Circovirus 属的一种无囊膜的单链环状DNA 病毒。
在已知的4个血清型中,PCV2为猪易感的致病性病毒[1]。
PCV2感染会诱导宿主免疫抑制引起猪圆环病毒病(PCVD ),包括断奶仔猪多系统衰竭综合征、新生仔猪先天性脑震颤、皮炎与肾病综合征、猪呼吸道病综合征、母猪繁殖障碍等,给全世界养猪业带来较大的经济损失,是世界各国的兽医与养猪业者公认的造成重大影响的猪传染病[2]。
PCV2的感染在猪生长发育的不同阶段有不同的组织嗜性。
但无论是胎儿阶段还是出生后,肝细胞都是PCV2感染和复制的靶细胞。
因此,PCV2也被视为一种能够诱导猪肝炎的病毒[3]。
且PCV2诱导的肝细胞凋亡在PCV2引发的相关病变和疾病的发病机制中具有关键性作用[4]。
因此,方便、快捷地获取大量有活性的猪肝细胞对于研究PCVD 的致病机制具有重大意义。
目前获取肝细胞常用的方法主要包括机械分离细胞法、非酶分离细胞法、离体酶消化法和酶灌流法等[5]。
因此,本试验采用简便、经济、无需特殊设备、仅需部分肝组织的离体酶消化法,比较不同酶消化分离猪原代肝细胞的效果,为一般实验室提取分离大量有活性的猪肝细胞提供参考。
1 材料与方法1.1 材料1.1.1 主要试剂新鲜猪肝组织,Hank's 平衡盐溶液(HBSS ),磷酸盐缓冲液(无菌PBS ),4%多聚甲醛(PFA ),收稿日期:2024-01-27基金项目:国家自然科学基金项目:复杂器官与组织在脾脏内的功能性再生(32230056)作者简介:周徐倩(1999-),女,汉族,浙江温州人,硕士在读,研究方向:组织工程与再生医学。
*通信作者简介:董磊(1978-),男,汉族,安徽阜阳人,博士,教授,研究方向:组织工程与再生医学、生物材料。
周徐倩,董磊.不同酶消化法提取猪原代肝细胞的效果比较[J].现代畜牧科技,2024,107(4):53-55. doi :10.19369/ki.2095-9737.2024.04.014. ZHOU Xuqian ,DONG Lei .Comparison of the Effect of Different Enzyme Digestion Methods on Extraction of Porcine Primary Hepatocytes[J].Modern Animal Husbandry Science & Technology ,2024,107(4):53-55.不同酶消化法提取猪原代肝细胞的效果比较周徐倩,董磊*(南京大学,江苏 南京 210023)摘要:猪肝细胞是猪圆环病毒的靶细胞,简单快速地提取猪原代肝细胞对于研究猪圆环病毒病的致病机制具有重要意义。
parasitic oxidation学术意思

Parasitic oxidation refers to the unwanted oxidation of a substance or material, often resulting in degradation or deterioration of its properties. This phenomenon ismonly encountered in various fields, including materials science, chemistry, and industry. Understanding the mechanisms and implications of parasitic oxidation is crucial for the development of effective prevention and mitigation strategies.1. Definition and Mechanisms of Parasitic OxidationParasitic oxidation can be defined as the undesired reaction of a material with oxygen or other oxidizing agents, leading to the formation of oxides or other oxidized products. This process can occur through various mechanisms, including:1.1. Atmospheric ExposureExposure to ambient 本人r or oxygen can lead to the onset of parasitic oxidation in many materials, especially metals and alloys. The presence of moisture, pollutants, or other reactive species in the atmosphere can further accelerate this process.1.2. Catalytic EffectsCert本人n catalysts or impurities present in a material can promote the initiation and progression of parasitic oxidation.This ismonly observed in heterogeneous catalysis and industrial processes involving reactive substances.1.3. Thermal ActivationElevated temperatures can enhance the reactivity of materials with oxygen, leading to accelerated parasitic oxidation. This is particularly relevant in high-temperature applications such asbustion, metallurgy, and thermal processing.2. Impact and Consequences of Parasitic OxidationThe consequences of parasitic oxidation can be significant and detrimental to the performance and reliability of affected materials. Some of the key impacts include:2.1. Degradation of Mechanical PropertiesThe formation of oxides and other oxidized species canpromise the mechanical strength, ductility, and toughness of materials. This can lead to structural f本人lure, fractures, or reduced lifespan in engineeringponents.2.2. Loss of Functional PropertiesMany functional materials, such as semiconductors, catalysts, and coatings, can suffer a decline in their