单项验证结果评价报告
单项验证结果评价报告
一、原料及成品检验结果分析
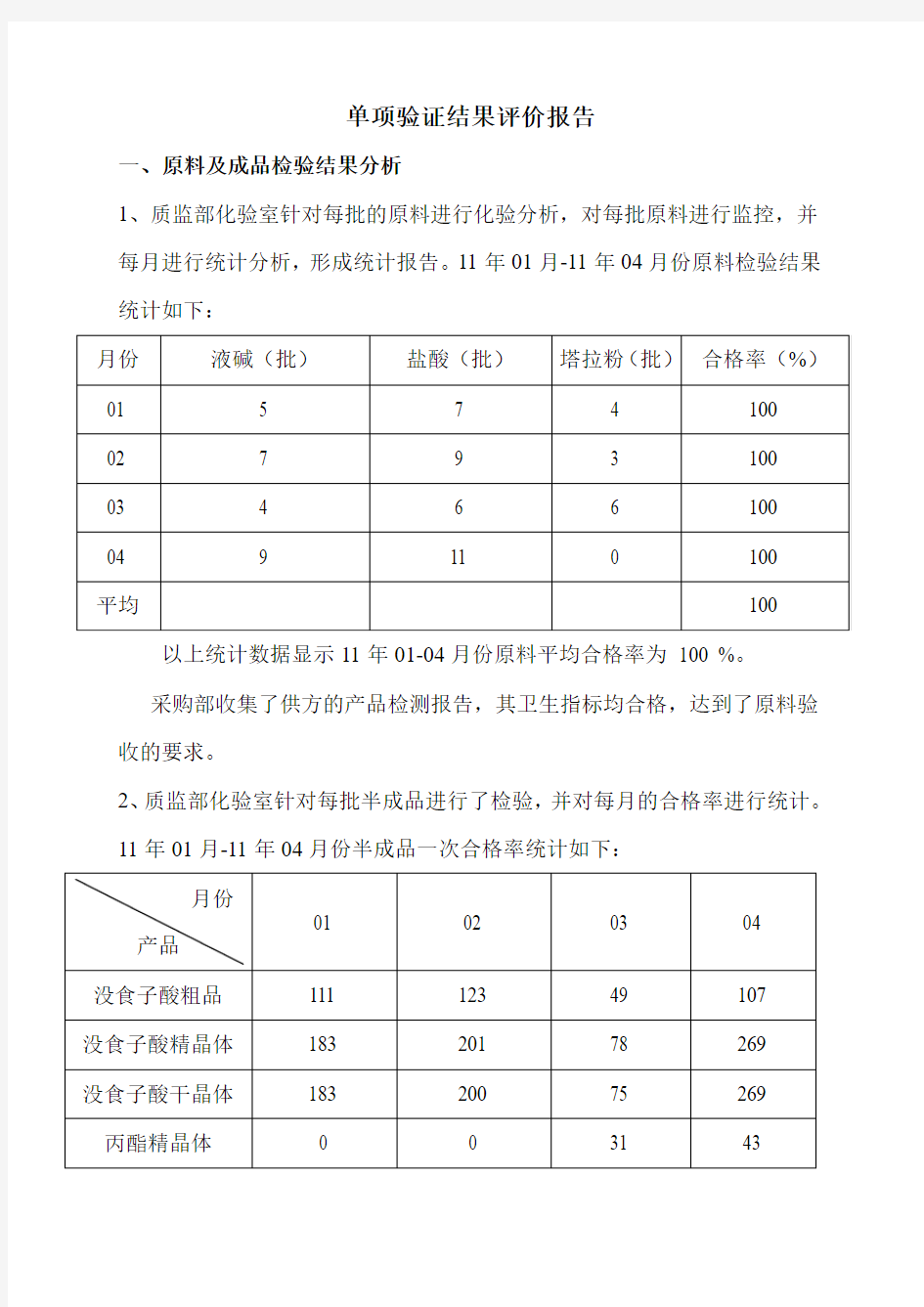
1、质监部化验室针对每批的原料进行化验分析,对每批原料进行监控,并每月进行统计分析,形成统计报告。11年01月-11年04月份原料检验结果统计如下:
以上统计数据显示11年01-04月份原料平均合格率为100 %。
采购部收集了供方的产品检测报告,其卫生指标均合格,达到了原料验收的要求。
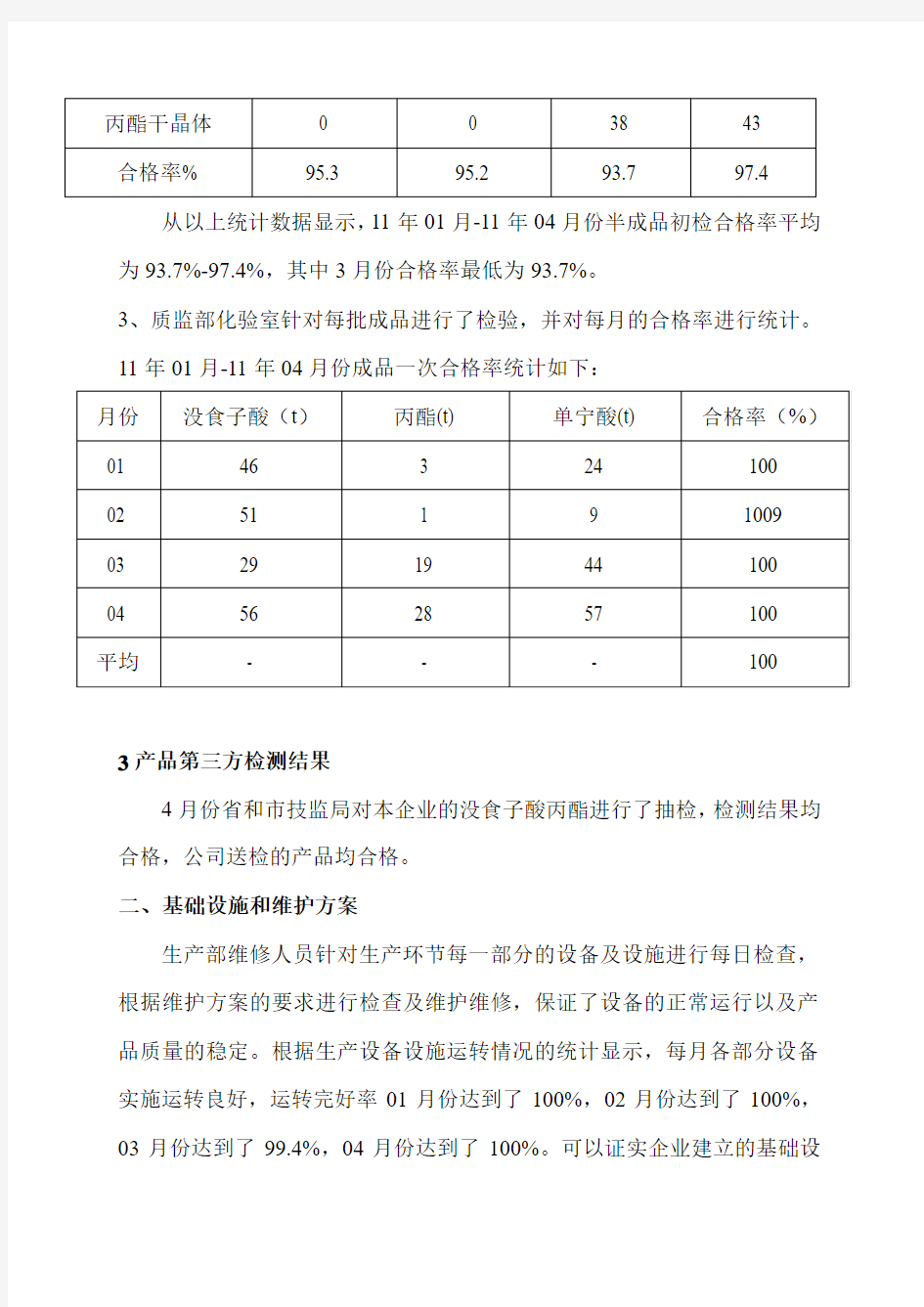
2、质监部化验室针对每批半成品进行了检验,并对每月的合格率进行统计。11年01月-11年04月份半成品一次合格率统计如下:
从以上统计数据显示,11年01月-11年04月份半成品初检合格率平均为93.7%-97.4%,其中3月份合格率最低为93.7%。
3、质监部化验室针对每批成品进行了检验,并对每月的合格率进行统计。11年01月-11年04月份成品一次合格率统计如下:
3产品第三方检测结果
4月份省和市技监局对本企业的没食子酸丙酯进行了抽检,检测结果均合格,公司送检的产品均合格。
二、基础设施和维护方案
生产部维修人员针对生产环节每一部分的设备及设施进行每日检查,根据维护方案的要求进行检查及维护维修,保证了设备的正常运行以及产品质量的稳定。根据生产设备设施运转情况的统计显示,每月各部分设备实施运转良好,运转完好率01月份达到了100%,02月份达到了100%,03月份达到了99.4%,04月份达到了100%。可以证实企业建立的基础设
施和维护方案基本得以实施并且有效。
三、危害水平低于可接受水平的验证
根据质监部对产品的检测结果表明,公司产品均合格,通过第3方检测结果表明,公司产品的卫生指标也均合格,主管部门监督抽查结果产品也均能满足要求,也未发生顾客投诉现象,证实企业产品的危害水平低于可接受水平。
四、其他程序的实施情况分析
1、根据年度培训计划,公司办公室对每一项培训内容实施了培训,并对每次的培训进行了评价,根据统计结果,培训实施率和合格率达到了100%,证实培训程序得以实施且有效。
2、根据采购控制程序的要求,对每个供应商进行了评价,对采购过程按照程序要求进行了控制。
3、根据内审和管理评审的要求,公司总经办于11年05月27日安排企业体系的内部审核,并在5月底安排了公司的管理评审会议。
根据以上分析能够证实企业的操作性前提方案和HACCP计划中的要素得以实施且有效,并已实施了基础设施和维护方案,生产的产品的危害水平低于确定的可接受水平,并且企业建立并要求的其他程序得以实施且有效。
评价人:HACCP小组
2011年05月18日
酒石酸美托洛尔片中间产品检验方法验证报告
分发部门:
目录 一、概述 二、验证前准备 三、验证记录与结果 四、漏项与偏差处理 五、评价与建议 六、验证结论 七、附件:相关记录
一、概述 为更好实现产品过程控制,我中控实验室依据《分析方法确认与验证管理规程》(文件编码:F0004-00)与《中华人民共和国药典》2010版附录中相关规定和相应指导原则的要求,制定出一系列检验方法验证实验来考察本产品中间产品检验方法的适用性,从而确保该方法能够可靠有效地用于控制药品的内在质量。 本中间产品检验标准操作规程中所列检验项目有:含量测定,溶出度,干燥失重。其方法验证参见相应成品检验方法验证,现主要对酒石酸美托洛尔含量测定检验项目进行方法验证。 验证实施时间:自年月日开始至年月日完成。 二、验证前准备 1、培训确认 2、所用仪器设备,包括电子分析天平、检验方法中规定的仪器设备已经校验,且在有效期内。 3、试验所用的玻璃计量器具需清洁,并经检定后符合要求。 4、相关对照品、试剂试药均符合《中国药典》要求。 三、验证结果 1、含量测定方法各项验证实验结果 含量——专属性验证结果 试验人/日期复核人/日期
含量——重复性验证结果 试验人/日期复核人/日期 含量——准确度验证结果 是否符合要求: 试验人/日期复核人/日期 含量——线性验证结果 试验人/日期复核人/日期2、含量测定方法验证小结 小结人:日期:
四、漏项与偏差处理 无 五、评价与建议 小结人:日期:六、验证结论 总结人:日期:七、附件 相应验证记录与图谱。
