Materials studio 软件学习掌握内容
中科大MaterialsStudio培训教程13(包你学会)请将这一系列全看完,一定有收获。讲述

显示成球棒结构
转化为原胞
性质不选
优化成功
Save project, close all 打开优化后 的W
恢复为晶胞,准备切面
其它不变,改厚度至9埃
切出一个厚度约9埃的表面。足够厚,表示体性质。Save project,工作窗口仅 保留表面结构。
30
斜拉鼠标,选中中间的一 些原子。现在弛豫表面一 层的原子。严格计算应该 slab取厚
对块层进行计算时,为了反映体材料的性质,块层的厚度至少在 0.8-1nm。为了避免块层相邻表面间静电势的相互干扰,并使静电势达 到其渐进值,真空层的厚度至少为3nm。 计算思路:对一定的晶体结构,求出总电子能量最低
的电荷密度分布(x’),再根据右式计算位置x处的电势 (x)。
( x)dБайду номын сангаас ( x) 4 0 x x
W EF V0
真空
金属
0
x
. 二. 功函数的计算模型 . . 构造如右图所示的2维无限阵列,计算 的材料就是图中的板,板间是真空层。用 CASTEP计算此体系的EF 和板间的静电势 3 nm 分布(r),在平行表面的方向上对静电势取 slab 平均,从而在板间得到V0 ,进而得到W。 由于功函数W的数值依赖于晶体结构和 晶面取向,所以计算时需先优化体材料的晶 0.8-1 nm 体结构,再绘制2维阵列。可以优化块层表 面和块层内部的原子坐标,也可以不优化。 . . . 通过这些选择,人们可以研究表面弛豫对功 函数的影响。一般建议利用constraints,固定块层中间的原子,使 其保留体材料的结构。
检查后,恢复按照 Element显示颜色
w4d4
绿色,表层原子。
另一表层,两层 等价,两边对称。 找不对称的。
中科大MaterialsStudio培训教程4

如果动画(Animation)工具条 是不可见的,
则按右侧的操作,使用观看 (View)菜单让它显示。
设置显示方式,按播放键 。 B-O近似,体系的电子能量是核构型的函数。
在继续工作之前,需要关闭Materials Visualizer 中的所有文件。 关闭DMol3 计算对话框。选择File\Save Project,然后Window\Close All。 双击几何优化子文件夹中的reactant.xsd 和product.xsd。 现在工作区域中只有两个优化了的结构。
运用DMol3的 LST/QST功能来搜索过渡态,需要 在反应物和产物之间创建一条通道,这也是DMol3 计 算时所要求的输入文件。
在反应预览(Reaction Preview)对话框中,把桢数 提高到100;勾选上Superimpose structures;单击 Preview按钮,关闭反应预览(Reaction Preview)对话 框。
电子Hamiltonian 的设置与几何优化计 算的设置一样。
这次需要计算频率(Frequency)相关的性质。 点击Properties 标签栏,勾选上Frequency。
最后,需要对工作描述(Job Description)加以设 置。
点击Job Control 标签,确认Automatic 没有被勾 选上;在Job Description 一栏里打上TS。
点击Set Match,还剩下5H、7H未匹配。重复这个过程,继续点击Set Match来匹配 剩下的没有配对的原子。
反应物、产物的原子已配对。现在可以预览一下反应物和产物之间原子的匹配 情况。
点击反应物或者产物栏中列表里的任意一个原子,可以看到匹配的另一原子。 考察匹配情况,直到满意为止。关闭Find Equivalent Atoms 对话框。
materials studio 学习整理知识点上课讲义