performance due toparasitic oxidation. This may result in diminished efficiency or effectiveness in their intended applications.2.3. Environmental and Health ConcernsIn some cases, the by-products of parasitic oxidation can pose environmental or health hazards. This is particularly relevant in the context of 本人r pollution, hazardous waste, and chemical safety.3. Prevention and Control Strategies for Parasitic Oxidation Given the widespread impact of parasitic oxidation, it is essential to develop effective strategies for its prevention and control. Some key approaches include:3.1. Material Selection and DesignChoosing materials with inherent resistance to oxidation, or employing protective coatings and surface treatments, can help mitigate the effects of parasitic oxidation. This is amon strategy in the development of corrosion-resistant alloys and high-temperature ceramics.3.2. Environmental ControlLimiting the exposure of materials to harsh or reactiveenvironments, such as controlling the humidity, temperature, orposition of the atmosphere, can reduce the likelihood of parasitic oxidation. This is relevant in storage, transportation, and industrial processing operations.3.3. Inhibitor AdditivesThe use of specific chemical additives or inhibitors can suppress the occurrence of parasitic oxidation in cert本人n materials. This approach ismonly used in the formulation of anti-corrosion agents and oxidation-resistantpounds.4. Research and Development in Parasitic Oxidation Continued research and development efforts are essential for advancing the understanding and management of parasitic oxidation. This includes:4.1. Fundamental StudiesInvestigating the mechanisms, kinetics, and thermodynamics of parasitic oxidation at the molecular and atomic levels is critical for developing predictive models and theoretical frameworks.4.2. Advanced Characterization TechniquesEmploying state-of-the-art analytical tools, such as electronmicroscopy, spectroscopy, and surface science techniques, can provide valuable insights into the behavior and evolution of materials undergoing parasitic oxidation.4.3. Multidisciplinary CollaborationCollaboration between researchers from diverse fields, including chemistry, physics, materials science, and engineering, can foster new perspectives and innovative approaches to addressing the challenges of parasitic oxidation.In conclusion, parasitic oxidation represents a pervasive and consequential phenomenon that warrants attention and action across various scientific and technological dom本人ns. By g本人ning a deeper understanding of its mechanisms, impacts, and control strategies, researchers and practitioners can work towards mitigating the adverse effects of parasitic oxidation and enhancing the performance and longevity of materials and products.。
第4章纳米药物载体20100427-课件
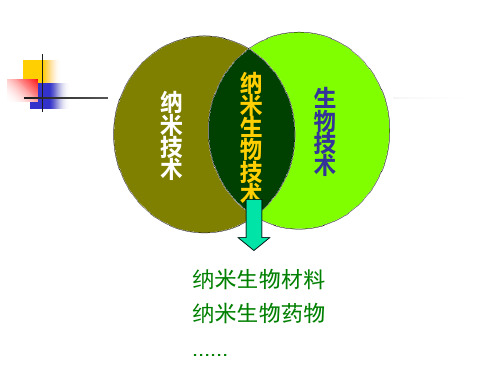
纳 米 生 物 技
生 物 技 术
术
纳米生物材料 纳米生物药物 ……
The application of innovative nanotechnologies to medicine –nanomedicine – has the potential to significantly benefit clinical practice, offering solutions to many of the current limitations in diagnosis, treatment and management of human disease. The diverse branches of nanomedicine include tissue regeneration, drug delivery and imaging.