酒石酸美托洛尔片中间产品检验方法验证记录 1 检验依据:《中国药典》2005版。 2 检验方法:取相当于本品20片的颗粒,精密称定,研细,精密称取适量(约相当于酒石酸美托洛尔0.12g),置100ml量瓶中,加2%氯化钠溶液适量,振摇使酒石酸美托洛尔溶解并稀释至刻度,摇匀,滤过,滤过,精密量取续滤液5ml,置50ml量瓶中,加水稀释至刻度,摇匀。照紫外-可见分光光度法(中国药典2005年版二部附录IV A),在274nm的波长处测定吸收度。另精密称取酒石酸美托洛尔对照品适量,加水制成每1ml中含0.12mg的溶液,同法测定,计算,即得。 3 验证记录: 3.1相关物料 3.2 仪器用具 3.3 验证步骤 3.3.1 专属性:分别取空白辅料混合物、空白溶剂,按照“2”项下所述方法配制空白溶液,在200~600nm处进行紫外扫描。 实验结果: 3.3.2 准确度:按处方比例配制3个不同浓度(80%、100%、120%)的试样,每个浓度的样品按照“2”
残留溶剂顶空分析报告方法验证方案设计模版2
方案批准 注:在方案批准部分签字表明签字者同意方案中规定的检测项目检测方法和记录要求。在执行本方案的过程中可能会出现影响严格执行本方案的偏差,对较小的偏差将通过偏差报告的形式来解决,对于关键性偏差,如对方法的调整、对参数或接受标准的调整必须制定出增补方案并按照原方案批准程序得到批准才能进行。所有的偏差报告和增补方案必须在提交验证报告供批准时一同提交。
目录 1.概述 (3) 2.参考资料 (4) 3. 职责 (4) 4. 色谱系统及色谱条件 (4) 5. 器材与试剂 (5) 6. 验证试验 (5) 6.1系统适应性 (5) 6.2专属性 (6) 6.3耐用性 (7) 6.4定量限 (8) 6.5检测限 (8) 6.6线性与围 (8) 6.7准确度 (9) 6.8精密度 (11) 7.再验证周期 (12) 8.偏差及纠正措施 (13) 9.最终审核和批准 (13) 药品残留溶剂顶空分析方法草案 (14)
1.概述 1.1根据ICH对药品中残留溶剂含量的要求及盐酸噻氯匹定生产工艺,必须控制盐酸噻氯匹定生产工艺中使用到的溶剂乙醇、丁酮、甲苯、N,N-二甲基甲酰胺(DMF)的残留量。限度分别为:乙醇≤5000ppm、丁酮≤5000ppm、甲苯≤890ppm、DMF≤880ppm。 1.2分析方法草案见附件。 1.3本分析方法属于杂质定量分析,因此需要验证的项目有:系统适应性、专属性、线性、 准确度、检测限、定量限、精密度、耐用性,具体参数及接受标准要求见下表:
2.参考资料 ICH Q3C (R3), November 2005. ICH Q2 (R1), November 2005. <467> Residual Solvents, United States Pharmacopoeia 31, November 2007. <20424> Residual Solvents, European Pharmacopoeia 6.0, June 2007. 3. 职责 4.1色谱系统
检验方法验证规程
目的:检验方法验证的目的是证明采用的方法适合于相应检测要求。 适用范围:在制定药品质量标准时所采用的检验方法应验证,药物生产方法变更、制剂组分变更、原检验方法进行修订时,检验方法应进行再验证。 责任者:化验室、研究开发实验室 内容: 1.验证项目药品检验方法包括化学检验、生物测定和仪器分析三种。验证项目应包括: 鉴别试验; 杂质定量或限度检查; 制剂中其他成分(如降解产物、防腐剂等)测定; 有效成分含量测定; 溶出度释放度检查中溶出量的测试方法。 2.一般步骤 2.1方案的起草及审批 通常由研究开发实验室提出,检验实验室会签,根据产品工艺条件、原辅料化学结构、中间体、分解产物查阅有关资料提出规格标准,确定检查项目,规定杂质限度和操作步骤,并经有关领导审批方可实施。 2.2仪器的确认验证过程中所用的仪器设备均应校验,对于新的大型
