小麦粉及其面粉处理剂中苯甲羟肟酸的测定bjs2020
高效液相色谱法快速测定小麦粉中过氧化苯甲酰

HP C. s l : h e z y eo i ewa h wn t el e ri h a g o 0 2 gml o 1 0 gml te l t fd tcin L Re u t T eb n o l r x d ss o b n a t er n e f m . / 0 / , h i ee t s p o i n r t mi o o
w s03 /gT erc vr fh to sb t e n9 .% a d9 .% . h rcso s( D) r s a % . h r s a .mgk .h o eyo emeh dwa ew e 41 n 90 T epe iin RS weel st n2 T eewa e t e h
Ke ywor : h g p rom a elq i hrm ao r p y; e o l r xde Dee i ai n  ̄ ih e fr nc i u d c o tg a h b nz y o i ; tr n to pe m
高效液相色谱快速测定小麦粉中过氧化苯甲酰含量

液相色谱法 , 该法 样 品前 处理 较复 杂 , 特 别 是 标 准 曲 线 的 制 备 分 别 取 8个 点 进 行 加 标 , 处理过程繁琐 , 完 成标 准 曲 线
3 . 2 流动相 ; 甲醇 : 乙酸铵溶液 ( O . 0 2 mo l / L ) 一5 : 9 5
・
应 用 技术研 究 ・
高 效液 相色谱快速测定小麦粉中过氧化苯 甲酰含量
粱 兰
( 宿州 市食 品 药 品 检 验 所 , 安徽 宿 州 2 3 4 0 0 0 ) 摘要: 过氧 化苯 甲酰俗名增 白剂 , 它 作 为食 品 添 加 剂 在 小 麦 粉 中使 用 , 可使 面粉 增 白, 改善 其 品质 , 但 加 入 量 不 得
高效 液 相 色 谱 仪 ( 日本 日 立 L 一7 1 O O ) 、 C H。 C H OH ( AR ) 、 6 0 KI水 溶 液 、 CHs OH ( 色谱 纯) 、 乙 酸 铵 溶 液
( 0 . 0 2 mo l / L ) : 称取 1 . 5 4 g乙 酸 铵 , 加水 至 1 0 0 0 mL, 溶解 ,
二、 仪 器 与试 剂
准确 称 取 约 3 g小 麦 粉 于 3 0 m L 比 色 管 中, 加 6 . 0
m L C H。 C H 。 O H, 3 mL 6 0 K I 溶液 , 混匀 放置 2 O分 钟 , 使 其 充
分反 应 。加 2 0 m L H O混 匀 , 3 0 0 0转 / 分钟离心 2 O分 钟 , 取 上
第1 4卷 第 3期
2 0 1 5年 6月
小麦粉及其面粉处理剂中苯甲羟肟酸的测定(BJS 202002)

附件2小麦粉及其面粉处理剂中苯甲羟肟酸的测定BJS 2020021 范围本方法规定了小麦粉及其面粉处理剂中苯甲羟肟酸的高效液相色谱测定方法。
本方法适用于小麦粉及其面粉处理剂中苯甲羟肟酸的测定。
2 原理试样中的苯甲羟肟酸用甲醇提取,采用高效液相色谱测定,外标法定量。
试样中检出苯甲羟肟酸后采用液相色谱-质谱/质谱法进行确证。
3 试剂和材料除非另有说明,本方法所有试剂均为分析纯,水为GB/T 6682规定的一级水。
3.1 试剂3.1.1 甲醇(CH3OH):色谱纯。
3.1.2 磷酸(H3PO4)。
3.1.3 甲酸铵(HCOONH4):色谱纯。
3.2 试液配制3.2.1 磷酸溶液(0.01%):准确量取1000 mL水,加入100 µL磷酸(3.1.2)得到pH在2.9±0.1之间的磷酸溶液。
3.2.2 甲酸铵溶液(5 mmol/L):称取甲酸铵0.26 g(3.1.3)(精确至0.001 g),加水溶解并定容至1000 mL。
3.3 标准品3.3.1 苯甲羟肟酸:纯度不低于98%的标准品或经国家认证并发布的有证标准物质。
其中文名称、英文名称、CAS号、分子式、相对分子质量、结构式见附录A。
3.4 标准溶液配制3.4.1 标准储备液:准确称取苯甲羟肟酸标准品10 mg(精确至0.01 mg),置于50 mL容量瓶中,加甲醇-水溶液(50/50,v/v)溶解并稀释至刻度,摇匀,得到浓度为200 μg/mL的标准储备液,4 ℃避光保存,有效期90天。
