119溶出度测定法

编制依据:药品生产质量管理规范(2010年修订)《中华人民共和国药典》2015年版四部120页(通则0931) 内容:
溶出度系指活性药物从片剂、胶囊剂或颗粒剂等普通制剂在规定条件下溶出的速率和程度,在缓释制剂、控释制剂、肠溶制剂及透皮贴剂等制剂中也称释放度.
仪器装置
第一法(篮法)
(1)转篮分篮体与篮轴两部分,均为不锈钢或其他惰性材料制成,其形状尺寸如图1所示.篮体
A 由方孔筛网(丝径为0.28mm±0.03mm,网孔为0.04mm±0.04mm)制成,呈圆柱形,转篮内径为
20.2mm±1.0mm,上下两端都有封边.篮轴B的直径为9.75mm±0.35mm,轴的末端连一圆盘,作为转篮的盖;盖上有一通气孔(孔径为2.0mm±0.5mm);盖边系两层,上层直径与转篮外径相同,下层直径与转篮内径相同;盖上的3个弹簧片与中心呈120°
(2)溶出杯一般由硬质玻璃或其他惰性材料制成的底部为半球形的1000ml杯状容器,内径为102mm±4mm,高为185mm±25mm;溶出杯配有适宜的盖子,盖上有适当的孔,中心孔为篮轴的位置,其他孔供取样或测量温度用.溶出杯置恒温水浴或其他适当的加热装置中.
(3)篮轴与电动机相连,由速度调节装置控制电动机的转速,使篮轴的转速在各品种项下规定转速的±4%范围之内.运转时整套装置应保持平稳,均不能产生明显的晃动或振动(包括装置所处的环境).转篮旋转时,篮轴与溶出杯的垂直轴在任一点的偏离均不得大于2mm,转篮下缘的摆动幅度不得偏离轴心1.0mm.
(4)仪器一般配有6套以上测定装置.
第二法(桨法)
除将转篮换成搅拌桨外,其他装置和要求与第一法相同.搅拌桨的下端及桨叶部分可涂适当的惰性材料(如聚四氟乙烯),其形状尺寸如图2所示.桨杆对称度(即桨轴左侧距桨叶左边缘距离与桨轴右侧距桨叶右边缘距离之差)不得超过 0.5mm,桨轴和桨叶垂直度 90°±0.2°;桨杆旋转时,桨轴与溶出杯的垂直轴在任一点的偏差均不得大于2mm;搅拌桨旋转时A、B两点的摆动幅度不得超过0.5mm.
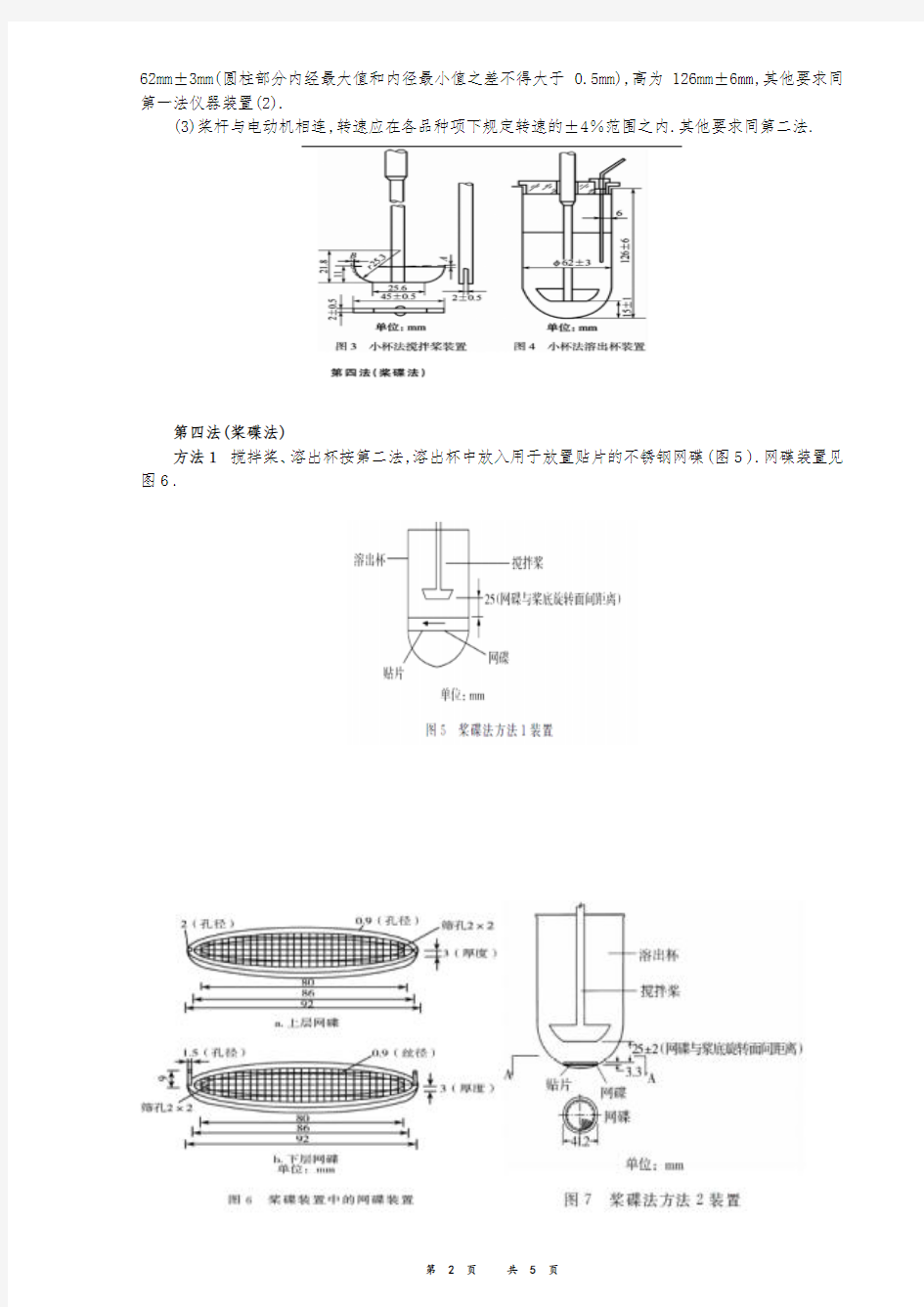
第三法(小杯法)
(1)搅拌桨形状尺寸如图4所示.桨杆上部直径为9.75mm±0.35mm,桨杆下部直径为 6.0mm±0.2mm;桨杆对称度(即桨轴左侧距桨叶左边缘距离与桨轴右侧距桨叶右边缘距离之差)不得超过0.5mm,桨轴和桨叶垂直度90°±0.2°;桨杆旋转时,桨轴与溶出杯的垂直轴在任一点的偏差均不得大于2mm;搅拌桨旋转时,A、B两点的摆动幅度不得超过0.5mm.
(2)溶出杯一般由硬质玻璃或其他惰性材料制成的底部为半球形的250ml 杯状容器,内径为
62mm±3mm(圆柱部分内经最大值和内径最小值之差不得大于0.5mm),高为126mm±6mm,其他要求同第一法仪器装置(2).
(3)桨杆与电动机相连,转速应在各品种项下规定转速的±4%范围之内.其他要求同第二法.
第四法(桨碟法)
方法1搅拌桨、溶出杯按第二法,溶出杯中放入用于放置贴片的不锈钢网碟(图5).网碟装置见图6.
方法2除将方法1的网碟换成图7所示的网碟外,其他装置和要求与方法1相同.
第五法(转筒法)
溶出杯按第二法,但搅拌桨另用不锈钢转筒装置替代.组成搅拌装置的杆和转筒均由不锈钢制成,其规格尺寸见图8.