精密仪器应进行确认,确保测试数据的准确可靠。大型精密仪器应制定确认方案,经安装确认、运行确认符合要求,有关领导批准确认报告后方可投入使用。 2.3适用性试验适用性试验内容包括:准确度试验、精密度测定、线性范围试验、选择性试验、检测限等。 2.3.1准确度试验准确度是指测量值与真值接近的程度,测量值与真值愈接近,测量值的误差愈小,测量就愈准确。在准确度试验中可用对照试验、回收试验和空白试验三种方法。准确度通常用回收率表示。 原料含量测定方法的准确度一般用已知含量的对照品或标准品作样品,以所用的方法对它进行定量测定,从分析结果与标准样品或纯净物的含量差值就可知道误差。 制剂含量测定方法的准确度一般用已知含量的原料和处方中相应的辅料按比例模拟配制成制剂进行测定。 杂质定量测定方法的准确度一般用加入已知量杂质进行测定,应能明确证明单一杂质或杂质总量相当于主成分的重量百分比。 在规定范围内,至少用9次测定结果进行评价,如制备3个不同浓度的样品,各测定3次,或把被测物浓度当作100%,用至少测定6次的结果进行评价。 2.3.2精密度测定精密度系指在规定的测试条件下,用同一方法对某一成份进行多次测定,所测得值彼此符合的程度,所以也称重现性,测定值彼此愈接近,测量值的偏差愈小,测量就愈精密。 精密度通常用相对标准偏差表示。 在规定的范围内至少用9次测定结果进行评价。 2.3.3线性范围试验在检验过程中取样量或样品浓度是允许在一定范围内变化的,测定的结果也应随取样量或样品浓度成正比的变化,这样的检验方法才能达到准确度和精密度的要求。取样量在一定范围内变化时,测得含量的结果也成正比的变化,这样的取样范围称之为线性范围。 线性范围的测试通常精密配制一系列供试样品(至少5份)进行测定,以测得的响应信号作为被测物浓度的函数,用最小二乘法进行线性回归。
分析方法验证报告2.2
分析方法验证报告 2019QQHJKX01 分析方法:《水质石油类的测定紫外分光光度法(试行)》(HJ970-2018) 验证人员: 验证时间:2019年01月23日—25日 乌拉特前旗环境保护监测站
1参加人员情况 2仪器、标准物质情况 3 工作曲线的测定 3.1 工作曲线的测定条件 分析日期:2019年01月23日 温度:20℃湿度:18% 测定波长:225nm 3.2 工作曲线的测定
3.2 标准曲线的绘制 4 方法检出限的测定 依据《环境监测分析方法标准制修订技术导则》(HJ168-2010)附录A 方法特性指标确定方法。 方法检出限的一般确定方法: 按照样品分析的全部步骤,重复n(≥7)次空白试验,将各测定结果换算为样品中的浓度,计算n次平行测定的标准偏差,按公式A-1计算,方法检出限按公式A-2计算。
标准偏差S= (A-1) 式中:n —— 样品的平行测定次数 X i —— 单次测定值 X —— 测定平均值 MDL=t (n-1,0.99)×S (A-2) 式中:MDL —— 方法检出限 n —— 样品的平行测定次数 t —— 自由度为n -1 ,置信度为99%时的t 分布 S —— n 次平行测定的标准偏差 ( ) 1 1 2 - ? ? ? ? ? - ∑ = n X x n i i
5 精密度、准确度测试 分别对标准浓度为0.4mg/L、0.8mg/L、1.0mg/L和有证标准物质BW022四个浓度进行6次测定,测定结果见表5-1。
6 评价与验证结论 6.1 评价 根据《水质石油油类的测定紫外分光光度法(试行)》(HJ970-2018)对本实验的检出限、精密度、准确度进行相关评价。 6.1.1 空白值最低检出限评价 根据《水质石油油类的测定紫外分光光度法(试行)》(HJ970-2018)中的检出限为0.01 mg/L,本实验石油类的检出限为0.004mg/L,符合标准方法要求。 6.1.2 精密度评价 本次实验分别对油浓度为0.4 mg/L、0.8 mg/L、1.0 mg/L和有证标准物质BW022进行测试,相对标准偏差分别为4.6%、2.9%、4.7%、5.0%,符合标准方法要求。 6.1.3 准确度评价 根据方法条件,本次实验测定的加标回收率为96%-104%,平均值为99.75%,对标准浓度为0.4 mg/L、0.8 mg/L、1.0 mg/L和有证标准物质BW022的测定,相对误差分别为 2.5%、-1.2%、0.0%、5.0%,其结果均在标准范围内。 6.2 结论 通过对上述指标的验证,证明本站具备按照《水质石油油类的测定紫外分光光度法(试行)》(HJ970-2018)进行监测的能力,该项目可在本监测站正常开展。 分析者:复核者:审核者: 报告编写时间:2019年01月26日
硅酸盐岩石化学分析方法 第30部分:44个元素量测定 方法验证报告
方法验证报告 检测项目:硅酸盐岩石中锂、铍、钪等44个元素 量测定 方法名称及编号: 《硅酸盐岩石化学分析方法第30部分:44个元素量测定》GB/T 14506.30-2010 二O二O年三月
