NMDS分析过程(分享借鉴)
两步孟德尔随机化原理

两步孟德尔随机化原理
两步孟德尔随机化(Two-step Mendelian Randomization, 2SMR)是一种基于遗传变异的因果推断方法,其基本原理是利用自然界中的随机分配的基因型对表型的影响来推断生物学因素对疾病的影响。
两步孟德尔随机化的主要目的是探讨中介变量(mediator)是否能介导暴露对结局的影响,一般适用于寻找从暴露到结局的潜在发生机制。
该方法分为两个步骤:
从暴露的GWAS研究结果中寻找显著的SNP,去掉存在连锁不平衡的SNP,然后在中介变量的GWAS结果中提取剩下来的SNP信息。
这里需要保证剩下来的SNP不能和混杂因素以及中介变量直接相关。
最后计算暴露到中介变量的因果效应。
从中介变量的GWAS研究结果中寻找显著的SNP,去掉存在连锁不平衡的SNP,然后在结局的GWAS结果中提取剩下来的SNP信息。
这里需要保证剩下来的SNP不能和混杂因素以及结局直接相关。
最后计算中介变量到结局的因果效应。
通过这两个步骤,我们可以得到三个beta值(beta0、beta1和beta2)。
根据这三个beta值的显著情况,我们可以推断出暴露、中介变量和结局之间的因果关系。
需要注意的是,孟德尔随机化方法虽然能够提供有关因果关系的线索,但其结果并不总是绝对的。
在实际应用中,还需
要结合其他证据和方法来综合评估因果关系的可能性。
平菇发酵培养料制备过程中黄曲霉毒素降解变化分析

平菇发酵培养料制备过程中黄曲霉毒素降解变化分析作者:崔筱胡素娟刘芹吴杰师子文张玉亭孔维丽来源:《中国瓜菜》2023年第11期摘要:为解析平菇发酵培养料制备过程中黄曲霉毒素降解的变化规律,采用非靶向代谢组学技术和宏基因组学技术,对平菇发酵培养料制备过程中5个时期黄曲霉毒素含量及相关微生物基因丰度进行关联分析。
结果表明,发酵初期(T1)培养料含有AFM1、AFG2两种黄曲霉毒素,升温阶段(T2)其含量均升至最高;第2次至第4次翻堆(T3至T5)时期未检测到黄曲霉毒素。
相关性分析表明,两种黄曲霉毒素含量与曲霉属的相对丰度呈显著正相关。
类芽胞杆菌属(Paenibacillus)和藤黄单胞菌属(Luteimonas)在发酵过程中是优势属,且具有降解黄曲霉毒素的能力;发酵过程中黄曲霉毒素含量变化与该2个属的相对丰度呈极显著负相关,推测该2个属可能在平菇培养料发酵过程中降解了黄曲霉毒素。
关键词:平菇;发酵培养料;黄曲霉毒素;降解中图分类号:S646 文献标志码:A 文章编号:1673-2871(2023)11-093-07Analysis of aflatoxin degradation during preparation of Pleurotus ostreatus compost substrateCUI Xiao, HU Sujuan, LIU Qin, WUJie, SHI Ziwen, ZHANG Yuting, KONG Weili(Institute of Edible Fungi, Henan Academy of Agricultural Sciences, Zhengzhou 450002,Henan, China)Abstract: In order to analyze the change of the degradation of aflatoxins during the compost substrate preparation process of Pleurotus ostreatus, non-targeted metabolomics and metagenomic sequencing techniques were used to analyze aflatoxin content and related microbial gene abundance in 5 periods during the compost process. The results showed that in the initial stage of compost(T1),the substrates contained two types of aflatoxins, AFM1 and AFG2, while in the temperature increasing stage(T2), their contents rose to the highest. No aflatoxin was detected during the second to fourth turnover(T3 to T5)period. Correlation analysis showed that the content of two aflatoxins were significantly positively correlated with the relative abundance of Aspergillus genus during the compost process. Paenibacilli and Luteimonas were dominant genera in the compost process and had the ability to degrade aflatoxin; it was found that there was a highly significant negative correlation between the aflatoxin content and the relative abundance of these two genera during the compost process by correlation analysis. Therefore, it is speculated that these two genera may have degraded aflatoxin during the compost process.Key words: Pleurotus ostreatus; Compost substrate; Aflatoxin; Degradation玉米芯是黃淮海地区栽培平菇的主要原料,秋季收获后的玉米芯存放不当易产生黄曲霉毒素。
nmds分析微生物代码