测定法
第一法和第二法
普通制剂测定前,应对仪器装置进行必要的调试,使转篮或桨叶底部距溶出杯的内底部25mm±2mm.分别量取溶出介质置各溶出杯内,实际量取的体积与规定体积的偏差应在±1%范围之内,待溶出介质温度恒定在37℃±0.5℃后,取供试品6片(粒、袋),如为第一法,分别投入6个干燥的转篮内,将转篮降入溶出杯中;如为第二法,分别投入6个溶出杯内(当品种项下规定需要使用沉降篮时,可将胶囊剂先装入规定的沉降篮内;品种项下未规定使用沉降篮时,如胶囊剂浮于液面,可用一小段耐腐蚀的细金属丝轻绕于胶囊外壳.沉降篮的形状尺寸如图9所示).注意避免供试品表面产生气泡,立即按各品种项下规定的转速启动仪器,计时;至规定的取样时间(实际取样时间与规定时间的差异不得过±2%),吸取溶出液适量(取样位置应在转篮或桨叶顶端至液面的中点,距溶出杯内壁10mm 处;需多次取样时,所量取溶出介质的体积之和应在溶出介质的1%之内,如超过总体积的1%时,应及时补充相同体积的温度为37℃±0.5℃的溶出介质,或在计算时加以校正),立即用适当的微孔滤膜滤过,自取样至滤过应在30秒内完成.取澄清滤液,照该品种项下规定的方法测定,计算每片(粒、袋)的溶出量.
缓释制剂或控释制剂照普通制剂方法操作,但至少采用三个取样时间点,在规定取样时间点,吸取溶液适量,及时补充相同体积的温度为37℃±0.5℃的溶出介质,滤过,自取样至滤过应在30秒内完成.照各品种项下规定的方法测定,计算每片(粒)的溶出量.
肠溶制剂按方法1或方法2操作.
方法1酸中溶出量除另有规定外,分别量取0.1mol/L 盐酸溶液 750ml 置各溶出杯内,实际量取的体积与规定体积的偏差应在±1%范围之内,待溶出介质温度恒定在37℃±0.5℃ ,取供试品6片(粒)分别投入转篮或溶出杯中(当品种项下规定需要使用沉降篮时,可将胶囊剂先装入规定的沉降篮内;品种项下未规定使用沉降篮时,如胶囊剂浮于液面,可用一小段耐腐蚀的细金属丝轻绕于胶囊外壳.),注意避免供试品表面产生气泡,立即按各品种项下规定的转速启动仪器,2小时后在规定取样点吸取溶出液适量,滤过,自取样至滤过应在30秒内完成.按各品种项下规定的方法测定,计算每片(粒)的酸中溶出量.
其他操作同第一法和第二法项下普通制剂。
缓冲液中溶出量上述酸液中加入温度为37℃±0.5℃的0.2mol/L 磷酸钠溶液250ml(必要时用
2mol/L 盐酸溶液或2mol/L 氢氧化钠溶液调节pH值至6.8),继续运转45分钟,或按各品种项下规
定的时间,在规定取样点吸取溶出液适量,滤过,自取样至滤过应在30秒钟内完成.按各品种项下规
定的方法测定,计算每片(粒)的缓冲液中溶出量.
方法2酸中溶出量除另有规定外,量取 0.1mol/L盐酸溶液 900ml,注入每个溶出杯中,照方法
1酸中溶出量项下进行测定.
缓冲液中溶出量弃去上述各溶出杯中酸液,立即加入温度为37℃±0.5℃的磷酸盐缓冲液(pH6.8)(取0.1mol/L 盐酸溶液和0.2mol/L 磷酸钠溶液,按3:1混合均匀,必要时用2mol/L 盐酸溶
液或2mol/L氢氧化钠溶液调节 pH 值至 6.8)900ml,或将每片(粒)转移入另一盛有温度为37℃±0.5℃的磷酸盐缓冲液(pH6.8)900ml 的溶出杯中,照方法1缓冲液中溶出量项下进行测定.
第三法
普通制剂测定前,应对仪器装置进行必要的调试,使桨叶底部距溶出杯的内底部15mm±2mm.分
别量取溶出介质置各溶出杯内,介质的体积150~250ml,实际量取的体积与规定体积的偏差应在±1%范围之内(当品种项下规定需要使用沉降装置时,可将胶囊剂先装入规定的沉降装置内;品种项
下未规定使用沉降装置时,如胶囊剂浮于液面,可用一小段耐腐蚀的细金属丝轻绕于胶囊外壳).以下
操作同第二法.取样位置应在桨叶顶端至液面的中点,距溶出杯内壁6mm 处.
缓释制剂或控释制剂照第三法普通制剂方法操作,其余要求同第一法和第二法项下,缓释制剂
或控释制剂.
第四法
透皮贴剂分别量取溶出介质置各溶出杯内,实际量取的体积与规定体积的偏差应在±1%范围
之内,待溶出介质预温至32℃±0.5℃;将透皮贴剂固定于两层碟片之间(方法1)或网碟上(方法2),
溶出面朝上,尽可能使其保持平整.再将网碟水平放置于溶出杯下部,并使贴剂与桨底旋转面平行,两
者相距25mm±2mm,按品种正文规定的转速启动装置.在规定取样时间点,吸取溶出液适量,及时补充
相同体积的温度为32℃±0.5℃的空白溶出介质.
其他操作同第一法和第二法项下缓释制剂或控释制剂.
第五法
透皮贴剂分别量取溶出介质置各溶出杯内,实际量取的体积与规定体积的偏差应在±1%范围
之内,待溶出介质预温至32℃±0.5℃除另有规定外,按下述进行准备,除去贴剂的保护套,将有黏性
的一面置于一片铜纺上,铜纺的边比贴剂的边至少大1cm.将贴剂的铜纺覆盖面朝下放置于干净的表面,涂布适宜的胶粘剂于多余的铜纺边.如需要,可将胶粘剂涂布于贴剂背面.干燥1分钟,仔细将贴
剂涂胶粘剂的面安装于转筒外部,使贴剂的长轴通过转筒的圆心.挤压铜纺面除去引入的气泡.将转
筒安装在仪器中,试验过程中保持转筒底部距溶出杯内底部25mm±2mm,立即按品种正文规定的转速
启动仪器.在规定取样时间点,吸取溶出液适量,及时补充相同体积的温度为32℃±0.5℃的空白溶出
介质.取样位置在介质液面与转筒顶部的中间位置,距溶出杯内壁10mm 处.同法测定其他透皮贴剂.
取样方法同第一法和第二法项下缓释制剂或控释制剂.
以上五种测定法中,当采用原位光纤实时测定时,辅料的干扰应可以忽略,或可以通过设定参比
波长等方法消除;原位光纤实时测定主要适用于溶出曲线和缓释制剂溶出度的测定.
结果判定
普通制剂符合下述条件之一者,可判为符合规定:
(1)6片(粒、袋)中,每片(粒、袋)的溶出量按标示量计算,均不低于规定限度(Q);
(2)6片(粒、袋)中,如有1~2片(粒、袋)低于Q,但不低于Q-10%,且其平均溶出量不低于Q;
(3)6片(粒、袋)中,有1~2片(粒、袋)低于Q,其中仅有1片(粒、袋)低于Q-10%,但不低于Q
-20%,且其平均溶出量不低于Q时,应另取6片(粒、袋)复试;初、复试的12片(粒、袋)中有1~3
片(粒、袋)低于Q,其中仅有1片(粒、袋)低于Q-10%,但不低于Q-20%,且其平均溶出量不低于Q.以上结果判断中所示的10%、20%是指相对于标示量的百分率(%).
缓释制剂或控释制剂除另有规定外,符合下述条件之一者,可判为符合规定:
(1)6片(粒)中,每片(粒)在每个时间点测得的溶出量按标示量计算,均未超出规定范围;
(2)6片(粒)中,在每个时间点测得的溶出量,如有1~2片(粒)超出规定范围,但未超出规定范围
的10%,且在每个时间点测得的平均溶出量未超出规定范围;
(3)6片(粒)中,在每个时间点测得的溶出量,如有1~2片(粒)超出规定范围,其中仅有1片(粒)超出规定范围的10%,但未超出规定范围的20%,且其平均溶出量未超出规定范围,应另取6片(粒)复试;初、复试的12片(粒)中,在每个时间点测得的溶出量,如有1~3片(粒)超出规定范围,其中仅有1片(粒)超出规定范围的10%,但未超出规定范围的20%,且其平均溶出量未超出规定范围.