一、方法依据: 根据GB/T 14506.30-2010电感耦等离子体质谱法测定硅酸盐岩石中锂、铍、钪等44个元素的含量。 二、方法原理 样品用氢氟酸和硝酸在封闭溶样器中溶解,电热板上蒸发赶尽氢氟酸,再用硝酸密封溶解,稀释后用ICP-MS外标法直接测定。 三、仪器、试剂及标准物质 3.1 仪器 电感耦合等离子体质谱仪--安捷伦7700 感量天平--赛多利斯科学仪器有限公司 烘箱—上海一恒科学仪器有限公司 3.2 试剂 3.3 标准物质
四、样品 4.1 样品采集和保存 按照HJ/T166的相关规定进行土壤样品的采样和保存,样品采集和保存应使用塑料或玻璃容器,采样量不少于500g,新鲜样品小于4℃时可保存180天。 4.2 样品的制备 将采集的土壤样品放置于风干盘中自然风干,适时压碎、翻动,检出砂砾、植物残体。 在研磨室将风干的样品倒在有机玻璃板上,用木锤敲打,压碎,过孔径2mm尼龙筛,过筛后的样品全部置于无色聚乙烯薄膜上,充分搅匀,用四分法取两份,一份留样保存,一份用作样品细磨。 用于细磨的样品混匀,再用四分法分成四份,取一份研磨到全部过孔径0.074mm筛,装袋待分析。 4.3 样品前处理 称取约0.025g(精确到0.0001g)样品,置于50ml聚四氟乙烯
(PTFE)消解罐中,加1ml氢氟酸,0.5ml硝酸,密封,将溶样器放入烘箱中,加热24h,温度控制在185℃。冷却后取出内罐,置于电热板上加热蒸至近干,再加入0.5ml硝酸蒸发尽干,重复操作此步骤一次。加入5.0ml硝酸,再次密封,放入烘箱中,130℃3h。冷却后取出消解罐,将溶液定量转移至塑料瓶中,用水稀释,定容至25ml,摇匀。此溶液直接用ICP-MS测定。 4.4 实验室空白试样:随同样品进行双份空白试验,所用试剂取自同一瓶,加入同等的量。 4.5 结果计算与表示 计算固体样品中待测物的量: m V c w0? -=) ρ (ρ ) ( 式中:W C—样品中待测元素的量,μg/g; ρ—试样中元素的质量浓度,μg/L; ρ0 —空白试样中元素的质量浓度,μg/L; V—消解后试样的定容体积,ml; m—被称取样品的质量,g; 五、校准曲线 5.1 锂、铍、钒、锰、钴、镍、铜、锌、镓、砷、锶、镉、钡、铊、铅、铋标准曲线 取 5.00ml 100mg/L 多元素混合标准溶液(GSB 04-1767-2004(196046-1))于100ml容量瓶,以1%硝酸定容,得5.00mg/L 标准中间液。取5ml 5.0mg/L的标准中间液浓度于100ml容量瓶中,用1%
产品包装验证报告
文件编号:产品包装材料验证报告 拟制日期年月日 审核日期年月日 批准日期年月日 版号生效日期年月日 有限公司
产品包装材料验证报告 一、总则 1 包装材料的要求 依据:YY/T0681.1、YY/T0313 、YZB/国《体》产品注册标准。 用作制造XXXX的包装材料原料是原始材料,应有原料的来源,明确其历史和可追溯性,并受到控制,以确保成品始终能满足要求。 2 包装材料的设计必须在满足原定用途的条件下,既能够确保内包装材料的符合性,又把对使用者或患者的安全造成危害的可能性降低到最小程度。 2.1 包装材料与XXXX的相容性(即包装与医疗器材相互无不良影响):主要考虑的有:包装材料的安全性毒性的要求,拟包装的医疗器械的大小和形状,对物理和其它防护的要求,医疗器械对特殊危险例如辐射、湿气、机械性撞击,静电放射的敏感性。 2.2 包装材料与标识方式的相容性:标识方法必须对包装材料与采用的灭菌过程的相容性无不良影响,印刷或书写所采用的油墨不会转移到XXXX产品上,也不会和包装材料起反应而影响包装材料的效用,也不会变色而使标识变的模糊不清,对固定在包装材料表面的标识,其附着方式必须能耐受灭菌过程的使用及制造厂规定的贮存和运输条件。 3 包装材料能够提供对物理、化学和微生物的防护。 3.1包装材料在使用场所与使用者撕开包装取出使用时的要求相容性(例如无菌的开封)。
3.2 在使用条件下,在灭菌前、中、后,包装材料不可释放已知是有毒的,其数量足以对健康危害的物质。 3.3无菌状态的保持:(即从其产品灭菌后,成为无菌之时起,直至规定的失效日期或使用时止),包装完整性及包装材料的微生物阻隔特性。 3.4 材料的毒性检测。
有机挥发性物质检测方法验证报告2
验证报告Validation Report
