转录调控-无参转录组测序最优解决方案
DNA的质量监测通常有两个方法

2)DNA的质量监测通常有两个方法:首先OD260/OD280比值应该在1.8左右(1.7-1.9),否则意味着DNA样品中存在大量的蛋白质或RNA污染。
其次,琼脂糖电泳分析时应主要以超螺旋条带为主。
最多不超过三条带(分别为超螺旋DNA,线性化DNA和环状DNA)。
否则意味质粒DNA的质量不高,应该重新制备。
2.限制性内切酶的活性1)限制性内切酶一般需要低温保存,而且反复的升降温过程对酶活性的损害很明显。
因而为了确保在有效期内的限制性内切酶不会失活,限制性内切酶的日常保存和使用应当很小。
2)建议购买具有保温功能的冻存盒保存限制性内切酶(-20度),而且取用限制性内切酶时,也应该使用具有保温功能的冻存盒,尽量防止酶的温度反复出现大的波动。
3.限制性内切酶的用量1)限制性内切酶的单位定义通常为:在合适的温度下,完全消化1ugDNA底物所需的酶量定义为一个单位。
2)在这个单位定义中,有几个不确定因素:首先是底物,不同的酶单位定义是选择的底物可能不同(常用的几个底物DNA包括:Lambda DNA ,AD2 DNA 和一些质粒DNA);第二个不确定因素是限制性内切酶在底物DNA上的酶切位点的个数。
由于单位定义中要求完全消化,因而底物上某个酶的酶切位点的个数的多少,就直接影响了该酶的单位定义。
3)因而,在进行酶切时,用1ul酶(一般10IU/ul)消化1ugDNA的通常做法是很不科学的,这也导致在实际工作中,大家要进行多次预实验才能确定最合适酶切条件。
4)以前,我推荐了一个在线的双酶切设计软件,double digestion designer, 可以精确地计算酶切时的限制性内切酶的用量。
使用中,能够注意到,用来进行双酶切的两个酶的用量有时竟然相差近20倍(EcoRI + NheI),而且发现,小片段PCR产物(100-500bp)进行酶切时,需要的酶量比质粒DNA酶切时用量多10倍以上。
5)该软件目前可以免费使用,用户名和密码都是test。
转录组测序方法

转录组测序方法
转录组测序方法:
① 从目标生物体中获取高质量RNA样品并使用DNase处理去除基因组DNA污染;
② 使用反转录酶将mRNA转化为cDNA第一链并合成双链cDNA 作为测序模板;
③ 对cDNA文库进行片段化处理通常使用超声波法得到长度均匀之片段;
④ 在片段两端加上通用接头序列便于后续PCR扩增与测序仪识别;
⑤ 通过PCR扩增增加文库拷贝数并富集接头修饰后之cDNA片段;
⑥ 使用生物信息学软件对文库进行质检如片段大小分布GC含量等;
⑦ 根据所用测序平台如Illumina PacBio等制备相应之测序文库;
⑧ 将文库装载入测序芯片并按照厂商说明书进行上机测序;
⑨ 获取原始序列数据后使用FASTQC等工具进行质量控制剔除低质量reads;
⑩ 使用比对软件如STAR HISAT2等将clean reads映射到参考基因组上;
⑪ 对比对结果进行统计分析鉴定差异表达基因新转录本可变
剪切事件等;
⑫ 根据研究目的进行下游功能注释富集分析网络构建等深入探讨基因调控机制。
无参考基因的转录组分析

无参考基因的转录组分析无参考基因的转录组分析是指在没有对应基因组序列的情况下,对生物体的转录组数据进行分析,从中获取信息并进行生物学研究。
在无参考基因组的情况下,无法直接对转录组数据进行比对和注释,因此需要采取一些策略和方法来解决这个问题。
1. 转录本组装:通过对转录组数据进行拼接,将转录本组装成单个完整序列,从而获得转录本信息。
这可以使用多个软件来实现,如Trinity、Cufflinks等。
通过对转录本进行定量分析,可以确定各个基因的表达水平。
2. 转录本定量:通过建立转录本的表达矩阵,可以对各个基因的表达水平进行比较和分析。
这可以使用软件如RSEM、eXpress等来完成。
3. 基因功能注释:虽然没有对应基因组序列,但可以利用已知物种的参考基因组信息来进行基因功能注释。
这可以使用一些在线数据库和工具,如Gene Ontology (GO)、KEGG、PANTHER等。
4. 差异表达基因筛选:通过比较不同样本组之间的转录本表达差异,可以筛选出差异表达基因。