3.4.2 标准中间液(20.0 μg/mL):吸取标准储备液(3.4.1)5 mL于50 mL容量瓶中,用甲醇-水溶液(50/50,v/v)稀释至刻度,摇匀,得到标准中间液,4 ℃避光保存,有效期60天。
3.4.3 标准系列工作液:吸取标准中间液(3.4.2)0.25 mL、0.50 mL、1.25 mL、2.5 mL、5.0 mL和12.5 mL于50 mL容量瓶中,用甲醇-水溶液(50/50,v/v)稀释至刻度,混匀,得到标准系列工作液。
高效液相色谱-串联质谱法同时测定面粉中7种非法添加剂

高效液相色谱-串联质谱法同时测定面粉中7种非法添加剂作者:王颖怡吴玉田邹璐孟春杨钟雪周贻兵来源:《食品安全导刊·下》2024年第03期摘要:目的:建立一种分析面粉中7种非法添加剂的高效液相色谱-串联质谱检测方法。
方法:试样经乙腈∶水(1∶1,V∶V)提取、超声、离心、氮吹浓缩后,经Agilent Eclipse Plus C18 RRHD色谱柱分离、0.1%甲酸-5 mmol·L-1甲酸铵水和甲醇为流动相进行等度洗脱,采用多反应监测模式分析。
结果:7种目标化合物在0.10~8.00 ng·mL-1呈良好的线性关系(R2>0.990),检出限为0.03~0.15 mg·kg-1,定量限为0.10~0.45 mg·kg-1;回收率在88.6%~104.3%,相對标准偏差在2.1%~5.8%(n=6)。
结论:该方法简便、灵敏、准确,可满足小麦粉中7种非法添加物的同时检测要求。
关键词:高效液相色谱-串联质谱法;小麦粉;非法添加剂;噻苯咪唑;苯甲羟肟酸Simultaneous Determination of 7 Illegal Additives in Flour by High Performance Liquid Chromatography-Tandem Mass SpectrometryAbstract: Objective: To establish a high performance liquid chromatography-tandem mass spectrometry method for the determination of 7 illegal additives in flour. Method: The samples were extracted by acetonitrile∶water (1∶1,V∶V), concentrated by ultrasound, centrifugation and nitrogen blowing, separated by Agilent Eclipse Plus C18 RRHD column, and then eluted with 0.1% formic acid -5 mmol·L-1 ammonium formate water and methanol as mobile phase, and analyzed by multi-reaction monitoring mode. Result: There was a good linear relationship (R2>0.990) between 0.10~8.00 ng·mL-1 for the 7 target compounds. The limit of detection was 0.03~0.15 mg·kg-1, and the limit of quantification was 0.10~0.45 mg·kg-1. The recoveries were88.6% ~104.3%, and the relative standard deviations were 2.1%~5.8%(n=6). Conclusion:The method is simple, sensitive and accurate, and can satisfy the simultaneous detection of 7 illegal additives in wheat flour.Keywords: high performance liquid chromatography tandem mass spectrometry; wheat flour; illegal additives; thiobenzimidazole; benzohydroxamic acid国内三大粮食作物之一的小麦是仅次于稻谷的第二大粮食作物,其含有大量碳水化合物和蛋白质,小麦粉(面粉)是其主要加工产品。