以上结果判断中所示超出规定范围的10%、20%是指相对于标示量的百分率(%),其中超出规定范围10%是指:每个时间点测得的溶出量不低于低限的-10%,或不超过高限的+10%;每个时间点测得的溶出量应包括最终时间测得的溶出量.
肠溶制剂除另有规定外,符合下述条件之一者,可判为符合规定:酸中溶出量 (1)6片(粒)中,每片(粒)的溶出量均不大于标示量的10%;
(1)6片(粒)中,有1~2片(粒)大于10%,但其平均溶出量不大于10%.
缓冲液中溶出量 (1)6片(粒)中,每片(粒)的溶出量按标示量计算均不低于规定限度(Q);除另有规定外,Q 应为标示量的70%;
(2)6片(粒)中仅有1~2片(粒)低于Q,但不低于 Q-10%,且其平均溶出量不低于Q;
(3)6片(粒)中如有1~2片(粒)低于Q,其中仅有1片(粒)低于 Q-10%,但不低于 Q-20%,且其平均溶出量不低于Q时,应另取6片(粒)复试;初、复试的12片(粒)中有1~3片(粒)低于Q,其中仅有1片(粒)低于 Q-10%,但不低于 Q-20%,且其平均溶出量不低于Q.
以上结果判断中所示的10%、20%是指相对于标示量的百分率(%).
透皮贴剂除另有规定外,同缓释制剂或控释制剂.
【溶出条件和注意事项】
(1)溶出度仪的适用性及性能确认试验除仪器的各项机械性能应符合上述规定外,还应用溶出度标准片对仪器进行性能确认试验,按照标准片的说明书操作,试验结果应符合标准片的规定.
(2)溶出介质应使用各品种项下规定的溶出介质,除另有规定外,室温下体积为900ml,并应新鲜配制和经脱气处理;如果溶出介质为缓冲液,当需要调节pH值时,一般调节pH值至规定pH值±0.05之内.
(3)取样时间应按照品种各论中规定的取样时间取样,自6杯中完成取样的时间应在1分钟内.
(4)除另有规定外,颗粒剂或干混悬剂的投样应在溶出介质表面分散投样,避免集中投样.
(5)如胶囊壳对分析有干扰,应取不少于6粒胶囊,除尽内容物后,置一个溶出杯内,按该品种项下规定的分析方法测定每个空胶囊的空白值,作必要的校正.如校正值大于标示量的25%,试验无效.如校正值不大于标示量的2%,可忽略不计.
溶解度的测定
硝酸钾溶解度得测定(方法1:结晶析出法) 实验原理: 先设计好不同溶质与溶剂得量,称量、混合、加热、搅拌使其溶解,降温并用温度计分别测定其开始析出晶体时得温度,即所得溶液为该温度下得饱与溶液,计算该温度下得溶解度。实验用品: 托盘天平(J0160,200g,0.2g),烧杯(J6124),大试管(J6104),玻璃棒(J6453),温度计(J6071,量程0~100℃),酒精灯(J6201),量筒(J6001,10ml),方座支架(J1102,带铁圈),石棉网(J6432),药匙(J6442),试管刷(J6471),硝酸钾(化学纯),蒸馏水。 实验步骤: 一、检查实验用品就是否齐全、完好。 二、硝酸钾得称取与溶解。 1、用托盘天平分别准确称取硝酸钾3.5g、1.5g、1.5g、2.0g、2.5g,称量过程详见分组实验三得步骤二。将称好得5份硝酸钾放在实验台上,并做标记。 2.在一支大试管中加入上面称取得3.5g硝酸钾。 3.用量筒准确量取10.0m1蒸馏水,加入大试管中。 4.在水浴中加热大试管,边加热边搅拌,至硝酸钾完全溶解(水浴温度不要太高,以刚好使硝酸钾溶解为宜,否则会使下一步结晶析出操作耗时过长) 三、硝酸钾得结晶。 1.自水浴中取出大试管,插入一支干净得温度计,用玻璃棒轻轻搅拌并摩擦试管壁,同时观察温度计得读数。当刚开始有晶体析出时,立即记下此时得温度t1,并填入下表中。 2.把试管再放入水浴中加热,使晶体全部溶解,然后重复两次上述实验步骤得操作,分别测定开始析出晶体时得温度t2、t3。将读数填入表格。 四、溶解度曲线得绘制。 1.依次向试管中再加入1.5g、1.5g、2.0g、2.5g硝酸钾(使试管中依次共有硝酸钾5.0g、6.5g、8.5g、11.0g),每次加入硝酸钾后都重复溶解、结晶实验步骤得操作,并将晶体开始析出时得温度读数填人表格。
紫外分光光度法计算
第20章 吸光光度法 思 考 题 1. 什么叫单色光复色光哪一种光适用于朗伯-比耳定律 答:仅具有单一波长的光叫单色光。由不同波长的光所组成光称为复合光。朗伯--比耳定律应适用于单色光。 2. 什么叫互补色与物质的颜色有何关系 答:如果两种适当的单色光按一定的强度比例混合后形成白光,这两种光称为互补色光。当混合光照射物质分子时,分子选择性地吸收一定波长的光,而其它波长的光则透过,物质呈现透过光的颜色,透过光与吸收光就是互补色光。 3. 何谓透光率和吸光度 两者有何关系 答:透光率是指透射光强和入射光强之比,用T 表示 T = t I I 吸光度是吸光物质对入射光的吸收程度,用A 表示,A εbc =,其两者的关系 lg =-A T 4. 朗伯-比耳定律的物理意义是什么 什么叫吸收曲线 什么叫标准曲线 答:朗伯--比耳定律是吸光光度法定量分析的理论依据,即吸光物质溶液对光的吸收程度与溶液浓度和液层厚度之间的定量关系。数学表达式为 lg A T εbc =-= 吸收曲线是描述某一吸光物质对不同波长光的吸收能力的曲线,即在不同波长处测得吸光度,波长为横坐标,吸光度为纵坐标作图即可得到吸收曲线。 标准曲线是描述在一定波长下,某一吸光物质不同浓度的溶液的吸光能力的曲线,吸光度为纵坐标,浓度为横坐标作图即可得到。 5. 何谓摩尔吸光系数质量吸光系数两者有何关系 答:吸光系数是吸光物质吸光能力的量度。摩尔吸光系数是指浓度为 mol ·L ,液层度为1cm 时,吸光物质的溶液在某一波长下的吸光度。用ε表示,其单位 11cm mol L --??。 质量吸光系数是吸光物质的浓度为1g 1L -?时的吸光度,用a 表示。其单位 11cm g L --?? 两者的关系为 εM a =? M 为被测物的摩尔质量。 6. 分光光度法的误差来源有哪些 答:误差来源主要有两方面,一是所用仪器提供的单色光不纯,因为单色光不纯时,朗伯—比耳定律中吸光度和浓度之间的关系偏离线性;二是吸光物质本身的化学反应,其结果同样
溶出度测定方法
影响因素试验的溶出度测定 测定方法参照美国药典盐酸二甲双胍缓释片质量标准。 照释放度测定法(中国药典2010年版二部附录X D第一法),采用溶出度测定法(中国药典2010年版二部附录XC第一法蓝法)的装置,以pH6.8磷酸二氢钾缓冲液(1000ml水中加入6.8g磷酸二氢钾,用0.2N的氢氧化钠溶液调pH为6.8 ± 0.1)1000ml为溶剂,转速为每分钟100转,按溶出度测定法依法操作。分别于预定时间取溶液5ml滤过(并及时向溶出杯中补充同温度的溶剂5ml),取续滤液用释放介质稀释至适当浓度,照紫外分光光度法(中国药典2010版二部附录IV A),在232nm处测定吸光度。另精密称取盐酸二甲双胍对照品适量,用释放介质配制成约5μg/ml浓度的溶液作为对照品溶液,计算出每片的释放度。 一、溶出介质的配制 用电子天平称量磷酸二氢钾(固体)xxxg,氢氧化钠(固体)xxxg,置1000ml 烧杯中,用800ml蒸馏水溶解后,倒入10L广口瓶中,再用蒸馏水稀释至10L,配得缓冲介质。 二、对照品溶液的配制 各置于100ml容量瓶中,用溶出介质溶解并定溶至刻度;用1ml移液管各精密量取1ml至50ml容量瓶中,用溶出介质定溶至刻度。. 各样品称量值自己列出。 三、试验过程 向溶出仪6个溶出杯中各加入1000ml已配好的溶出介质,加热,待溶出杯中溶液温度达到37℃后,将6片药片同时放到6个溶出杯中后,立即开始搅拌并计时。在1h、3h、5h、7h、10h时,用10ml的注射器各取样5ml,同时向溶出杯中补加同温度溶出介质5ml。 1h、3h样品取出后,过0.45um微孔滤膜,弃去2ml初滤液,取3ml续滤液;1h样品稀释25倍后测其吸光度;3h样品稀释50倍后测其吸光度。 四、实验结果见下表 计算公式:(1)校正因子f f=(f1+f2+f3)/3 f1=C1/A1; f2=C2/A2; f3=C3/A3 C1、C2、C3:三份对照品的浓度 A1、A2、A3:三份对照品的吸光度 (2)累积释放度 result=(f*A*n*v+C1h*5+ C3h*5+…..)*v*100/m