nmds分析微生物代码sample<-read.table(file.choose(),sep="\t",s=1)#第一行为列名group<read.table(file.choose(),sep="\t",header=T,row.na mes=1)#其中header=T表示将文件中第一行设为列名字。
s=1表示第一列设为行名group1<-group[match(rownames(sample),rownames(group)),]#匹配行名,很重要group=group[rownames(sample),]#nmds分析nmds1<-metaMDS(sample,distance='bray',k=3)summary(nmds1)#提取数据nmds1.stress<-nmds1$stressnmds1.point<-data.frame(nmds1$point)nmds1.species<-data.frame(nmds1$species)sample_site<-nmds1.point[1:2]sample_site$names<-rownames(sample_site)colnames(sample_site)[1:2]<-c('NMDS1','NMDS2')#合并分组数据sample_site<-cbind(sample_site,group1)#给分组排序sample_site$Depth<-factor(sample_site$Depth,levels=c('0m','25m','50m','117m',' 150m','200m',ordered=FALSE))sample_site$Particlesize<-factor(sample_site$Particlesize,levels=c("0.2-0.6μm","0.6-1.2μm","1.2-2μm","2-20μm",">20μm",ordered=FALSE))View(sample_site)#NMDS图绘制windowsFonts(TNM=windowsFont("TimesNewRoman"))#设置字体nmds_plot<-ggplot()+geom_point(data=sample_site,aes(NMDS1,NMDS2,color=Parti clesize,shape=Depth),size=5,alpha=0.8)+#可在这里修改点的透明度、大小scale_shape_manual(values=c(15,17,18,19,20,79))+#可在这里修改点的形状scale_color_manual(values=c("#99CCFF","#FF9999","#FFCC3 3","#99CC99","#996699"))+#可在这里修改点的颜色。
科学网——精选推荐

科学⽹做过16s测序的⼩伙伴们都知道测完之后会拿到⼀份结果报告但这并不代表可以开始写⽂章了看似⼀⼤堆数据图表却不知如何下⼿这是很多⼈头疼的地⽅那么怎样给报告中的数据赋予灵魂让它真正成为对你有帮助的分析呢?今天我们来详细解读下。
⼀⽂扫除困惑⾸先什么是16S rRNA?16S rRNA 基因是编码原核⽣物核糖体⼩亚基的基因,长度约为1542bp,其分⼦⼤⼩适中,突变率⼩,是细菌系统分类学研究中最常⽤和最有⽤的标志。
16S rRNA基因序列包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系,⽽可变区序列则能体现物种间的差异。
16S rRNA基因测序以细菌16S rRNA基因测序为主,核⼼是研究样品中的物种分类、物种丰度以及系统进化。
⼆代⾼通量测序原理⽬前⼆代测序是⼀个边合成边测序的过程,使⽤的是荧光可逆终⽌⼦。
每个可逆终⽌⼦的碱基3’端都有⼀个阻断基团,⽽在侧边带有⼀种荧光。
由于有4种不同的碱基(ATCG),因此也会有对应4种不同颜⾊的荧光。
开始扩增每次结合上⼀个碱基,DNA的扩增便会停⽌,此时能收到⼀种荧光信号。
然后放试剂除去阻断基团,进⾏下⼀个碱基的结合,以此类推得到⼀连串的荧光信号组合序列。
⽽根据荧光的颜⾊我们便可以确定每⼀个位点的基因型,即可以得到这⼀段DNA⽚段的序列。
环境样品⾼通量分析需要重复么?在进⾏实验设计前,这是有些⼩伙伴⾯临的⼀个问题。
环境样本由于来源和条件不完全可控,每个样品之间会存在很⼤的差异,即便是相同样本的不同取样时间和部位也会存在⼀定的差异。
基于⾼通量测序主要是为了了解样品的菌群构成和功能分析,以及寻找不同环境之间的差异,包括菌和功能基因以及代谢。
如果仅做单⼀样本,很可能结论只能代表这个单⼀取样样本的信息,⽆法排除不同样本重复之间的差异,也就可能得不到真正代表环境差异的结果。
所以环境样品不仅要重复⽽且还应该以分组⽅式取尽量多的样本以全⾯的代表⼀个环境条件下的各种变异情况。
NMDS分析过程