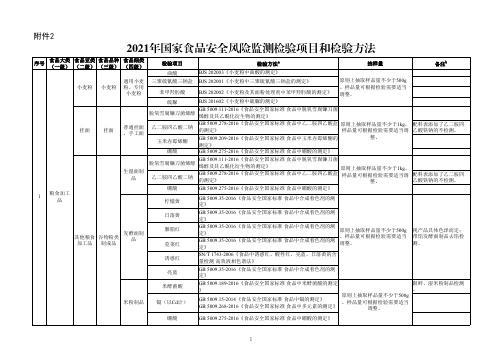
2021年国家食品安全风险监测检验项目和检验方法
限鲜、湿米粉制品检测
》
。
GB 5009.15-2014《食品安全国家标准 食品中镉的测定》
原则上抽取样品量不少于500g 。样品量可根据检验需要适当
GB 5009.268-2016《食品安全国家标准 食品中多元素的测定》
调整。
硼酸
GB 5009.275-2016《食品安全国家标准 食品中硼酸的测定》
附件2
2021年国家食品安全风险监测检验项目和检验方法
序号 1
食品大类 (一级)
粮食加工 品
食品亚类 食品品种 (二级) (三级)
小麦粉 小麦粉
挂面
挂面
其他粮食 谷物粉类 加工品 制成品
食品细类 (四级) 通用小麦 粉、专用
小麦粉
普通挂面 、手工面
生湿面制 品
发酵面制 品
米粉制品
检验项目
检验方法a
抽样量
邻苯二甲酸二丁酯 GB 5009.271-2016《食品安全国家标准 食品中邻苯二甲酸酯的
(DBP)
测定》
菜籽油
邻苯二甲酸二(2-乙 GB 5009.271-2016《食品安全国家标准 食品中邻苯二甲酸酯的
基)己酯(DEHP) 测定》
原则上小包装产品(净含量<
辣椒素总量(天然辣 椒素、二氢辣椒素、 BJS 201801《食用油脂中辣椒素的测定》
(DBP)
测定》
花生油
邻苯二甲酸二(2-乙 GB 5009.271-2016《食品安全国家标准 食品中邻苯二甲酸酯的
基)己酯(DEHP) 测定》
辣椒素总量(天然辣 椒素、二氢辣椒素、 BJS 201801《食用油脂中辣椒素的测定》
合成辣椒素)
原则上小包装产品(净含量< 25L(kg),抽样数量不少于1L (kg),且不少于2个独立包
高效液相色谱法测定小麦粉中过氧化苯甲酰及其还原产物苯甲酸

山东农业科学 2009,1:97~98Shandong Agricultural Sciences收稿日期:2008-09-08作者简介:高艾英(1977-),女,山东泰安人,硕士研究生,目前从事食品检测工作。
E -mail:aygao@1261com 3通讯作者,E -mail:yuhongjin79@1631com高效液相色谱法测定小麦粉中过氧化苯甲酰及其还原产物苯甲酸高艾英1,张 昊1,许 芬2,吴爱民1,金玉红33(11泰安市产品质量监督检验所,山东泰安 271000;21泰山医学院,山东泰安 271000;31山东农业大学食品科学学院,山东泰安 271018) 摘 要:本试验直接用甲醇提取小麦粉中的过氧化苯甲酰和苯甲酸进行高效液相色谱测定,结果样品中两者的分离效果较好,无杂峰及拖尾峰现象,且前处理简单。
过氧化苯甲酰及苯甲酸的加标回收率分别为9610%~9913%、9113%~9517%,准确度和灵敏度较高。
关键词:小麦粉;过氧化苯甲酰;苯甲酸;高效液相色谱法中图分类号:O65717+2;TS211.7 文献标识号:A 文章编号:1001-4942(2009)01-0097-02 小麦粉增白剂———过氧化苯甲酰又称面粉改良剂,对面粉有明显的增白和促进熟化的效果,它主要通过与水分作用产生具有强氧化性的游离原子氧[O ],氧化小麦粉中类胡萝卜素等多烯类色素使其褪色,从而使面粉变白。
过氧化苯甲酰在面粉中适量添加,可有效增加面粉的白度和筋力,大大改善面粉的加工性能;其还原产物苯甲酸有防腐、防霉的效果,可改善面粉的储藏性;而且适宜的剂量,对人体也安全无害。
但过量使用对人体却是非常有害。
因此,我国《食品添加剂使用卫生标准》[1]规定其最大使用量为0106g/kg 。
但仍有少数小麦粉生产企业因工艺落后,在面粉生产中添加过量增白剂,个别生产企业有意添加过量增白剂增加面粉的白度,以劣质或低等级面粉冒充高档面粉,严重危害消费者的身体健康。
食品补充检验方法数据(截止2020年9月)

20
食品中二甲双胍等非食品用化学物质的测定
BJS201901
2019年第4号公告