这可以使用软件如DESeq2、edgeR等来完成。
5. 寻找新基因:在无参考基因组的情况下,还可以利用转录组数据寻找新基因。
这可以通过比对转录组序列到已知物种的参考基因组上,找出不在参考基因组上的序列,进而预测出新基因。
这可以使用软件如TransDecoder、CPC等来完成。
6.功能富集分析:通过对差异表达基因进行功能富集分析,可以了解这些基因在功能上的特点。
这可以使用一些在线工具和数据库,如DAVID、GSEA等。
7.转录因子分析:转录因子在调控基因的转录过程中起到重要的作用。
通过分析转录因子在转录组中的表达情况,可以了解其在调控过程中的参与情况。
这可以使用一些软件和数据库,如JASPAR、MEME等。
8. 代谢通路分析:通过对差异表达基因进行代谢通路分析,可以了解不同样本组之间在代谢水平上的差异。
这可以使用一些在线工具和数据库,如KEGG、MetaboAnalyst等。
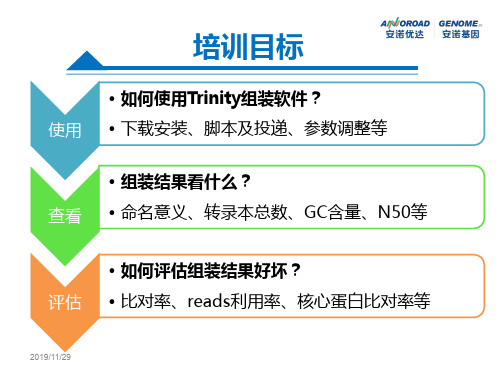
无参转录组序列组装及实际操作
2019/11/29
实际操作
核心蛋白比对率评估:
• mkdir assemblyevaluation #创建结果存放目录 • vi cegmer.sh #生成脚本
2019/11/29
Trinity组装习题
请将以下真菌数据拷贝到自己的PMO下(自己任意命名一 个文件夹即可),根据本节所学习到的知识完成数据的组装, 并对各项指标做统计。 数据路径:/home/chenxi/Trinity/practice 数据量大小:每个样本数据大小1M
框移错误导 致的缺口以 及过早终止
的比例。
2019/11/29
组装评估
判断标准: ① 无外源物种污染。
② 比对率大于80%。
组装评估
物种近缘性 良好 CDS序列相 对完整 60%以上
注释比率 核心蛋白 比对率
80%以上
准确性
2019/11/29
Stop Codon比率20%以下
Trinity参数调整
cat /home/chenxi/Trinity/clean/ Sp.ds.right.fq /home/chenxi/Trinity/ clean/ Sp.hs.right.fq> /home/chenxi/Trinity/fq/reads_2.fq
2019/11/29
实际操作
• 生成组装的shell:vi triniy.sh
物种。 优点:不依赖任何的参考基因组。 缺点:假阳性问题。
2019/11/29
组装效果统计
2019/11/29
Trinity简介
• Trinity是一款高效且稳定的以RNA-Seq为基础从头组装 转录组的软件。
• Trinity包含三个独立的软件模块: a. Inchworm(C++) b. Chrysalis(C++) c. Butterfly(Java) • 通过有秩序的对大规模的RNA-seq reads 数据进行读取,
全基因组和转录组测序技术

全基因组和转录组测序技术1.引言1.1 概述全基因组和转录组测序技术是当今生命科学领域中的重要研究工具。
随着测序技术的不断发展和成熟,我们已经能够对生物体的基因组和转录组进行高效准确的测序。
全基因组测序技术是指对一个生物体的全部基因组进行测序的技术。
它可以揭示出一个物种的全部遗传信息,包括基因的组成、位置和功能等。
全基因组测序技术的出现,使得我们可以更加深入地研究生物体的遗传变异、进化历程以及与生物特征和疾病相关的基因变异等。
同时,全基因组测序也为基因组学、遗传学、进化生物学等研究领域提供了丰富的数据资源。
转录组测序技术是指对一个生物体的转录过程中所产生的mRNA进行测序的技术。
转录组测序可以揭示出一个生物体在特定条件下的基因表达模式和调控机制。
通过分析转录组数据,我们可以了解到哪些基因在特定生理状态下被激活、哪些信号通路被调控,从而揭示出生物体的生理过程和响应机制。
转录组测序技术已经广泛应用于生物医学研究、生物工程领域以及作物育种等领域。
全基因组测序技术和转录组测序技术的发展,为我们了解生物体的基因组结构和功能提供了有力的工具。
这些测序技术的不断创新和完善,不仅提高了测序速度和准确性,还降低了测序成本。