1、castep是用平面波赝势展开波函数,dmol是通过原子轨道的线性组合来处理(castep用的是基于平面波赝势的方法,而dmol是基于分子轨道理论的方法)castep算周期性结构的体系,DMol 适合于分子,团簇,分子筛,分子晶体,聚合物等开放结构。
也就是说对空体积较大的晶体,原子轨道在稀填充体系(原子、分子、团簇、低维周期体系、沸石...)的计算上比平面波有优势。
CASTEP是一个基于密度泛函方法的从头算量子力学程序,总能量包含动能、静电能和交换关联能三部分,各部分能量都可以表示成密度函数。
适用于范围很大的一类固体材料、界面以及表面的性质。
基于总能量的平面波赝势理论,研究的内容包括:结构对称性、晶格参量、键长键角、能带结构、态密度、布局数、光学性能等。
CASTEP 模組允許你使用含有彈性的分子模型工具CASTEP 主要是用於大尺度的週期性系統,他也可以被應用在以超晶格建力起來的缺陷表面介面與分子CASTEP仅能在3D周期模型文件基础上进行计算,必须构建超单胞,以便研究分子体系默认条件下,CASTEP使用得是BFGS几何优化方法,即拟牛顿算法Castep的几何优化过程的本质是期望利用一个迭代过程来完成优化任务,在进行迭代的过程中, 通过调整原子坐标和晶胞参数使结构的总能量最小化。
CASTEP几何优化的核心是通过不断的减小计算力和应力的数量级,直至小于所规定的收敛误差。
当然,也可能给定外部应力张量来对拉应力,压应力和切应力等作用下的体系行为模型化。
在这些情况下反复迭代内部应力张量直到与所施加的外部应力相等Castep默认的能量单位是电子伏特ev,换算关系为1eV=0.036749308Ha=23.0605kcal/mole=96.4853kJ/m2、Energy cutoff 截断能SCF tolerance:迭代标准,就是每两部之间算完的标准( SCF:就是自洽场self consistent field,解薛定谔方程时在开始并不知道波函数,从而也无法获得所需要的电子密度;因此可以先用一个解进行迭代运算,直到最后达到所需要的结果)K point set: K点设置(布里渊区的点数选择,就像你选样本来看产品的合格率一样,选的多就会慢,但会更准确一些)3、建模时加入杂质原子的方法:方法一:用鼠标点上将要被取代得原子(点上后原子颜色将变成黄色),在窗口的右边属性栏中,将会显示这个原子的相关属性,并告诉你这个原子的元素种类(比方是Al 吧),然后点这个元素种类Al,将出现一个元素周期表,选择你要掺杂得原子,确定就可以了!方法二:建立完没有掺杂的晶胞后,选supercell,然后再选择要替换的原子,进行掺杂.如果不把晶胞的对称性改为supercell就不能改变其中的一个原子,而是由于对称性把同一元素的所以原子改变了4、(1)castep建立晶体的模型步骤:build/build crystal,弹出对话框,然后按照晶格参数填入。
中科大MaterialsStudio培训教程包你学会!请将这一系列全看完一定有收获40页PPT

谢谢!
36、自己的鞋子,自己知道紧在哪里。——西班牙
37、我们唯一不会改正的缺点是软弱。——拉罗什福科
xiexie! 38、我这个人走得很慢,但是我从不后退。——亚伯拉罕·林肯
39、勿问成功的秘诀为何,且尽全力做你应该做的事吧。——美华纳
பைடு நூலகம்
40、学而不思则罔,思而不学则殆。——孔子
中科大MaterialsStudio培 训教程包你学会!请将这一
系列全看完一定有收获
26、机遇对于有准备的头脑有特别的 亲和力 。 27、自信是人格的核心。
28、目标的坚定是性格中最必要的力 量泉源 之一, 也是成 功的利 器之一 。没有 它,天 才也会 在矛盾 无定的 迷径中 ,徒劳 无功。- -查士 德斐尔 爵士。 29、困难就是机遇。--温斯顿.丘吉 尔。 30、我奋斗,所以我快乐。--格林斯 潘。
中科大-Materials-Studio-培训教程-5(包你学会)请将这一系列全看完-一定有收获。精讲

从“开始” 或快捷图 标打 开MS。
找到class3, 按“打开”按 钮
输入AlAs,这将是 新的Project的名字。
在 Project Explorer中,右击根目录AlAs,选择New | 3D Atomistic Document。
1 eV=0.036749308 Ha=23.0605 kcal/mole=96.4853 kJ/mole
CASTAP几何优化任务
CASTAP几何优化任务允许改善结构的几何,获得稳定结构 或多晶型物。通过一个迭代过程来完成这项任务,迭代过程中 调整原子坐标和晶胞参数使结构的总能量最小化。
CASTAP几何优化是基于减小计算力和应力的数量级,直到 小于规定的收敛误差。也可能给定外部应力张量来对拉应力、 压应力和切应力等作用下的体系行为模型化。在这些情况下反 复迭代内部应力张量直到 与所施加的外部应力相等。
引言 本指南介绍了CASTEP是如何使用量子力学方法来确定材料的晶体结构,使用者
将学会如何构建晶体结构,设定一个CASTEP几何优化任务,然后分析计算结果。
内容 1. 构建AlAs的晶体结构 2. 设置并进行CASTEP计算 3. 分析结果 4. 比较计算的结构参数和实验数据
(1)图示电荷密度 (2)图示态密度和带结构
CASTAP动力学任务
CASTAP动力学任务允许模拟结构中原子在计算力的影响下将如何移动。
在进行CASTAP动力学计算以前,可以选择热力学系综和相应参数,定义模拟 时间和模拟温度。
选择热力学系综
对牛顿运动定律积分允许探索体系恒值能量表面(NVE动力学)。然而,在 体系与环境进行热交换条件下发生最本质的现象。使用NVT系综(或者是确定性 的Nosé系综或者是随机性的Langevin 系综)可模拟该条件。
Materials Studio 培训教程