2019-01-31
21
食品中5种α-受体阻断类药物的测定
BJS201808
2018年第28号公告
2018-11-26
22
肉制品中刚果红的测定
BJS201807
2018年第17号公告
2018-07-09
2019年第9号公告
2019-03-04
17
食品中多种动物源性成分检测实时荧光PCR法
BJS201904
2019年第4号公告
2019-01-31
18
食品中二苯乙烯类阴离子型荧光增白剂的测定
BJS201903
2019年第4号公告
2019-01-31
19
小麦粉及其制品中氨基脲的测定
BJS201902
2019年第4号公告
23
土豆及其制品中α-茄碱和α-卡茄碱的测定
BJS201806
2018年第17号公告
2018-0Biblioteka -0924食品中那非类物质的测定
BJS201805
2018年第14号公告
2018-07-02
25
畜肉中卡拉胶的测定
BJS201804
2018年第10号公告
2018-06-11
26
饮料中γ-丁内酯及其相关物质的测定
BJS201709
2017年第114号公告
2017-09-18
39
食用植物油中乙基麦芽酚的测定
BJS201708
2017年第97号公告
2017-08-17
40
植物蛋白饮料中植物源性成分鉴定
小麦粉中曲酸的测定(BJS 202003)

小麦粉中曲酸的测定BJS 2020031 范围本标准规定了小麦粉中曲酸的高效液相色谱测定方法。
本标准适用于小麦粉中曲酸的测定。
2 原理用纯水提取试样中曲酸,采用配有二极管阵列检测器或紫外检测器的高效液相色谱仪检测,外标法定量。
3 试剂和材料除另有规定,本方法所用试剂均为分析纯,水为GB/T 6682规定的一级水。
3.1 试剂3.1.1甲醇(CH3OH):色谱纯。
3.1.2 磷酸二氢钾(KH2PO4)。
3.1.3 磷酸(H3PO4)。
3.2 试剂配制磷酸二氢钾溶液(0.02 mol/L,pH 3.7):称取2.722 g磷酸二氢钾(3.1.2),加水溶解并定容至1000 mL,加入磷酸(3.1.3)调节pH在3.7 ± 0.1之间,经水相微孔滤膜(0.45 μm)过滤后备用。
3.3 标准品曲酸标准品英文名称、CAS号、分子式、相对分子质量、结构式见附录A,纯度≥99%。
3.4 标准溶液配制3.4.1 标准储备液(1000 mg/L):准确称取曲酸标准品100 mg(精确至0.0001 g),加水溶解并转移至100 mL容量瓶中,定容至刻度,摇匀,制成浓度为1000 mg/L标准储备液,4℃避光保存,有效期3个月。
3.4.2 标准使用液(10.0 mg/L):准确吸取标准储备液(3.4.1)1.0 mL于100 mL容量瓶中,用水稀释至刻度,摇匀,制成浓度为10.0 mg/L的标准使用液,4℃避光保存,有效期3个月。
3.4.3 标准系列工作溶液:准确吸取标准使用液(3.4.2)适量,用水配制成质量浓度为0.1 mg/L、0.2 mg/L、0.5 mg/L、1.0 mg/L、5.0 mg/L,或依仪器响应和实际情况配制适当浓度的标准系列工作溶液。
4 仪器与设备4.1 液相色谱仪:配有二极管阵列检测器或紫外检测器。
4.2 分析天平:感量分别为0.01 g和0.0001 g。
4.3 pH计:精度0.01。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附件2
小麦粉及其面粉处理剂中苯甲羟肟酸的测定
BJS 202002
1 范围
本方法规定了小麦粉及其面粉处理剂中苯甲羟肟酸的高效液相色谱测定方法。
本方法适用于小麦粉及其面粉处理剂中苯甲羟肟酸的测定。
2 原理
试样中的苯甲羟肟酸用甲醇提取,采用高效液相色谱测定,外标法定量。
试样中检出苯甲羟肟酸后采用液相色谱-质谱/质谱法进行确证。
3 试剂和材料
除非另有说明,本方法所有试剂均为分析纯,水为GB/T 6682规定的一级水。
3.1 试剂
OH):色谱纯。
CH3.1.1 甲醇(33.1.2 磷酸(HPO)。
433.1.3 甲酸铵(HCOONH):色谱纯。
43.2 试液配制
3.2.1 磷酸溶液(0.01%):准确量取1000 mL水,加入100 μL磷酸(3.1.2)得到pH在2.9±0.1之间的磷酸溶液。
.