目录CONTENTS 报告总结Summary Report 1、目的Purpose 2、验证范围Scope 3、验证依据Validation Basis 4、责任者Person Responsibility 5、接受标准Acceptance Criterion 6、有机挥发性物质的测定Determination of Organic Volatile Impurities 6.1有机挥发性物质的限度Limit of Organic Volatile Impurities 6.2溶液配制Preparation 6.3色谱系统Chromatographic System 6.4系统适应性试验System Suitability Test 6.5测定Procedure 6.6结果计算Calculate 6.7样品测定Sample Determination 7、仪器和试药Instruments and Reagents 7.1仪器Instruments 7.2试药Reagents 8、验证内容Validation Contents 8.1专属性(定位)Specificity 8.2检测限Limit of Detection 8.3精密度Precision 8.4线性范围Linearity and Range 8.5准确度/回收率Accuracy/Recovery 9、变更和偏差调查Change and Deviation Investigation 10、结果分析、结论和评价Comprehensive, Conclusion and Assessment 11、附录A ppendix
工艺验证报告模板
工艺验证报告 文件编码: 起草人:姓名: 部门: 日期: 审核人:姓名: 部门: 日期: 批准人:姓名: 部门: 日期:
目录 1.介绍 (2) 2.验证目的 (2) 3.验证范围 (2) 4.验证类型 (2) 5.验证日期与相关批号 (2) 6.验证小组成员及职责 (2) 7.简单工艺描述(略) (2) 8.胺化工艺验证 (3) 8.1.工艺参数 (3) 8.2.验证人员及日期 (3) 8.3.验证标准、分析方法 (3) 8.4.验证数据 (3) 8.5.验证结果分析、评价及建议 (4) 9.纯化工艺验证 (4) 9.1.工艺参数 (4) 9.2.验证人员及日期 (5) 9.3.验证标准、分析方法 (5) 9.4.验证数据 (5) 9.5.验证结果分析、评价及建议 (6) 10.成盐工艺验证 (7) 10.1.工艺参数 (7) 10.2.验证人员及日期 (8) 10.3.验证标准 (8) 10.4.分析方法 (8) 10.5.验证数据 (8) 10.6.验证结果分析、评价及建议 (9) 11.验证结果批准、会签及日期 (10)
1.介绍 在验证生产过程中发生偏差与异常情况,我们按“偏差处理程序--SOP-ZL-9004-02”进行了处理。 2.验证目的 证明本工艺路线在预定的工艺参数范围内运行能持续有效地生产出高质量的合格的硫酸羟基氯喹。 3.验证范围 4.验证类型 前瞻性验证 5.验证日期与相关批号 验证日期:2007 年03 月20 日至2007 年04 月20 日验证批号:070402、070403、070404 6.验证小组成员及职责 7.简单工艺描述(略) 详见工艺验证方案SMP-ZL-7244-00。
钯元素测定分析方法验证报告
Palladium Analytical Method Validation Report 钯元素测定分析方法验证报告Effective Day 生效日期:
TABLE OF CONTENTS 目录 1 PURPOS E 的 .................... 2 SCOP 范围 ...................... 3 RESPONSIBILITIES 职责 ............... 4 ABBREVIATION 缩略语................. 5 REGULATIONS AND GUIDELIN 法规和指南 ..... 6 REFERENCE DOCUME 参考St 件 ......... 7 CONFIRMATION PREREQUISIT E 决条件确认...... 8 CONFIRM THE TEST RESUL 确认检测结果汇总 ... 9 DEVIATION HANDIN 偏差处理总结 .......... 10 SUMMARY AND CONCLUSION 与结论 ........ 11 ADVICE (IF ANY) 建议(如有) .. (12) ATTACHMENT LIS 附件清单 ............ 1 Purpose 目的 本验证报告的目的是通过记录在案的测试, 证明原子吸收分光光度法适用于 原料药(API )中钯元素残留进行定量分析。证明此方法适用于盐酸伐昔洛 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签 错误!未定义书签