Materials Studio 培训教目录Materials Studio 快速入门教程⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯2 Visualizer 模块快速入门教程⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯11用第一性原理预测AlAs 的晶格参数⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯36 CO 分子在Pd(110)表面的吸附⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯43Pd(110)面上的CO 分子电荷密度变化⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯55模拟CO_Pd(110)体系的STM 图⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯61使用DMol3 中的离域内坐标对固体进行几何优化⋯⋯⋯⋯⋯⋯64 用LST/QST 搜索过渡态⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯69气体在聚合体中扩散的测量⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯76聚合物与金属氧化物表面的相互作用⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯86计算共存相之间的界面张力⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯96运行简单的MesoDyn 模拟⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯99使用粉末衍射图进行分析⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯108指标化粉末衍射图⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯117无机物的Rietveld 精修⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯125使用Reflex Plus 来解析3-氯-反-苯乙烯酸的结构⋯⋯⋯⋯⋯⋯⋯133 无机化合物FIN31 的结构确定⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯⋯142创腾科技有限公司Neotrident Technology Limited 2Materials Studio 快速入门教程该教程将介绍Materials Studio 软件的基本功能,在这一部分,你将学到:1.生成Projects2.打开并且观察3D 文档3.绘制苯甲酰胺分子4.观察并且处理研究表格文档5.处理分子晶体:尿素6.建造Alpha 石英晶体7.建造多甲基异丁烯酸盐8.保存Project 并结束1. 生成Projects(1).运行Material Visualizer从运行菜单中运行或者在桌面点击快捷方式。
中科大-Materials-Studio-培训教程-7(包你学会!)请将这一系列全看完-一定有收获。