3.2.2 甲酸铵溶液(5 mmol/L):称取甲酸铵0.26 g(3.1.3)(精确至0.001 g),加水溶解并定容至1000 mL。
3.3 标准品
3.3.1 苯甲羟肟酸:纯度不低于98%的标准品或经国家认证并发布的有证标准物质。
其中文名称、英文名称、CAS号、分子式、相对分子质量、结构式见附录A。
3.4 标准溶液配制
3.4.1 标准储备液:准确称取苯甲羟肟酸标准品10 mg(精确至0.01 mg),置于50 mL容量瓶中,加甲醇-水溶液(50/50,v/v)溶解并稀释至刻度,摇匀,得到浓度为200 μg/mL的标准储备液,4 ℃避光保存,有效期90天。
3.4.2 标准中间液(20.0 μg/mL):吸取标准储备液(3.4.1)5 mL于50 mL容量瓶中,用甲醇-水溶液(50/50,v/v)稀释至刻度,摇匀,得到标准中间液,4 ℃避光保存,有效期60天。
3.4.3 标准系列工作液:吸取标准中间液(3.4.2)0.25 mL、0.50 mL、1.25 mL、2.5 mL、5.0 mL 和12.5 mL于50 mL容量瓶中,用甲醇-水溶液(50/50,v/v)稀释至刻度,混匀,得到标准系列工作液。
浓度分别为0.10 μg/mL、0.20 μg/mL、0.50 μg/mL、1.0 μg/mL、2.0 μg/mL和5.0 μg/mL,4 ℃避光保存,有效期30天。
4 仪器和设备
4.1 高效液相色谱仪:配有二极管阵列检测器。
4.2 电子天平:感量分别为1 mg和0.01 mg。
4.3 pH计:精确至0.01。
4.4 涡旋振荡器。
4.5 超声波发生器。
4.6 高速离心机:转速不低于8000 r/min。
1
4.7 滤膜:有机相,孔径0.22 μm。
5 分析步骤
5.1 试样制备
称取具代表性小麦粉或面粉处理剂样品约200 g,混合均匀,贮存于洁净广口容器中,干燥避光保存备用。
5.2 试样处理
准确称取试样2 g(精确至0.001 g),置于50 mL具塞离心管中,准确加入10 mL甲醇(3.1.1),手动振荡20次,涡旋1 min,超声提取10 min,在4 ℃下8000 r/min离心5 min,取离心后上清液5
mL,于45 ℃氮气吹至近干,甲醇-水溶液(50/50,v/v)定容至1 mL,涡旋混匀后过0.22 μm 有机相微孔滤膜(4.7),弃去2~5滴初滤液,取续滤液作为待测试样液。
5.3 液相色谱参考条件
a)色谱柱:C色谱柱(4.6 mm×150 mm,5 μm)或性能相当者。
18b)流动相:A为0.01%的磷酸溶液(3.2.1),B为甲醇(3.1.1),梯度洗脱程序见表1。
c)流速:1.0 mL/min。
d)检测波长:228 nm。
e)柱温:35 ℃。
f)进样量:20 μL。
苯甲羟肟酸标准溶液的液相色谱图见附录B。
表1 流动相梯度洗脱程序
时间(min)A(%) B (%)
8 0.0 92
8 92 2.0
50 14.0 50
50 15.0 50
8 15.1 92
8
20.0
92
2
5.4 标准曲线的制作
将标准系列工作液(3.4.3)分别注入高效液相色谱仪中,测定相应的峰面积,以标准系列工作液的浓度为横坐标,以峰面积为纵坐标,绘制标准曲线。
5.5 试样溶液的测定
将待测试样液注入高效液相色谱仪中,得到苯甲羟肟酸的峰面积,根据标准曲线计算得到试样液中苯甲羟肟酸的浓度。
试样液中苯甲羟肟酸的浓度应在标准曲线线性范围内,超出线性范围应稀释试样液后重新进行分析。
5.6 定性测定