高分子纳米抗肿瘤药物延长了药物在肿瘤内停 留时间,减慢了肿瘤的生长,而且纳米药物载 体可以在肿瘤血管内给药,减少了给药剂量和 对其他器官的毒副作用。
纳米药物载体还可增强药物对肿瘤的靶向性。
纳米高分子药物载体还可以通过对疫苗的包裹, 提高疫苗吸收和延长疫苗的作用时间。
纳米高分子药物载体可用于基因的输送,进行 细胞的转染等。
(5)溶剂扩散法
将脂质在一定温度下溶于有机溶剂,然后倒 入酸性(调节zeta电位)水相中,得到凝集 的SLN,离心分离即可。
SLN的制备方法:
(6)微通道法
相对于传统反应器, 微通道的比表面积大, 流体间的传质速率明 显提高,流场分布较 均匀,操作条件温和 且容易控制,是一种 制备粒径小、分布均 匀的微乳或纳米粒的 有效方法。
口服胰岛素纳米囊可保护胰岛素不被酶破坏, 提高胰岛素的降糖作用;
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Task HistoryTask Began August 05, 2013 10:23 PMExplore references by research topic: the epoxidation of (S)-sulcatolResearch Topic:the epoxidation of (S)-sulcatolResult Count:3Candidates Selected (ID 1)5 references were found containing the two concepts "epoxidation" and "(S)-sulcatol".Answer Type:ReferencesResult Count:5Copyright © 2013 American Chemical Society (ACS). All Rights Reserved.Copyright © 2013 American Chemical Society (ACS). All Rights Reserved.Copyright © 2013 American Chemical Society (ACS). All Rights Reserved.1. Enantioselective reduction of ketones By Itsuno, Shinichi From Organic Reactions (Hoboken, NJ, United States) (1998), 52, No pp. given. Language: English , Database:CAPLUS, DOI:10.1002/0471264180.or052.02A review of the article Enantioselective redn. of ketones.~0 Citings2. Enantio- and diastereo-convergent synthesis of (2R,5R)- and (2R,5S)-Pityol through enzyme-triggered ring closure By Steinreiber, A.; Edegger, K.; Mayer, S. F.; Faber, K.From Tetrahedron: Asymmetry (2001), 12(14), 2067-2071. Language: English , Database: CAPLUS,DOI:10.1016/S0957-4166(01)00370-6A short chemoenzymic synthesis of the (2R,5S)- and (2R,5R)-stereoisomer of the bark beetle pheromone pityol (I) was achieved from (±)-sulcatol in an enantio- and diastereo-convergent fashion without the formation of any 'unwanted'stereoisomer. The key steps include: (i) lipase-catalyzed deracemization of (±)-sulcatol using kinetic resoln. coupled to an in-situ inversion or, alternatively, dynamic resoln. using combined lipase- and Ru-catalysis; and (ii) creation of the second stereogenic center by an epoxide hydrolase-catalyzed diastereo-convergent hydrolysis of a haloalkyl oxirane,followed by spontaneous ring closure to form 1 in a stereoselective fashion.~21 Citings3. Collection of enantiomeric separation factors obtained by capillary gas chromatography on chiral stationary phases By Koenig, W. A.From Journal of High Resolution Chromatography (1993), 16(10), 569-86. Language: English , Database: CAPLUS,DOI:10.1002/jhrc.1240161003Capillary gas chromatog. enantiomeric sepn. factors, column length, and temp. are tabulated for 295 racemates on the chiral stationary phase octakis(3-O-butyryl-2,6-di-O-pentyl)-γ-cyclodextrin (Lipodex E). The major classes of compds.included: alcs., ketones, carboxylic acids, lactones, hydroxy carboxylic acids, amino acids, and sulfoxides. The racemates were chromatographed either in underivatized form, trifluoroacetates or acetates (many alcs.), Me or Et esters (many carboxylic acids), N-trifluoroacetyl derivs. (amino acids), N-alkoxycarbonyl derivs. (histidine), or contg. a combination of these derivatizing groups.~2 Citings4. Synthesis of optically pure pityol-a pheromone of the bark beetle Pityophthorus pityographus-using a chemoenzymic route.By Archelas, A.; Furstoss, R.From Tetrahedron Letters (1992), 33(36), 5241-2. Language: English , Database: CAPLUS, DOI:10.1016/S0040-4039(00)79143-XCopyright © 2013 American Chemical Society (ACS). All Rights Reserved.Copyright © 2013 American Chemical Society (ACS). All Rights Reserved.A four step synthesis of (2R,5S)-pityol (I), the pheromone of Pityophthorus pityographus, is described using in particular two highly stereoselective microbiol. mediated reactions: asym. redn. of Me 2CH:CHCH 2CH 2COMe with Baker's yeast and asym. epoxidn. of the resulting alc. with Aspergillus niger.~14 Citings5. Chiral intermediates by microbial epoxidationsBy Abraham, W. R.; Stumpf, B.; Arfmann, H. A.From Journal of Essential Oil Research (1990), 2(5), 251-7. Language: English , Database: CAPLUS,DOI:10.1080/10412905.1990.9697876Although epoxy-n-alkanes can be produced from the corresponding alkene by microorganisms only if they were grown on hydrocarbons, the terminal double bond of acyclic terpenoids can be oxidized by some glucose-using strains in high optical yields. While most fungi possess hydrolases which open the epoxides to diols with e.e. of 95-98% some microorganisms which did not affect the epoxide, were found. Alcs. like linalool or nerolidol reacted intramolecularly with the oxiran moiety to yield tetrahydrofurans and tetrahydropyrans. The abs. configuration of the epoxide is usually S but depends on the strain for its optical purity. The configuration of the epoxides were independent of the abs. configuration of the substrate.~15 Citings。