(新)检验方法的验证及确认
检验方法是指实验室用于实施检验检测工作所依据的标准检验方法和技术规范。检验方法是实验室实施检验工作的主要依据,是开展检验检测工作所必须的资源,如果方法及程序不同就会造成结果不同。本文就来聊聊如何对检验方法进行确认。文章为原创大赛往期作品回顾,在此仅作为对大家的启发之用。欢迎批评指正。 <<实验室资质认定评审准则>> 条款中规定:“实验室应确认能否正确使用所选用的新方法。如果方法发生了变化,应重新进行确认。实验室应确保使用标准的最新有效版本。”在条款中也有相应的规定。 实验室采用的检测方法包括样品的抽取、处理、运输、存储和制备等各个环节,确认时应当记录确认所获得的结果、使用确认的程序、确认对方法是否适合于预期的用途等,必要时还应包括不确定度和分析数据的统计学处理技术。 ? 下面谈谈就方法发生了变更时或颁布新标准时,对方法如何进行确认: 1.在首次对外出具数据之前应确认(证实)标准方法已被正确的运用。 2.标准方法发生了变化应重新确认。 3.对标准方法定期清理或者查新,以确保最新有效版本。 一、检测方法的选择及使用要求 实验室资质认定(或认可)现场考核时确定的检测项目的依据是国家标准、行业标准和地方标准。所以说,当没有国际、国家、行业、地方规定的检验方法时,实验室应尽可能选择已经公布或由知名的技术组织或有关科技文献或杂志上公布的方法,但应经实验室技术主管确认。如是在实验室计量认证或认可批准业务范围内,因客户的特殊要求而发生的情况,其检验结果和报告上应有明确的说明。 另外需要使用非标准方法时,这些方法应征得委托方同意,并形成有效文件,使出具的报告为委托方和用户所接受。这是指必须在实验室计量认证或认可批准业务范围内使用,所谓有效文件是指甲乙双方对使用非标准方法检测达成协议,一般来说应有双方签字盖章,也可以在检测委托(协议)书上注明,实验室在检测报告中也必需加以说明。 因此,在检测方法的选择上,优先使用国家标准,然后是行业标准、地方标准,非标准方法仅限于委托方同意才使用。 对于实验室完成的每一项或每一系列检验的结果,均应按照检验方法中的规定,准确、清晰、明确、客观地在检验证书或报告中表述,应采用法定计量单位。证书或报告中还应包括为说明检验结果所必需的各种信息采用方法所要求的全部信息。除上述明确的要求外,检测报告中必需有检测数据和结论。 所以说,检测方法选择的核心就是方法有效性,要特别注意的是:要使用最新有效版本的方法。
消毒方法及效果验证报告
消毒方法及效果验证报告
消毒方法及效果验证报告 验证报告编号: 起草人: 日期:年月日审核人:日期:年月日批准人:日期:年月日
目录 1验证目的 2验证范围 3验证组及职责 4验证进度计划 5验证引用的文件 6验证内容 7再验证周期确认 8偏差或变更说明 9结果分析及评价
1.验证目的: 确认消毒剂浓度和用量及消毒效果,以保证生产环境卫生的消毒质量。 2.验证范围 精烘包洁净车间。 3.验证组及职责 年月日至年月日 5. 验证引用的文件 检查人:复核人: 日期:年月日
6. 验证内容 6.1 验证的准备工作 6.1.1验证所需文件资料 进行消毒方法及效果验证前,应检查证明以下文件齐全。 6.1.2检查“验证所需仪器、仪表、量具清单及校正”情况应符合规程,见附件1。 6.1.3检查“验证所需试验条件”落实齐备、符合要求,见附件2。 6.1.4检查微生物检定室空气净化系统各验证数据符合净化要求。 检查结果:见微生物检定室空气净化系统验证报告。 检查人:日期: 6.1.5 检查XYJ型医用净化工作台验证结果符合规定。 检查结果:见XYJ型医用净化工作台验证报告。 检查人:日期: 6.1.6人员: 列出参加消毒方法及效果验证的人员名单,查阅培训档案,确认是否经过了微生物基础知识及微生物污染的防范培训和岗位技术操作及安全防护后的培训并考察合格,是否参加了健康查体,有健康证。 检验人员培训、查体、情况表