NMDS分析过程NMDS(Nonmetric Multidimensional Scaling)是一种用于分析多元数据的统计方法,通过将样本点在多维空间中的位置转换为相似性或距离矩阵,进而进行可视化和聚类分析。
下面将详细介绍NMDS的分析过程。
1.数据准备首先需要准备一组多元数据,这些数据可以是样本的特征、属性、变量等,可以是连续型、二值型或多值型的数据。
将这些数据整理成一个数据矩阵,每一行代表一个样本,每一列代表一个变量。
2.相似性或距离矩阵计算NMDS需要将样本间的相似性或距离转化为矩阵形式。
如果数据是连续型变量,可以使用距离度量方法(如欧氏距离、曼哈顿距离等)计算样本间的距离矩阵;如果数据是多值型变量,可以使用相关系数、Jaccard 系数等计算样本间的相似性矩阵。
3.NMDS模型拟合在进行NMDS分析之前,需要选择一个合适的距离或相似性测度,并定义一个合适的模型。
常用的模型有几何模型、非几何模型等,每个模型都有自己的假设和性质。
通常,可以首先使用几何模型进行分析,如果结果不理想时,再尝试其他模型。
4.初始点计算NMDS需要指定初始样本点的位置,在多维空间中,这些点将代表样本。
初始点的选择对结果的稳定性和准确性有一定的影响。
常见的初始点选择方法有随机选择、主坐标估计等。
5.迭代计算NMDS采用迭代算法(如梯度下降法)通过不断调整样本点在多维空间中的位置,使得样本点之间的距离或相似性与原始数据矩阵中的距离或相似性最为接近。
迭代过程中,可以设置一定的停止准则,当达到停止准则时,迭代计算停止。
6.结果评价在迭代计算完成后,可以通过各类统计指标来评价NMDS的结果。
常见的评价指标包括压缩比、应力值、Stress图等。
压缩比表示新的坐标与原始距离矩阵的匹配度,应力值越小说明结果越好。
Stress图可以用来观察迭代过程中误差的减小情况。
7.可视化和聚类分析最后,可以利用NMDS的结果进行多维空间的可视化和聚类分析。
NMDS非度量多维尺度分析—基于微生物群落

NMDS⾮度量多维尺度分析—基于微⽣物群落今天,看到赖江⼭⽼师在博客中分享了vegan中的⼀些函数的中⽂帮助⽂件,翻译专业,可读性强,这本材料是我们熟悉vegan原理和提⾼内涵的有⼒学习途径。
(末尾有彩蛋)本⽂主要做NMDS分析并做⼀张完善的⾼质量图⽚,提取stress值,推荐适合NMDS结果的差异分析并通过命令展⽰在图形上,最后加上置信区间椭圆。
⾮度量多维尺分析( NMDS)是⼀种很好的排序⽅法,因为它可以使⽤ 具有⽣态学意义的⽅法来度量群落差异 。
⼀个好的 相异性测度与环境梯距离具有很好的秩 关系。
因为NMDS只使⽤秩信息,并且映射的在有序空间 上是⾮线性的, 故它能处理任意 类型 的⾮线性物种 矩阵 ,并能有效、稳健地找 到潜在梯度。
NMDS分析,⽹络上已近有很多相关教程分享其原理,与其他排序(PCA、PCoA、CCA、RDA) ⽅法的不同之处,简单来讲NMDS也是⼀种使⽤物种组成数据的排序称作⾮限制性排序;NMDS基于距离算法,优于PCA、PCoA、CCA、RDA的地⽅在于当样本或者物种数量过多的时候使⽤NMDS会更加准确;vegan 的ordiplot()函数可以⽤来绘制NMDS 的结果:plot(vare.mds, type = "t")vegan 包中的metaMDS()函数不需要单独计算相异矩阵,直接 将原始数据矩阵作为输⼊。
结果⽐以前更丰富 ,除了奥杜尔包中isoMDS()结果中 的成分外还有很多其他结果输出:nobj, nfix, ndim, ndis, ngrp,diss, iidx, jidx, xinit, istart, isform, ities, iregn, iscal, maxits, sratmx, strmin, sfgrmn, dist, dhat, points, stress, grstress, iters, icause, call,model, distmethod, distcall, data, distance, converged, tries,engine, species。
微生物分析用图的看图说明
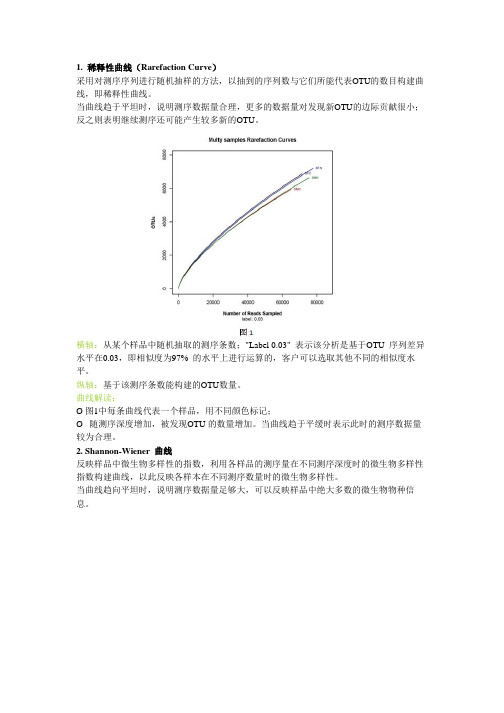
1. 稀释性曲线(Rarefaction Curve)采用对测序序列进行随机抽样的方法,以抽到的序列数与它们所能代表OTU的数目构建曲线,即稀释性曲线。
当曲线趋于平坦时,说明测序数据量合理,更多的数据量对发现新OTU的边际贡献很小;反之则表明继续测序还可能产生较多新的OTU。
横轴:从某个样品中随机抽取的测序条数;"Label 0.03" 表示该分析是基于OTU 序列差异水平在0.03,即相似度为97% 的水平上进行运算的,客户可以选取其他不同的相似度水平。
纵轴:基于该测序条数能构建的OTU数量。
曲线解读:Ø 图1中每条曲线代表一个样品,用不同颜色标记;Ø 随测序深度增加,被发现OTU 的数量增加。
当曲线趋于平缓时表示此时的测序数据量较为合理。
2. Shannon-Wiener 曲线反映样品中微生物多样性的指数,利用各样品的测序量在不同测序深度时的微生物多样性指数构建曲线,以此反映各样本在不同测序数量时的微生物多样性。
当曲线趋向平坦时,说明测序数据量足够大,可以反映样品中绝大多数的微生物物种信息。
横轴:从某个样品中随机抽取的测序条数。
纵轴:Shannon-Wiener 指数,用来估算群落多样性的高低。
Shannon 指数计算公式:其中,S obs= 实际测量出的OTU数目;n i= 含有i 条序列的OTU数目;N = 所有的序列数。
曲线解读:Ø 图2每条曲线代表一个样品,用不同颜色标记,末端数字为实际测序条数;Ø 起初曲线直线上升,是由于测序条数远不足覆盖样品导致;Ø 数值升高直至平滑说明测序条数足以覆盖样品中的大部分微生物。
3.Rank-Abundance 曲线用于同时解释样品多样性的两个方面,即样品所含物种的丰富程度和均匀程度。
物种的丰富程度由曲线在横轴上的长度来反映,曲线越宽,表示物种的组成越丰富;物种组成的均匀程度由曲线的形状来反映,曲线越平坦,表示物种组成的均匀程度越高。
扩增子β多样性分析