试样液和浓度接近的标准溶液同时进样分析,试样液中被测物质的保留时间与标准溶液中对应的保留时间偏差在±2.5%之内,则可初步认定试样中存在苯甲羟肟酸,需按附录C方法进行确证。
5.7 空白试验
除不加试样外,均按试样同法操作。
6 分析结果的表述
试样中被测组分的含量按式(1)计算:
(1)
式中:
X——试样中苯甲羟肟酸的含量,单位为毫克每千克(mg/kg);
c——由标准曲线计算得到的待测试样液中苯甲羟肟酸的浓度,单位为微克每毫升(μg/mL);V——试样提取液体积,单位为毫升(mL);1V——试样经提取浓缩后的最终定容体积,单位为毫升(mL);3V——用于浓缩分取的试样提取液体积,单位为毫升(mL);2m——试样质量,单位为克(g);
1000 ——单位换算系数。
3
计算结果保留两位有效数字。
7 精密度
在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
8 准确度
本方法在0.3 mg/kg ~3.0 mg/kg添加浓度范围内,苯甲羟肟酸的回收率在70%~110%之间。
9 方法检出限和定量限
当取样量为2 g,提取溶液体积为10 mL,并浓缩5倍时,本方法中苯甲羟肟酸的检出限为0.1 mg/kg,定量限为0.3 mg/kg。
4
A 附录
苯甲羟肟酸相关信息
5
附录B
苯甲羟肟酸高效液相色谱图及紫外吸收光谱图
苯甲羟肟酸标准溶液(1.0 μg/mL)的液相色谱图见图B.1。
苯甲羟肟酸
)的高效液相色谱图μ苯甲羟肟酸标准溶液(B.1 图1.0 g/mL6
附录C
苯甲羟肟酸液相色谱-质谱/质谱确证试验
C.1 液相色谱参考条件
a)色谱柱:C色谱柱(2.1 mm×50 mm,1.7μm)或性能相当者。
18b)流动相:A为5 mmol/L 甲酸铵溶液(3.2.2),B为甲醇(3.1.1),梯度洗脱程序见表C.1。
c)流速:0.3 mL/min。
d)柱温:35 ℃。
e)进样量:5 μL。
表C.1 流动相梯度洗脱程序
时间(min)A(%) B (%)
8 92 0
80 5.0 20
8 5.1 92
8
92
7.0
C.2 质谱参考条件
a) 离子源:电喷雾离子源(ESI源)。
b) 检测方式:多反应监测(MRM)。
c) 扫描方式:负离子模式扫描。
d) 电喷雾电压:-2.5 KV。
e) 脱溶剂温度:500 ℃。
f) 干燥气(氮气)流速:700 L/h。
g) 碰撞气(氩气)流速:0.12 mL/min。
h)定性离子对,定量离子对,锥孔电压和碰撞能量见表C.2。
表C.2 苯甲羟肟酸质谱参数
7
所列仪器条件仅供参考,采用不同质谱仪器时,仪器参数可能存在差异,测定前应将C*代表定量离子对。
附录注:质
谱参数优化到最佳。
C.3 定性判定试的条件测定试样液和标准溶液。
试样液和浓度接近的标准溶液同时进样分析,按照C.1和C.2之内;且试样液中被测组分样液中被测物质的保留时间与标准溶液中对应的保留时间偏差在±2.5%定性离子的相对丰度与浓度接近的标准溶液中对应的定性离子的相对丰度进行比较,偏差不超过表 C.3规定的范围,则可判定为试样液中存在苯甲羟肟酸。
定性确证时相对离子丰度的最大允许偏差表C.3
)离子色谱图MRM 图C.1 苯甲羟肟酸标准溶液的多反应监测(
8
本方法负责起草单位:中国计量科学研究院方法验证单位:中国检验检疫科学研究院综合检测中心、山东省食品药品检验研究院、河北省食品检验研究院、河南省产品质量监督检验院、南京市食品药品监督检验院、国贸食品科技(北京)有限公司实验室。
方法主要起草人:国振、李秀琴、周霞、赵光亮、赵博、李先江、张庆合、李红梅
9。