6.1.7消毒剂 6.1. 7.1 75%酒精,按照75%酒精配制规程进行配制。 6.1. 7.2 0.3%新洁尔灭,按照0.3%新洁尔灭配制规程进行配制。 75%酒精配制记录 0.3%新洁尔灭配制记录 6.2 验证内容: a、消毒剂(75%酒精和0.3%新洁尔灭)浓度及用量确认。 b、消毒剂(75%酒精和0.3%新洁尔灭)消毒周期确认。 以上消毒剂的消毒效果及消毒周期的确认,选择D级洁净区厂房中操作间的顶棚、墙壁和工序中的生产设备、容器等及操作人员的手为验证对象,每项实验连续进行三次。 6.2.1消毒剂(75%酒精与0.3%新洁尔灭溶液)浓度及用量确认。 6.2.1.1消毒前检验:验证检验人员需先将所验证对象(顶棚、墙壁、设备、容器)分别取样做一次消毒前微生物含量检查。用事先经过灭菌的生理盐水湿润的适当大小的灭菌纱布或灭菌脱脂棉充分擦拭,然后放入广口瓶中加一定量的灭菌生理盐水振摇5分钟,再对浸出液进行细菌培养,计算统计细菌数。(CFU/25cm2) 车间洁净区消毒前物体表面取样检验结果统计表
xiSS检测方法验证报告
悬浮物测定的方法验证报告 1.本方法依据参照:GB11901-89(重量法) 2.测定方法原理 用0.45m滤膜过滤水样,留在滤料上并于103-105℃烘至恒重的固体,经103~105℃烘干后得到SS含量。 3.仪器 3.1、烘箱 3.2、分析天平 3.3、干燥器 3.4、孔径为0.45μm滤膜、直径45~60mm。 3.5、玻璃漏斗 3.6、真空泵 3.7、内径为30-50㎜称量瓶 3.8、无齿扁嘴镊子 3.9、蒸馏水或同等纯度的水 4.测定步骤
4.1用无齿扁嘴镊子将滤膜放在称量瓶中,打开瓶盖,移入烘箱中于(103~105℃)烘干2h后取出置于干燥器内冷却至室温,称其重量。反复烘干、冷却、称量,直至恒重(两次称量相差不超过0.5mg) 4.2去除悬浮物后震荡水样,量取充分混合均匀的试样100ml抽吸过滤。使水分全部通过滤膜。再以每次10ml蒸馏水连续洗涤三次,继续吸滤以去除痕量水分。如样品中含有油脂,用10ml石油醚分两次淋洗残渣。 4.3停止吸滤后,仔细取出载有SS的滤膜放在原恒重的称量瓶里,移入烘箱中于103~105℃下烘干2h后移入干燥器中,使冷却到室温,称其重量,反复烘干、冷却、称量,直至两次称量的重量差≤0.4mg为止。 5.计算: 悬浮固体(mg/L)=[(A-B)×1000×1000]/V 式中:A——悬浮固体+滤膜及称量瓶重(g) B——滤膜及称量瓶重(g) V——水样体积 6.讨论 6.1方法适应范围 本方法适用于废水中SS的测试。 6.2精密度(重复性)的讨论。
重复性:实验室同一分析人员对同一浓度水平样品取样7次测试,用所得结果的标准偏差(RSD)来表示其精密度;RSD≤5%符合要求。 表1重复性 结论以上各项的讨论均符合要求,即表明方法满足本实验室检测需求,确认予以使用
工艺验证报告模板
验证文件 2013年XX月 6.验证报告起草、审核与批准 6.1验证报告起草 6.2 再验证报告审核 6.3 再验证报告批准
目录 1. 验证概述 2. 验证目的 3. 验证范围 4. 再验证依据标准 5. 机构与职责 5.1 验证机构 5.2 验证职责 6. 验证方式 7. 验证准备 7.1 设备设施准备 7.2 仪器试剂准备 7.3 原辅物料准备 7.4 文件与培训 8. 验证时间与计划 9. 验证实施 9.1 产品的工艺流程图 9.2产品的工艺验证: 9.2.1称量备料 9.2.1.1目的 9.2.1.2文件 9.2.1.3检查项目及结果9.2.2 配制 9.2.2.1 目的 9.2.2.2 文件 9.2.2.3 评估项目 9.2.2.4 评估方法
9.2.2.5 取样方法 9.2.2.6配制试验数据 9.2.3 灌装封尾 9.2.3.1 目的 9.2.3.2文件 9.2.3.3评估项目 9.2.3.4评估方法 9.2.3.5灌装封尾检查数据9.2.4 成品抽样检验 9.2.4.1 目的 9.2.4.2 文件 9.2.4.3 评估项目 9.2.4.4 评估方法 9.2.4.5产品检验报告复印件 10. 偏差与处理. 11. 结果与分析 11.1 验证数据汇总 11.2 存在问题与措施 11.3 风险与预防 12. 验证结论 12.1 验证结论 12.2 验证评价与建议 13. 验证周期 14. 附件 15.参考或引用文件