溶出度检查法美国药典USP-711
<711> DISSOLUTION 溶出度 (USP39-NF34 Page 540) General chapter Dissolution <711> is being harmonized with the corresponding texts of the European Pharmacopoeia and/or the Japanese Pharmacopoeia. These pharmacopeias have undertaken to not make any unilateral change to this harmonized chapter. 通则<711>溶出度与欧盟药典和日本药典中的相应部分相统一。这三部药典承诺不做单方面的修改。 Portions of the present general chapter text that are national USP text, and therefore not part of the harmonized text, are marked with symbols to specify this fact. 本章中的部分文字为本国USP内容,并没有与其他药典统一。此部分以()标注。 This test is provided to determine compliance with the dissolution requirements where stated in the individual monograph for dosage forms administered orally. In this general chapter, a dosage unit is defined as 1 tablet or 1 capsule or the amount specified. Of the types of apparatus designs described herein, use the one specified in the individual monograph. Where the label states that an article is enteric coated and a dissolution or disintegration test does not specifically state that it is to be applied to delayed-release articles and is included in the individual monograph, the procedure and interpretation given for Delayed-Release Dosage Forms are applied, unless otherwise specified in the individual monograph. 本测试用于检测药品口服制剂的溶出度是否符合各论中的规定。本章中,除另有规定外,单位制剂定义为1片或1粒胶囊。对于本章中所述多种仪器,使用各论中规定的种类。除各论中另有规定外,如果检品是肠溶衣片且各论中的溶出度或崩解时限检查项下没有特别指出适用迟释剂的,使用本章中适用于迟释剂的流程和解释。 FOR DOSAGE FORMS CONTAINING OR COATED WITH GELATIN涂有或包含明胶的剂型 If the dosage form containing gelatin does not meet the criteria in the appropriate Acceptance Table (see Interpretation, Immediate-Release Dosage Forms, Extended-Release Dosage Forms, or Delayed-Release Dosage Forms) because of evidence of the presence of cross-linking, the dissolution procedure should be repeated with the addition of enzymes to the medium, as described below, and the dissolution results should be evaluated starting at the first stage of the appropriate Acceptance Table. It is not necessary to continue testing through the last stage (up to 24 units) when criteria are not met during the first stage testing, and evidence of cross-linking is observed. 如果剂型中含有明胶,其不符合验收表中的标准(见判断,速释制剂,延释制剂,缓释制剂),因为存在明胶交联结合作用,它的溶解过程与外加的媒介酶是重复的,见下面的描述,并且溶解结果可以通过适当的验收表的开始的第一阶段标准进行评估。如果溶出结果不满足第一阶段的测试标准,那么就没有必要继续测试到最后阶段,并且也证明了明胶交联结合作用的存在。
实验6 电导法测定难溶盐的溶解度
实验10 电导法测定难溶盐的溶解度 一、实验目的 1. 掌握电导法测定难溶盐溶解度的原理和方法。 2. 学会电导率仪的使用方法。 二、基本原理 第二类导体导电能力的大小,常以电阻的倒数表示,即电导: (10.1) 式中G称为电导,单位是西门子S、 导体的电阻与其长度成正比,与其截面积成反比,即: (10.2) 是比例常数,称为电阻率或比电阻。根据电导与电阻的关系,则有: (10.3) k称为电导率或比电导,它相当于两个电极相距1m,截面积为导体的电导,其单位是。 对于电解质溶液,若浓度不同,则其电导亦不同。如取1mol电解质溶液来量度,即可在给定条件下就不同电解质来进行比较。1mol电解质全部置于相距为1m的两个电极之间,溶液的电导称之为摩尔电导,以Λ表示之。如溶液的浓度以C表示,则摩尔电导可以表示为: (10.4) 式中Λm的单位是;C的单位是。Λm的数值常通过溶液的电导率k,经(10.4)式计算得到。而k与电导G有下列关系,由(10.3)式可知: (10.5) 对于确定的电导池来说,是常数,称为电导池常数。电导池常数可通过测定已知电导率的电解质溶液的电导(或电阻)来确定。