扩增子-β多样性分析美格基因一、关于β多样性分析β多样性(Beta Diversity)是指不同样品间的生物多样性的比较,是对不同样品间的微生物群落构成进行比较。
β多样性分析通常由计算环境样本间的距离矩阵开始,对群落数据结构进行自然分解,并通过对样本进行排序(Ordination),从而观测样本之间的差异。
β多样性与α多样性一起构成了总体多样性或一定环境群落的生物异质性。
β多样性分析中通常采用以下几种算法:bray_curtis、euclidean、abund_jaccard、unweighted_unifrac、weighted_unifrac等计算任意两个样本间的距离从而获得样本距离矩阵,这些算法主要分为两大类:加权(如Bray-Curtis和Weighted Unifrac)与非加权(如Jaccard和Unweightde Unifrac)。
利用非加权的计算方法,主要比较的是物种的有无,如果两个群体的β多样性越小,则说明两个群体的物种类型越相似。
而加权方法,则需要同时考虑物种有无和物种丰度两个层面。
Bray curtis 距离基于物种的丰度信息计算,是生态学上反应群落之间差异性常用的指标之一。
Weighted Unifrac 距离是一种同时考虑各样品中微生物的进化关系和物种的相对丰度,计算样品的距离,而(Unweighted Unifrac)则只考虑物种的有无,忽略物种间的相对丰度差异。
Uweighted Unifrac 距离对稀有物种比较敏感,而Bray curtis 和Weighted Unifrac 距离则对丰度较高的物种更加敏感。
最后,基于以上的距离矩阵,通过多变量统计学方法主坐标分析(PcoA,Principal co-ordinatesAnalysis),非加权组平均聚类分析(UPGMA,UnweightedPair-groupMethod with Arithmetic Means)等分析,进一步从结果中挖掘各样品间微生物群落结构的差异和不同分类对样品间的贡献差异。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
以微量热指标数据为例
1.数据输入格式
其中,Number指的是土样个数;Species指测定指标;Excel文件格式为.xls(因为PCORD5只识别这个格式)。
2.安装并打开PCORD5软件
软件界面:
导入数据:File—Import Matrix—Main Matrix
我们文件类型是Excel所以就选择Excel Spreadsheet选项,点击OK。
选择所需要的文件 。
3.数据分析
Ordination菜单栏—NMS/NMDS选项。
Autopilot:三种模式,根据需要选择模式。
Distance Measure:根据数据选择合适的方法
常用的是Sorensen(Bray-Curtis)、Relative Sorensen、Relative Euclidean
不用修改
点击OK。
4.结果输出
5.作图
(1)
(2)二维图结果
(3)Statistics——correlationswith Main Matrix,点击OK。
(4)Statistics——Percentof Variance In Distance Matrix。
点击OHale Waihona Puke 。Origin8作散点图:
由于PCORD5做出来的图不好修改,所以将数据拷贝出来用Origin8来做图。