随着测序技术的推广应用和触角延伸,我们有望在生命科学领域取得更多的突破和进展。
因此,本文将重点介绍全基因组测序技术和转录组测序技术的原理与方法,并探讨它们在不同领域的应用。
同时,文章还将总结全基因组和转录组测序技术的意义,并展望未来的发展方向。
通过深入了解这些测序技术,我们相信能够更好地理解生物体的遗传特征和调控机制,为生命科学研究和应用提供更加有力的支持。
1.2 文章结构本篇文章将分为四个主要部分介绍全基因组和转录组测序技术。
首先,在引言部分,将对全文进行概述,并说明文章的目的。
然后,在第二部分将详细介绍全基因组测序技术,包括其原理与方法以及在不同领域的应用。
接着,在第三部分将重点介绍转录组测序技术,包括其原理与方法以及应用领域。
基因转录调控研究思路

基因转录调控研究思路基因转录调控是指在细胞内调控基因转录的过程,对于理解生物体发育、生长、代谢和适应环境的调控机制具有重要意义。
在过去的几十年里,科学家们通过不断探索和研究,逐渐揭示了基因转录调控的一些重要机制。
本文将围绕基因转录调控的研究思路展开讨论。
研究基因转录调控的第一步是确定感兴趣的基因和转录因子。
转录因子是一类能够结合到DNA上特定序列的蛋白质,它们可以促进或抑制基因的转录。
通过生物信息学方法,可以获取到基因组中所有的基因和转录因子的信息。
研究者可以根据自己的研究兴趣和研究对象,筛选出一些具有潜在调控作用的基因和转录因子进行后续的实验研究。
其次,研究者可以通过实验手段,如染色质免疫共沉淀、染色质免疫沉淀测序(ChIP-seq)等技术,来确定转录因子与目标基因的结合位点。
这些技术可以帮助研究者确定转录因子与DNA的相互作用,从而揭示基因转录调控的分子机制。
通过ChIP-seq技术,可以获得全基因组范围内的转录因子结合位点的信息,进一步研究这些结合位点在基因调控中的功能和作用机制。
随后,研究者可以使用RNA测序技术(RNA-seq)来研究转录因子对基因转录的影响。
通过RNA-seq技术,可以获得不同转录因子作用下的基因表达谱,从而分析基因转录调控的整体模式。
研究者可以比较不同条件下的基因表达谱,筛选出受到特定转录因子调控的基因。
进一步分析这些基因的功能和调控机制,可以帮助我们更好地理解基因转录调控的过程。
此外,研究者还可以利用基因组编辑技术,如CRISPR-Cas9系统,来研究转录因子在基因转录调控中的作用。
通过敲除或过表达转录因子基因,可以观察基因转录的变化情况。
这些实验可以帮助研究者验证转录因子的功能和作用机制,进一步揭示基因转录调控的调控网络。
最后,综合以上的实验结果,研究者可以构建基因转录调控的调控网络模型。
这个模型可以用来描述转录因子与目标基因之间的相互作用关系,以及这些相互作用对基因表达的调控效应。
无参转录组序列组装及实际操作
2019/11/29
Trinity使用—输入及输出
输入文件: fa或者fq文件
创建一个文件存放输出结果的目录:
mkdir assemble
框移错误导 致的缺口以 及过早终止
的比例。
2019/11/29
组装评估
判断标准: ① 无外源物种污染。
② 比对率大于80%。
组装评估
物种近缘性 良好 CDS序列相 对完整 60%以上
注释比率 核心蛋白 比对率
80%以上
准确性
2019/11/29
Stop Codon比率20%以下
Trinity参数调整
2019/11/29
实际操作
本地运行:sh triniy.sh
任务运行
本地挂起运行:nohup sh triniy.sh &
投递运行:qsub –cwd –l vf=10G –l p=5 triniy.sh
任务查看:qstat/qstat –j job_number/jobs
2019/11/29
full_cleanup
只保留组装结果文件,并以Trinity.fasta命 名。
group_pairs_distance 双端reads比对的最大长度(超过该长度认为 没有比对上)
min_kmer_cov
最小k-mer覆盖值。
2019/11/29
Trinity使用—任务及运行
生成组装任务脚本:vi trinity.sh
转录本数目过多,但是N50低,怎么办? 数据量太大,如何提高组装速度? 物种类型是真菌,参数需要注意什么?