溶液的电导常用惠斯顿电桥来测定,线路如图10.1所示。其中S为信号发生器;R1、R2和R3是三个可变电阻,R x为待测溶液的阻值;H为检流计,C1是与R1并联的一个可 变电容,用于平衡电导电极的电容。测定时,调节R1、R2、R3和C1,使检流计H没有电流通过。此时,说明B、D两点的电位相等,有下面的关系式成立: (10.6) Rx的倒数即为该溶液的电导。 本实验测定硫酸铅的溶解度。直接用电导率仪测定硫酸铅饱和溶液的电导率(K溶液)和配制溶液用水的电导率(K水)。因溶液极稀,必须从溶液的电导率(K溶液)中减去水的电导率(K水),即为: K硫酸铅=K溶液-K水(10.7) 根据10.4式,得到: (10.8) 式中:C是难溶盐的饱和溶液的浓度。由于溶液极稀,Λm可视为Λm∞。因此: (10.9) 硫酸铅的极限摩尔电导可以根据数值求得。因温度对溶液的电导有影响,本实验在恒温下测定。 电导测定不仅可以用来测定硫酸铅、硫酸钡、氯化银、碘酸银等难溶盐的溶解度,还可以测定弱电解质的电离度和电离常数,盐的水解度等。 三、仪器和试剂 仪器:恒温槽,电导率仪,电炉一个,锥形瓶两只,试管三支,电导电极。 试剂:二次蒸馏水配制 四、操作步骤
实验1 高吸光度示差分析法
实验二高吸光度示差分析法 一、目的: 通过标准曲线的绘制及试样溶液的测定,了解高吸光度示差分析法的基本原理,方法优点。掌握721型分光光度计的使用方法。 二、原理: 普通吸光光度法是基于测量试样溶液与试剂空白溶液(或溶剂)相比较的吸光度,从相同条件下所作的标准曲线来计算被测组份的含量,这种方法的准确度一般不会优于1~2%,因此,它不适合于高含量组份的测定。 为了提高吸光光度法测定的准确度,使其适合于高含量组分的测定,可采用高吸光度示差分析法。示差法与普通吸光光度法的不同之处,在于用一个待测组份的标准溶液代替试剂空白溶液作为参比溶液,测量待测量溶液的吸光度。它的测定步骤如下: (1)在仪器没有光线通过时(接受器上无光照射时)调节透光率为0,这与比色法或普通分光光度法相同。 (2)将一个比待测溶液(浓度为C+△C)稍稀的参比溶液(浓度为C)放在仪器光路中,调节透光率为100%。 (3)将待测量溶液(或标准溶液)推入光路中,读取表现吸光度A f。 表观吸光度A f实际上是由△C引起的吸收大小,可表达为: A f=ab△c 上式说明,待测溶液(或标准溶液)与参比溶液的吸光度之差与这两次溶液的浓度差成正比。 无论普通吸光度或高吸光度示差法,只要符合比尔定律,而且测量误差仅仅是由于透光率(或吸光度)读数的不确定所引起的,则可以方便地计算出分析的
误差。 仪器刻度上透光率读数改变数(dT )所引起的浓度误差dc 为绝对误差,它与透光率有关,其关系式容易由比耳定律推得: A f =ab △c=k △c lgT=-A f =-k △c 0.43lnT=-k △c KT dc 43 .0 ·dT 式中k 为标准曲线(A ~C )的斜率。实验中三条曲线的三个k 很接近。根据k 值及上述关系可以计算出实验中各点的绝对误差(假设透光率读数误差为l%,即dT=0.01)。 对于化学工作者来说,更有意义的是浓度的相对误差(c dc ),或者相对百分误差(c dc ×100)。浓度相对百分误差与参比溶液的浓度关系密切。随着有色参比溶液浓度的增加(或A 的增加),相对百分误差也随之减小。当所用参比溶液的A=1.736时,最低的相对百分误差也可减小至0.25%。由此可见了,差示法中高吸光度法可达到容量分析和重量分析的准确度。 三、仪器与试剂 721型分光光度计(附2只1厘米比色皿) 0~10ml 微量滴定管1支(刻度准确至0.005ml ) 25ml 容量瓶×16 0.2500M Cr (NO 3)3 四、实验步骤
实验室溶出度测定法规程
实验室溶出度测定法规程
实验室溶出度测定法规程 目的:建立溶出度测定法标准操作规程。 适用范围:溶出度测定。 责任:质检员实施本操作规程,检验室主任负责监督本规程正确执行。 程序: 1.简述 1.1溶出度(中国药典2000年版二部附录X C)是指药物从片剂或胶囊剂等口服固体制剂在规定溶剂中溶出的速度和程度。它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。 1.2溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮(或烧杯)中,在37.0±0.5℃恒温下,在规定的转速、溶剂中依法操作,在规定的时间内测定其溶出的量。 1.3本方法适用于片剂、胶囊剂及颗粒剂的测定。 1.4中国药典2000年版收载三种测定方法,第一法转篮法第二法桨法及第三法小杯法。 1.5凡检查溶出度的制剂,不再进行崩解时限的检查。 2.仪器与用具 2.1溶出度仪 2.1.1仪器的组成溶出度仪主要由电动机、恒温水 浴、篮体、篮轴、搅拌桨、圆底烧杯及杯盖组成,详见中国药典2000年版二部附录X C。 2.1.2仪器的装置与使用按仪器使用说明书及中国药
典的规定进行安装与使用。 2.1.3仪器的校正为使同一药物的溶出度测定得到良好的再现性,应对新安装的溶出度仪采用溶出度校正片进行校正,对已使用过的仪器也应定期(或在出现异常情况时)进行校正。 2.1. 3.1溶出度校正片分崩解型和非崩解型两种,崩解型为泼尼松片,非崩解型为水杨酸片。目前国内仅有非崩解型校正片。 2.1. 3.2校正前,应先调式所用仪器。 2.1. 3.3溶剂:磷酸盐缓冲液(PH7.4)。配制方法见中国药典2000年版二部附录XV D,要求PH值为7.40±0.05,临用前脱气。 2.1. 3.4对照品溶液的制备取溶出度校正用水杨酸片1片,精密称定,置乳体中,研细,精密称取适量(约相当于水杨酸10mg),置100ml量瓶中,加乙醇1ml,摇匀,加溶剂适量,经超声处理30分钟,使水杨酸溶解,加溶剂到刻度,摇匀,经滤纸(不宜使用滤膜)滤过,取续滤液为对照品溶液。(对照应做2份平行试验) 2.1. 3.5校正溶液的制备取溶剂各900ml,分别注入每个操作容器中,温度保持在37±0.5℃,按规定(桨法为50转/分钟;篮法为100转/分钟)调整转速。取溶出度校正用水杨酸片6片,分别精密称定,分置6个容器中,自药片接触溶出介质时,开始计时,并分别在10、15、20、25和30分钟时取样(连续取样不停机),每次抽取2ml(及时补充溶剂2ml),各自经滤纸滤过(六个小漏斗和六张滤纸,连续使用,每次滤过后,漏斗底部应无液体存在),取续滤液为校正溶液。 2.1. 3.6测定法精密吸取对照品溶液及校正溶液各
片剂溶出度实验报告数据
竭诚为您提供优质文档/双击可除片剂溶出度实验报告数据 篇一:片剂溶出度的测定 片剂溶出度的测定 转篮法 1.仪器装置 中国药典收录的转篮法装置如图10—11所示。 (1)转篮分篮体与篮轴两部分,均为不锈钢等金属材料制成。篮体A由不锈钢丝网(丝径为0.254mm,孔径0.425mm)焊接而成,呈圆柱形,内径为20.2±0.1mm,上下两端都有金属边缘。篮轴b的直径为9.4~10.1mm,轴的末端连一金属片,作为转篮的盖;盖上有通气孔(孔径 2.omm);盖边系两层,上层外径与转篮外径同,下层直径与转篮内径同;盖上的三个弹簧片与中心呈120o。转篮旋转时摆动幅度不得超过±1.omm。 (2)操作容器为1000ml的圆底烧杯,内径为98~106mm,高160~175mm,烧杯上有一有机玻璃盖,盖上有2孔,中心孔为篮轴的位置,另一孔供取样或测温度用。为使操作容器