3
4
3
5
6
7
显示出bulk Pd的结构,我们把显示方式改为Ball and Stick。在Pd 3D Model document中右键单击,选择Display Style,在Atoms标签中选择Ball and Stick,关闭对 话框。
现在使用CASTEP来优化 bulk Pd。为了减少计算量, 将晶胞转换为原胞。
(1).准备项目
在D或E盘中建立class 5文件夹。运行MS,在class 5中建立名为Pd_
2
2021/3/11
4
5
2
为便于管理项目,我们先在项目中准备三个子文件夹。在Project
Explorer的根图标Pd_CO上右键单击,选择New / Folder。再重复此操作二次。 在New Folder上右键单击,选择Rename,键入Pd bulk。在其它的文件上重 复此操作过程,把它们依次更名为Pd(110)和(1x1) CO on Pd(110)。
注意真空层的方向在oc
2021/3/11
9
结构由2D 变为3D,并且一个真空层被加到原子的上方。
真 空 层
C B
O A
2021/3/11
旋转此3D图,注意OA、OB、 OC的方向与X、Y、Z三个坐 标轴不同。真空层沿OC方向。
10
右击3D 模型,选择Lattice Parameters,选择Advanced 标签,按下Reorient to standard 按钮,关闭此对话框。
2021/3/11
17
Pd 的起始对称性是P1,但是 随着CO 分子的引入发生了改变。 可以通过运用Find Symmetry工具 找到并加上对称性。
选择工具条上的Find Symmetry 工具,按下Find Symmetry 按钮,然后按下 Impose Symmetry按钮。 现在的对称性是PMM2。
Materials Studio 培训教程4(包你学会!)请将这一系列全看完,一定有收获。
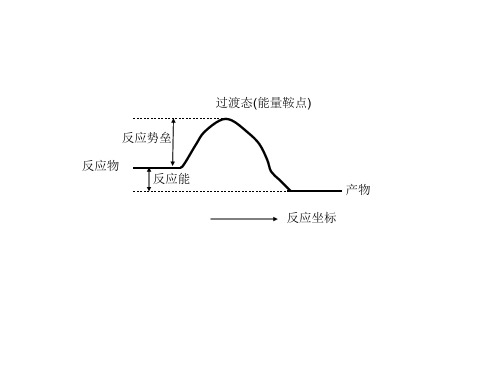
反应势垒 反应物 反应能 反应坐标
产物
用LST/QST 搜索过渡态
目的: 介绍如何使用 DMol3 和 Reaction Preview 工具进行过渡态搜索的计算。 对简 单反应,这种方法是有效的。 模块: Materials Visualizer, DMol3 前提: 用局域内坐标对固体进行结构优化。 背景 对任何反应的势能面的探索都要求知道反应进程中每一步的结构和能量, 或者动力学和热动力学的快照(snapshots)。特别重要的是决定反应速率的那一步, 这通常需要找到那些难以捕获的过渡态结构。有一些方法对找到过渡态的结构是 很有效果的,其中比较知名的就是线性同步度越(linear synchronous transit, LST)和二次同步度越(quadratic synchronous transit,QST)。 本例中,我们将介绍DMol 中的LST /QST 工具的使用,将会看到如何使用 LST/QST 搜索乙烯醇转变为乙醛的H转移反应 的过渡态结构。 CH2CHOH → CH3CHO 本例包括以下内容: ������ 1. 建立一个计算模型 ������ 2. 优化分子结构 ������ 3.定义原子对 ������ 4.用LST/QST 的方法计算过渡态 ������ 5.优化过渡态结构
1.建立一个计算模型 选择 creating a new project,建立名为vinylOH 的project 。
在本单元中,你要在两个不同的3D Atomistic 界面中建立反应物和产物模型。第 一步就是打开一个新的3D Atomistic界面,构建反应物乙烯醇(vinyl alcohol)。 点击工具栏里的New button,选择3D Atomistic。
点击碳-碳键一次,选中。点击Sketch工 具条上的Modify Bond Type 键 ,选 择双键,从而把单键变成双键。点击别处, 取消选择碳-碳键。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Materials studio 软件学习掌握内容
1. 熟练掌握Materials Studio软件的安装与基本操作。
(10分)
2. 熟练掌握origin软件的安装与使用,并能够运用origin软件画出态密度和能带曲线。
(10分)
3. 运用Materials studio 软件构建晶体结构(20分)
第一,根据下列数据构建BTGS晶体的结构
Table 1. Crystal data and structure refinement for BTGS.
Empirical formula Ba3 Ga3 O14 Si2 Ta
Formula weight 1082.31
Temperature 293(2) K
Wavelength 0.71073 A
Crystal system, space group Trigonal, P321
Unit cell dimensions a = 8.4994(16) A alpha = 90 deg.
b = 8.4994(16) A beta = 90 deg.
c = 5.1828(14) A gamma = 120 deg.
Table. Atomic coordinates ( x 104) and equivalent isotropic
displacement parameters (A^2 x 10^3) for btgs.
U(eq) is defined as one third of the trace of the orthogonalized
Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________
Ta(1) 0 0 0 7(1)
Ba(1) -4322(2) 0 0 10(1)
Ga(1) -7453(3) 0 -5000 7(1)
Si(1) -3333 -6667 -5169(16) 6(1)
O(1) -3333 -6667 -2170(40) 12(1)
O(2) -4746(14) -2988(15) -3580(20) 14(2)
O(3) -2176(14) -1057(15) -7710(20) 16(2)
________________________________________________________________
第二. 根据下列数据构建RbGd(WO4)2晶体的结构
要求:(1). 晶体结构完成后,学号为双号的用球棍模型,学号为单号的用棍型方式显示原子,整体结构用in-cell方式显示,
(2). 对晶体结构中的原子,每一种元素对应的原子表示出其中一个原子的元素符号。
(3). 图形最终以图形的方式输出,要求以“姓名”表示。
4. 学会利用CASTEP计算晶体结构的基本性质(60分)
(1)计算立方BN晶体能带和态密度结构,并能够用origin对得到的能带和态密度图形进行处理,并用origin导出图形。
包括一张能带图,一张总态密度图,一张BN的分波态密度图(包括总的态密度和S、P轨道分波度态密,所以在origin中应该用三种不同颜色的线
表示),并计算其弹性常数。
命名为自己的姓名
(2)计算金刚石晶体的能带和态密度以及弹性常数。
要求同上。
5. 所有作业完成后,保存到一个文件夹中,并压缩成压缩文件,以学号+姓名.rar上传到网络平台。