生物大数据技术解析转录调控网络的机制

生物大数据技术解析转录调控网络的机制转录调控是细胞内基因表达调控的重要过程。
在生物体发育、分化和响应环境刺激中,转录调控网络起着关键作用。
生物大数据技术作为一种有力工具,为我们深入理解转录调控网络的复杂机制提供了新的途径。
本文将以生物大数据技术为基础,解析转录调控网络的机制。
转录调控网络由转录因子和DNA结合位点组成。
转录因子是调控基因表达的蛋白质,能与DNA结合,促进或阻碍基因的转录。
DNA结合位点是转录因子与DNA结合的特定区域,通过识别DNA序列上的特定模式,转录因子能够选择性结合到目标基因。
生物大数据技术可以通过转录组学、表观基因组学和遗传学等手段来解析转录调控网络的机制。
其中,转录组学是研究细胞内所有转录过程的总和,通过测定RNA的表达水平来研究基因调控。
通过高通量测序技术,我们可以获取大规模的转录组数据,并使用生物大数据平台进行分析。
在转录组学研究中,差异表达基因分析是一个重要的方法。
通过比较不同条件下的转录组数据,我们可以鉴定出在不同生理状态下表达量发生变化的基因。
进一步分析这些差异表达基因的调控因子和结合位点,可以揭示转录调控网络的机制。
例如,通过转录组数据分析,研究人员发现一些转录因子在不同癌症类型中表达异常,进而发现它们与肿瘤发生和发展密切相关。
这为研究人员提供了新的治疗靶点。
表观基因组学是研究DNA甲基化、组蛋白修饰等表观遗传调控的一门学科。
生物大数据技术可以帮助我们分析表观基因组学数据,揭示转录调控网络的表观调控机制。
例如,研究人员发现一些甲基化酶的突变会导致肿瘤发生,这与表观基因组学的调控失衡有关。
通过分析表观基因组学数据,我们可以了解转录因子和DNA结合位点在不同组织和疾病中的表观调控模式,为研究疾病的发生和发展提供线索。
此外,遗传学也是解析转录调控网络机制的重要手段。
生物大数据技术可以帮助我们研究基因突变对转录调控网络的影响。
例如,通过分析大规模基因组数据,研究人员可以发现一些与某种疾病相关的基因突变,进而推断这些基因突变对转录因子与DNA结合位点之间的相互作用产生影响。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
微生物基因组测序16S/18S/ITS等扩增子测序细菌基因组 de novo 测序真菌基因组 de novo 测序微生物重测序宏基因组测序
动植物基因组测序全基因组su rvey 全基因组 de novo 测序泛基因组测序变异检测BSA性状定位遗传图谱全基因组关联分析群体进化Hi-C测序
人类基因组测序全基因组测序外显子测序目标区域测序单细胞基因组测序
建库测序
建库测序
版权所有:北京诺禾致源科技股份有限公司
转录调控测序 真核有参转录组测序医学转录组测序真核无参转录组测序比较转录组与泛转录组测序原核转录组测序宏转录组测序单细胞转录组测序LncRNA测序cir cRNA测序small RNA测序ChiP-seq RIP-seq
全基因组甲基化测序
分析内容
样本要求文库类型测序平台数据量
三代测序样品类型:total 样品总量:RIN :≥8cDNA 文库
(1-2K 、2-3K 、3-6K )
Pa cBio RS Ⅱ
推荐8个 SMRT cell /sam p l (约6G 数据量)
图1 Trinity拼接基因 ATP5J 和 GABPA 的转录本
Corset 应用优势
在无参转录组项目中,利用主流软件 Trinity 进行 De novo 拼接转录本,
而后选取最长的转录本作为 unigene 进行后续分析。
但是研究表明,完全以 unigene 作为基因的替身,有失恰当。
因为,拼接出来的最长转录本会掩盖掉本应真实的较短转录本(基因的 isoform)所具备的参考序列意义。
Corset 是 Trinity 官方推荐软件,可对拼接得到的转录本进行过滤和聚类,
获得更接近真实的“gene”,突破了传统“unigene”概念。
Corset 应用案例
ATP 5J 和 GABPA 两个基因有一段重叠的部分,当使用无参拼接时,会得到8条转录本,其中3条最长的转录本为拼接引起的假阳性转录本(如 C lust er b 中的转录本)。
若使用 uni gene 的方法,根据 uni gene 最长转录本原则,会选取假阳性转录本进行后续分析,这并不准确。
而使用 Cors et 聚合“G ene”的方法,可以将这些真实的转录本分离出来(如 C lust e r a 和 C lus ter d )。
无参转录组拼接升级
Corset 让“基因”概念更准确
策略
1
Trinity
Corset
or No Clustering
Unigene
*.fasta
transcript.fasta
基因分类注释
参考文献
[1] Davidson, N.M. and A. Oshlack, Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol, 2014. 15(7): p. 410. 阅读原文 >>
[2] Xu Z, Peters R, Weirather J. Full ‐length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis[J]. Plant Journal, 2015, 82(6): 951-961. 阅读原文 >>
首页 科技服务 测序指南 NGS项目文章
提高参考序列准确性 提高差异表达基因检出率 准确检测差异表达变化
无参转录组测序
最优研究策略
分析方法全新升级
三代测序大招辅助&。