保持恒温,应外套水浴,水浴的温度应能使容器内溶剂的温度保持在37±0.5℃。转篮底部离烧杯底部的距离为25±2mm。 (3)电动机与篮轴相连,转速可任意调节在每分钟50~200转,稳速误差不超过±4%。运动时整套装置应保持平稳,不得晃动或振动。 (4)仪器应装有6套操作装置,可一次测定6份供试品。取样点位置应在转篮上端距液面中间,离烧杯壁lomm处。 (5)转篮防腐涂料不得在测定用溶剂中溶蚀。 2.测定法 除另有规定外,量取经脱气处理的溶剂900ml,注人每 个操作容器内,加温使溶剂温度保持在37±o.5℃,调整转 速使其稳定。取供试品6片(个),分别投入6个转篮内,将转篮降入容器中,立即开始计时,除另有规定外,至45min 时,在规定取样点吸取溶液适量,立即经0.8/μm微孔滤 膜滤过,自取样至滤过应在30s内完成。取续滤液,照各药品项下规定的方法测定,计算每片(个)的溶出量。 第三节溶出度测定法-page2 3.结果判断 6片(个)中每片(个)的溶出量,按标示含量计算,均应 不低于规定限度(Q)。如6片(个)中仅有1~2片(个)低于规定限度,但不低于Q-10%(百分数均依标示量为基数),且其平均溶出量不低于规定限度时,仍可判为符合规定。如6片
溶出度与释放度的区别
溶出度与释放度的区别公司内部档案编码:[OPPTR-OPPT28-OPPTL98-OPPNN08]
溶出度系指药物从片剂或胶囊剂等固体制剂在规定溶剂中溶出的速度和程度。释放度系指口服药物从缓释制剂、控释制剂,肠溶制剂及透皮贴剂等在规定溶剂中释放的速度和程度。溶出度一般是针对普通制剂而言,看药物在一定的时间内是否能够释放出来。一般测一个点。释放度主要针对特殊制剂(包括缓控释制剂),测试时最少测三个点,第一个点看药物有没有突释,第二个点是药物释放一半左右的点,主要考察药物释放的特征,第三个点则是考察药物释放是否完全。难溶药物检查溶出度,易溶药物检查崩解时限,检查溶出度的药物就不需要再检查崩解时限。 1. 对于确定的药物,如何选择“崩解时限”与“溶出度”在上篇指导原则中介绍了固体口服制剂是否建立溶出度的判断方法:①如果制剂设计为修饰释放,则需建立释放度的标准(包括缓释、控释、胃溶和肠溶等)②如果制剂没有设计为修饰释放,则做如下考察: 考察一次剂量的原料药在37±0.5℃,pH1.2-6.8范围内, 在不多于 250ml水中是否完全溶解。如果不溶解,则建立单时间点的溶出度检查标准,如果溶解,则继续考察③以上考察的意义在于原料药的溶解性是综合剂量和胃容量来考虑的, 即验证一次服用量的原料药在胃中 (250ml)是否完全溶解。这使一些溶解性能并不好、但剂量小, 在 250ml中可以完全溶解的药品可选择做崩解时限而不做溶出度检查。 ③该制剂在15分钟内,在pH1.2、4.0、6.8条件下能否达到80%以上的溶出量。如果达不到80%的溶出量,则建立单时间点的溶出度检查标准;如果能达到80%的溶出量,则继续考察④③步考察的意义为在考察原
实训5 药物溶解度测定
实训5 药物溶解度测定 一、目的要求 1.了解药典对药物近似溶解度的规定。 2.理解药物结构特点(极性)与溶解性的关系。 3.了解CTC的形成对药物溶解度的影响及CTC在药剂学中的应用。 二、实验原理 药物的溶解度是指在一定的温度下,在一定体积的溶剂中药物形成饱和溶液时的浓度。溶解度的大小,表明一种药物在某一种溶剂中被分散的难易程度。药物溶解时,药物的分子结构不会改变,是一种物理性质。 溶剂一般分为三类:以水为代表的极性溶剂、以甲醇和乙醇为代表的亲水性有机溶剂和以苯、石油醚为代表的亲脂性有机溶剂。溶解的经验规则:相似相溶。 为了适应某种制剂的要求而将药物制成盐或加入助溶剂形成电子转移复合物(CTC),这是增加药物在水中溶解度的常用方法。 三、实验方法 (一)不同药物在水中的溶解度测定 1.“极易溶”药物的溶解:称取1.50克的药物于合适的试管中,加入纯化水1.0~1.5毫升,室温下每隔5分钟振摇30秒,30分钟后观察溶解情况。记录溶剂用量。 2.“易溶”药物的溶解:称取1.0克的药物于合适的试管中,加入纯化水1.0~10毫升,室温下每隔5分钟振摇30秒,30分钟后观察溶解情况。记录溶剂用量。 3.“溶解”药物的溶解:称取0.1克的药物于合适的试管中,加入纯化水1.0~3.0毫升,室温下每隔5分钟振摇30秒,30分钟后观察溶解情况。记录溶剂用量。 4.“略溶”药物的溶解:称取0.1克的药物于合适的试管中,加入纯化水3.0~10.0毫升,室温下每隔5分钟振摇30秒,30分钟后观察溶解情况。记录溶剂用量。 5.“微溶”药物的溶解:称取0.1克的药物于合适的试管中,加入纯化水10.0~100.0毫升,室温下每隔5分钟振摇30秒,30分钟后观察溶解情况。记录溶剂用量。 (注:以上实验是根据药典对药物溶解度定义设计的,药物在所加的溶剂范围内均可溶解,实验时原则上加入最小溶剂量即可,如果出现不溶的现象,则可能是药物的纯度差、药物的称量和溶剂的取量不准确等因素引起。将实验结果折算为标准溶解度。) (二)同一种药物在不同溶剂中的溶解度测定 1.取三支试管,一支加入0.01克的维生素C,加入乙醚10.0毫升,另两支均加入0.1克的维生素C,再分别加入10.0毫升乙醇和1.0毫升纯化水,室温下每隔5分钟振摇30秒,30分钟后观察溶解情况。记录溶剂用量。 2.取三支试管,一支加入0.1克的水杨酸,加入纯化水10.0毫升,另两支均加入0.1克的水杨酸,再分别加入1.0毫升乙醇和1.0毫升丙酮,室温下每隔5分钟振摇30秒,30分钟后观察溶解情况。记录溶剂用量。 思考题: 1.药物的极性与药物在水中的溶解性有什么关系? 2.什么是药物溶解度? 3.简述药典对药物近似溶解度的规定和溶解度的实验方法。 1
差示分光光度法测定高含量的二氧化硅
差示分光光度法测定高含量的二氧化硅 (作者:余建华,毛杏仙本信息发布于2009年08月11日,共有183人浏览) [字体:大中小] 二氧化硅是水泥及原材料化学分析的常检项目,由于材质、含量差别很大,因此关于二氧化硅的测定方法很多。根据二氧化硅含量的不同分为三类,含SiO2量较高(Wsio2≥95%)的材质,多采用重量法;含SiO2为常量(Wsio25%~95%)的,多采用容量法;含SiO2量较低(Wsio2<5%)的,一般采用硅钼蓝比色法测定。这三种方法各有特点,重量法和容量法理论上准确度较高方法可靠,但是整个操作流程相对较复杂,费时费力测定周期长;用比色法测定,适用范围很小。 用硅钼蓝光度法测定高含量SiO2,难于准确测定,主要是由于随SiO2含量的升高在制取母液时硅酸易产生聚合,标准曲线易产生弯曲等,使测定结果受到影响。在这种情况下,应用差示分光光度法,可使测定的准确度大为提高。这一方法的实质,是用已知浓度的标准溶液代替常用的水或空白溶液作参比来绘制工作曲线,也就是借增加参比液的吸光度提高待测溶液的吸光度读数的准确度,从而降低光度法的测定误差。本试验根据待测试样的SiO2含量估算范围不同,采取分段比色、减少称样量、浸取试样时以盐酸逆酸化法避免硅酸聚合、选取2~3个基体成分尽量与试样相近,二氧化硅含量比试样稍低和稍高的标样为参比校准标准曲线等多种手段,消除或减少测量误差,提高测量的准确性和稳定性,实现了常量二氧化硅的快速测定。 1 试验部分 1.1主要试剂与仪器 721型分光光度计;容量瓶;镍坩埚;马弗炉等; 氢氧化钾(分析纯);无水乙醇(分析纯);盐酸(V/V):1/1; 钼酸铵溶液(50g/L):量取500ml蒸馏水于塑料杯中,加入25g钼酸铵,搅拌至完全溶解并过滤,贮于塑料瓶中备用; 钼蓝显色剂:将30g草酸、30g硫酸亚铁铵溶于500ml水中,搅拌溶解后,缓缓的加入l00ml浓硫酸,用水稀释至l000ml,搅拌,备用。 1.2测定方法原理 测定时,调节吸光度至∞;吸光度为零的点用浓度C1稍低于试样溶液的标准溶液来调定。然后测定一系列大于Cl的已知溶液的标准溶液的吸光度,并按浓度与吸光度的对应关系,绘制工作曲线和测定试样溶液的吸光度。 设透过空白溶液、第一个标准溶液(C1)和第二个标准溶液(C2)的光强度依次为I0、I1和I2,对应于C1和C2的吸光度为A1,A3,ε为摩尔吸光系数,根据比耳定律:
溶出度测定法
1.目的 建立溶出度测定法操作规程。 2.适用范围 本规程适用于溶出度测定法。 3.编制依据 《药品生产质量管理规范(1998年修订)》国家药品监督管理局(1999)4.责任 QC主管、QC质检员对本规程的实施负责。 5.正文 5.1简述 5.1.1溶出度(中国药典2010年版二部附录X C)是指药物从片剂\胶囊剂或颗粒剂等固体制剂在规定条件中溶出速率和程度。它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。 5.1.2溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮(或烧杯)中,在37.0±0.5℃恒温下,在规定的转速、溶剂中依法操作,在规定的时间内测定其溶出量。 5.1.3中国药典2010年版收载三种测定方法,第一法转篮法,第二法桨法及第三法小杯法。 5.1.4除另有规定外,凡检查溶出度的制剂,不再进行崩解时限的检查。 5.2仪器与用具 5.2.1溶出度仪 5.2.1.1仪器原组成溶出度仪主要由电动机、恒温水浴、篮体、篮轴、搅拌桨、圆底烧杯及杯盖组成,详见中国药典2010年版二部附录X C。 5.2.1.2仪器的装置与使用按仪器使用说明书及中国药典的规定进行安装与使用。5.2.1.3仪器的校正为使药物的溶出度测定结果准确、可靠,应对新安装的溶出度仪采用溶出度校正片进行校正,对已使用过的仪器也应定期(或在出现异常情况时)进行校正。 5.2.1.4仪器的调试 5.2.1.4.1检查仪器水平及转动轴的垂直度与偏心度(使用水平以检查仪器是否处于水平状态;转轴的垂直程度应与容器中心线相吻合,用直角三角板检查转动轴与溶出杯平
溶出度检查方法及进展 PPT课件
溶出度检查方法及进展 定义:指药物在规定介质中,在一定条件下从片剂或胶囊等固体制剂中溶出的速度和程度。 药物溶出度直接影响药物在体内的吸收和利用,是评价药物质量的一个重要内在指标,也是评价制剂品质和工艺水平的一种有效手段,还是评价制剂活性成分生物利用度和制剂均匀度的一种有效标准,能有效区分同一种药物生物利用度的差异,因此是制药工业质量控制必检项目之一。从理论上讲,药物的体内试验(包括体内药物动力学的研究和临床试验) 才是评价药物最根本和最可靠的依据。20 世纪80 年代以来,生物利用度成为衡量药品质量的一个重要参数,但生物利用度实验工作量大、成本高,从药物生产的实际情况来看,不可能对每一个药物样品都采用体内实验来筛选评定。药物溶出度检查是一种模拟口服固体制剂在胃肠中溶出过程的体外实验法。尽管体内检验和体外检验结果不会完全一致,但具有一定相关性,而且溶出度的体外检验较体内检验简单易行,仍不失为一个经济有效的质量检测、控制手段。 药物制剂发展 制剂可分为四代,第一代为一般制剂或常规制剂,在崩解度试验水平,第二代一般为长效缓慢制剂或肠溶制剂,在溶出度试验水平,第三代为精密的控释制剂,药物输送系统,透皮吸收治疗系统,第四代为靶向制剂。 近年来,药物制剂研究向着“三效”(高效、速效、长效)和“三小”
(毒性、副作用、剂量)方向发展。 国外药典从70年代就相继收载了溶出检查法,我国在1985年版药典中正式收载了溶出度检查,这些年来,各国药典收载溶出度检查的品种呈上升趋势。2010年版药典加大了对药物溶出度的检查。这也本身是对药品质量标准的提高。也是多数人写论文的一个不错的方向。 溶出度在药品生产检验、临床疗效考察、药品稳定性检验、新药研制、处方筛选、工艺改进等许多方面都要作为考察指标。在美国、英国、日本,溶出度检查实际是指药物固体制剂按照各国药典规定的方法,在一定时间内从固体制剂溶入介质的累计百分率(以被测试剂标示量计算) 。 中国药典(2010 年版) 规定,片剂或硬胶囊制剂45min 的溶出度应大于70 %。一般认为下列药物,必须测定溶出度:(1) 难溶或难吸收的药物;(2) 治疗量与中毒量接近的药物;(3) 要求缓释、控释或长效的药物;(4) 用于治疗严重疾病的药物;(5) 急救、抢救用的药物。但现在溶出度检查已经逐渐普及到一般的药物。 我国药品监管部门对于药品溶出度的要求,是一个时间点、一个溶出介质、一个限度点,与欧美发达国家相比,存在不小差距,日本要测定药物在某一种或两种介质中的溶出度曲线,这也是导致国产药品、尤其仿制药质量参差不齐的一个主因。目前,国内市场上劣药已不多见,所以我们国家也正在考虑提高药品质量标准,来向发达国家靠齐。
溶解度的测定
硝酸钾溶解度的测定(方法1:结晶析出法)实验原理: 先设计好不同溶质和溶剂的量,称量、混合、加热、搅拌使其溶解,降温并用温度计分别测定其开始析出晶体时的温度,即所得溶液为该温度下的饱和溶液,计算该温度下的溶解度。 实验用品: 托盘天平(J0160,200g,0.2g),烧杯(J6124),大试管(J6104),玻璃棒(J6453),温度计(J6071,量程0~100℃),酒精灯(J6201),量筒(J6001,10ml),方座支架(J1102,带铁圈),石棉网(J6432),药匙(J6442),试管刷(J6471),硝酸钾(化学纯),蒸馏水。 实验步骤: 一、检查实验用品是否齐全、完好。 二、硝酸钾的称取和溶解。 1. 用托盘天平分别准确称取硝酸钾3.5g、1.5g、1.5g、 2.0g、 2.5g,称量过程详见分组实验三的步骤二。将称好的5份硝酸钾放在实验台上,并做标记。 2.在一支大试管中加入上面称取的3.5g硝酸钾。 3.用量筒准确量取10.0m1蒸馏水,加入大试管中。 4.在水浴中加热大试管,边加热边搅拌,至硝酸钾完全溶解(水浴温度不要太高,以刚好使硝酸钾溶解为宜,否则会使下一步结晶析出操作耗时过长) 三、硝酸钾的结晶。 1.自水浴中取出大试管,插入一支干净的温度计,用玻璃棒轻轻搅拌并摩擦试管壁,同时观察温度计的读数。当刚开始有晶体析出时,立即记下此时的温度t1,并填入下表中。
2.把试管再放入水浴中加热,使晶体全部溶解,然后重复两次上述实验步骤的操作,分别测定开始析出晶体时的温度t2、t3。将读数填入表格。 四、溶解度曲线的绘制。 1.依次向试管中再加入1.5g、1.5g、2.0g、2.5g硝酸钾(使试管中依次共有硝酸钾 5.0g、6.5g、8.5g、11.0g),每次加入硝酸钾后都重复溶解、结晶实验步骤的操作,并将晶体开始析出时的温度读数填人表格。 2.根据所得数据,以温度为横坐标,溶解度为纵坐标,绘制溶解度曲线图。 五、整理实验用品。 1.用试管刷清洗玻璃仪器。 2.整理实验用品,恢复实验前的摆放位置。 注意事项: 1.为了使测量结果准确,称取硝酸钾晶体的质量和量取倒入试管的蒸馏水的体积应尽量准确。 2.水浴加热时,烧杯里的水面不能低于试管里的液面。温度计应插在溶液的中部,使所示的温度具有代表性。 3.使试管里的液体升温时应采用水浴加热,而不能用酒精灯直接加热。
差示分光光度法
4.5 分光光度测定方法 中文词条名:差示分光光度法 英文词条名:differential spectrophotometry 分光光度法中,样品中被测组分浓度过大或浓度过小(吸光度过高或过低)时,测量误差均较大。为克服这种缺点而改用浓度比样品稍低或稍高的标准溶液代替试剂空白来调节仪器的100%透光率(对浓溶液)或0%透光率(对稀溶液)以提高分光光度法精密度、准确度和灵敏度的方法,称为差示分光光度法。差示分光光度法又可分高吸光度差示法,低吸光度差示法,精密差示分光光度法等。 4.5.2 差示分光光度法 吸光度A在0.2-0.8范围内误差最小。超出此范围,如高浓度或低浓度溶 液,其吸光度测定误差较大。尤其是高浓度溶液,更适合用差示法。 一般分光光度测定选用试剂空白或溶液空白作为参比,差示法则选用一已知浓度的溶液作参比。该法的实质是相当于透光率标度放大。 高吸收法在测定高浓度溶液时使用。选用比待测溶液浓度稍低的已知浓度溶液作标准溶液,调节透光率为100%。
低吸收法在测定低浓度溶液时使用。选用比待测液浓度稍高的已知浓度溶液作标准溶液,调节透光率为0。 最精密法是同时用浓度比待测液浓度稍高或稍低的两份已知溶液作 标准溶液,分别调节透光率为0或100%。 设试样浓度为,以溶剂作参比时,其透光率为,吸光度为。若选浓度为(其以溶剂为参比时的透光率为,吸光度为)的已知溶液作参比,调节透光率为100%。根据吸收定律,有: 溶剂作参比时,;(4.14) ;(4.15) 差示法,用已知浓度的溶液作参比时, (4.16) ,(4.17) (4.16)式为差示分光光度法的基本关系式。
溶出度测定法标准操作规程
溶出度测定法标准操作规程 目的:建立溶出度测定法标准操作规程。 适用范围:溶出度测定。 责任:质检员实施本操作规程,检验室主任负责监督本规程正确执行。 程序: 1.简述 1.1溶出度(中国药典2000年版二部附录X C)是指药物从片剂或胶囊剂等口服固体制剂在规定溶剂中溶出的速度和程度。它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。 1.2溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮(或烧杯)中,在37.0±0.5℃恒温下,在规定的转速、溶剂中依法操作,在规定的时间内测定其溶出的量。 1.3本方法适用于片剂、胶囊剂及颗粒剂的测定。 1.4中国药典2000年版收载三种测定方法,第一法转篮法第二法桨法及第三法小杯法。 1.5凡检查溶出度的制剂,不再进行崩解时限的检查。 2.仪器与用具 2.1溶出度仪 2.1.1仪器的组成溶出度仪主要由电动机、恒温水浴、篮体、篮轴、搅拌桨、圆底烧杯及杯盖组成,详 见中国药典2000年版二部附录X C。 2.1.2仪器的装置与使用按仪器使用说明书及中国药典的规定进行安装与使用。 2.1.3仪器的校正为使同一药物的溶出度测定得到良好的再现性,应对新安装的溶出度仪采用溶出度校正片进行校正,对已使用过的仪器也应定期(或在出现异常情况时)进行校正。 2.1. 3.1溶出度校正片分崩解型和非崩解型两种,崩解型为泼尼松片,非崩解型为水杨酸片。目前国内仅有非崩解型校正片。 2.1. 3.2校正前,应先调式所用仪器。 2.1. 3.3溶剂:磷酸盐缓冲液(PH7.4)。配制方法见中国药典2000年版二部附录XV D,要求PH值为7.40±0.05,临用前脱气。 2.1. 3.4对照品溶液的制备取溶出度校正用水杨酸片1片,精密称定,置乳体中,研细,精密称取适量
119溶出度测定法
编制依据:药品生产质量管理规范(2010年修订)《中华人民共和国药典》2015年版四部120页(通则0931) 内容: 溶出度系指活性药物从片剂、胶囊剂或颗粒剂等普通制剂在规定条件下溶出的速率和程度,在缓释制剂、控释制剂、肠溶制剂及透皮贴剂等制剂中也称释放度. 仪器装置 第一法(篮法) (1)转篮分篮体与篮轴两部分,均为不锈钢或其他惰性材料制成,其形状尺寸如图1所示.篮体 A 由方孔筛网(丝径为0.28mm±0.03mm,网孔为0.04mm±0.04mm)制成,呈圆柱形,转篮内径为 20.2mm±1.0mm,上下两端都有封边.篮轴B的直径为9.75mm±0.35mm,轴的末端连一圆盘,作为转篮的盖;盖上有一通气孔(孔径为2.0mm±0.5mm);盖边系两层,上层直径与转篮外径相同,下层直径与转篮内径相同;盖上的3个弹簧片与中心呈120° (2)溶出杯一般由硬质玻璃或其他惰性材料制成的底部为半球形的1000ml杯状容器,内径为102mm±4mm,高为185mm±25mm;溶出杯配有适宜的盖子,盖上有适当的孔,中心孔为篮轴的位置,其他孔供取样或测量温度用.溶出杯置恒温水浴或其他适当的加热装置中. (3)篮轴与电动机相连,由速度调节装置控制电动机的转速,使篮轴的转速在各品种项下规定转速的±4%范围之内.运转时整套装置应保持平稳,均不能产生明显的晃动或振动(包括装置所处的环境).转篮旋转时,篮轴与溶出杯的垂直轴在任一点的偏离均不得大于2mm,转篮下缘的摆动幅度不得偏离轴心1.0mm. (4)仪器一般配有6套以上测定装置. 第二法(桨法) 除将转篮换成搅拌桨外,其他装置和要求与第一法相同.搅拌桨的下端及桨叶部分可涂适当的惰性材料(如聚四氟乙烯),其形状尺寸如图2所示.桨杆对称度(即桨轴左侧距桨叶左边缘距离与桨轴右侧距桨叶右边缘距离之差)不得超过 0.5mm,桨轴和桨叶垂直度 90°±0.2°;桨杆旋转时,桨轴与溶出杯的垂直轴在任一点的偏差均不得大于2mm;搅拌桨旋转时A、B两点的摆动幅度不得超过0.5mm. 第三法(小杯法) (1)搅拌桨形状尺寸如图4所示.桨杆上部直径为9.75mm±0.35mm,桨杆下部直径为 6.0mm±0.2mm;桨杆对称度(即桨轴左侧距桨叶左边缘距离与桨轴右侧距桨叶右边缘距离之差)不得超过0.5mm,桨轴和桨叶垂直度90°±0.2°;桨杆旋转时,桨轴与溶出杯的垂直轴在任一点的偏差均不得大于2mm;搅拌桨旋转时,A、B两点的摆动幅度不得超过0.5mm. (2)溶出杯一般由硬质玻璃或其他惰性材料制成的底部为半球形的250ml 杯状容器,内径为
溶解度的测定
实验2 溶解度的测定 37 一 目的 藉由不同温度下测定物质的溶解度,以了解温度与溶解度之间的关系,并以图形表达之。 二 实验原理 溶质的溶解度会受到许多因素的影响,如溶质的本性、溶剂的种类、温度…等。即使是在同一种溶剂中,如图E2-1所示,不同的溶质在水中的溶解度也各不相同,硝酸钾在约22℃以下,其溶解度小于氯化钠,但高于此温度时,其溶解度则远大于氯化钠。大部分的固体溶质,其溶解度随着温度的增高而变大,但是如下图所示有些变化较大,有些则变化较小。 图E2-1中的各条曲线是如何画出来的?我们可以在高温下配制数支不同浓度的不饱和溶液,然后依序让试管内溶液的温度徐徐降低,直至溶液中有碎屑开始出现时,记录当时的温度,将其浓度换算即可得知该温度的溶解度,将数点不同温度下的溶解度在图形中相连,即可得相似的曲线。 三 实验器材 每組 器材(规格) 数量 器材(规格) 数量 天平 共享 中型试管(18 mm 口径) 4支 试管夹 1支 烧杯(600 mL ) 1个 量筒(25 mL ) 1个 电热板和磁搅拌子(或其他加热装置) 1组 温度计 1支 末端有环的铁丝(可自制) 1支 试管架 1座 溶解度的测定 如何使更多的固体溶到水中? 2 连结课本P.116 图E2-1 各种固体溶解度与温度关系
36高中化学(全)实验活动手册 四实验试药 每組 药品份量药品份量 水约20 mL 硝酸钾(KNO3)约14 g 五实验步骤 1 取600 mL烧杯,装热水 半满并置于电热板上,开 启电源,把火力调至最 小,加热烧杯内的水。 2 称取质量为2.0 g、3.0 g、 4.0 g和 5.0 g的硝酸钾倒入 四支试管。 3 再各加入5.0 g水于四支 试管。 4 将4支试管放入装水烧 杯中,以水浴法加热。 5 注意观察各试管内固体。 6 依序用试管夹将固体已 溶解的试管取出(其先后 顺序应为加了2.0 g、3.0 g、4.0 g和5.0 g硝酸钾 固体的试管),先进行下 一步骤,直到所有试管均 取出为止,关闭电热板的 电源。