1.概述: 复方醋酸地塞米松乳膏为我司生产多年的乳膏剂品种,自2009GMP再认证以来,乳膏剂生产线生产所用关键设备、生产工艺及工艺参数没有改变,为了验证在正常的生产条件和GMP文件管理体系下能生产出符合预定的规格及质量标准的产品,根据验证管理文件的要求,我们对复方醋酸地塞米松乳膏的生产工艺进行再验证。 2.目的: 在现行的GMP文件管理体系下,生产三批复方醋酸地塞米松乳膏进行工艺再验证: (1)确认关键工序质量监控点是否符合质量要求; (2)确认该产品质量是否符合预定成品的标准。 3.验证范围: 本次验证对复方醋酸地塞米松乳膏,依据工艺规程的各项参数设定指标,并认真按方案组织了实施,仅验证该品种工艺参数设定的科学性符合性。 4.再验证的依据与标准: 《药品生产质量管理规范》(2010版)、《复方醋酸地塞米松乳膏生产工艺规程》、《复方醋酸地塞米松乳膏中间产品内控质量标准》、《复方醋酸地塞米松乳膏成品内控质量标准》。 5 .机构与职责: 1.机构:在公司验证委员会的指导下,成立验证小组负责工艺验证的具体工作。验证小组的成员包括车间和生产部人员,QA和QC人员,工程设备人员、注册部人员。 2.职责:参与人员的职责
分析方法验证标准操作规程完整
NO. 编号:Analytical Method Validation Standard Operation Procedure 分析方法验证标准操作规程 Table of Contents
目录 1 Purpose 目的 (2) 2 Scope 范围 (3) 3 EHS 环境健康安全 (3) 4 Definition 定义 (3) 5 Responsibility 职责 (5) 6 Flow Chart 流程图 (5) 7 Procedure 程序/要求 (6) 7.1 Requirement 要求 (6) 7.2 Procedure 步骤 (9) 8 References 参考 (18) 9 Annexures 附件索引 (18) 10 Revision History 版本历史 (18) 1.Purpose 目的 描述分析方法的验证步骤,确认分析方法达到使用要求。为以下试验的验证提供指导和参考。 ?鉴别试验; ?杂质的定量测试或限度检查; ?原料药和制剂中有效成分含量测定; ?制剂中其它成分(如防腐剂等)的测定;
?制剂溶出度、释放度等检查中,其溶出量等; ?微生物限度。 2.Scope 范围 本程序适用的化学方法和微生物方法的验证,这些检测方法用于以下目的: 2.1 进行稳定性研究从而建立产品的有效期; 2.2 商业用途及临床用途的产品及其生产过程、清洁过程的放行检测。 另外,根据需要,本程序也可适用于在使用的上市产品、赋形剂、和原料产品的新方法的开发和验证。3.EHS 环境健康安全 不适用。 4.Definition 定义 4.1空白样品 不含被测物的特定样品。 4.2赋形剂 在剂型配方中除原料药以外的其他成分。 4.3安慰剂 和药品组成基本相同但不含活性成分或用惰性成分替代活性成分的制剂,或由药品赋形剂组成的混合物,其用量等同于药品制剂中的用量。 4.4准确度 真实值或认可的参考值与测量值之间的相近程度。 4.5检测限(DL) 一个分析规程的检测限是指样品中的被测物能被检测到的最低量,但此最低量不一定要定量到某一确定的数值。等同于检测限度(LOD)。 4.6定量限(QL) 分析规程的定量限是指样品中的被分析物能被精确和准确定量的最低量。定量限是样品组成中低水平化合物的定量分析的参数,尤其应用于杂质和/或降解产物的测定。等同于最低定量限(LOQ)。 4.7专属性(选择性) 指在一些可能存在的组分,如杂质、降解物、基质等存在时,对被分析物准确可靠测定的能力。如果一个分析方法缺乏专属性,可由其他辅助的分析方法作补充。 4.8线性 分析规程的线性是指(在给定的范围)所获得的测试结果与样品中被分析物浓度(数量)呈比例关系的能力。 4.9范围 分析规程的范围是样品中被分析物最高和最低浓度(数量)之间(包含最高和最低浓度)的间隔,已证明在此范围内分析规程具有适宜的精密度、准确度和线性。 4.10精密度 分析方法的精密度是指在规定条件下自均质样品的多次取样所获得的一系列测量之间的接近程度。精密度可从三个水平考虑:重复性、中间精密度和重现性。采用均质的,可信的样品进行精密度的研究。
清洁验证检验方法验证
生产设备清洁后取样方法和检验方法验证方案目录 概述 目的 验证小组成员与职责 验证小组成员 验证小组职责 4. 验证正文 4.1 验证前确认 4.2 验证方法描述 4.3 验证内容 检测方法验证部分 综合回收率验证 偏差总结 再验证情况 补充与修定 评价与结论 附录 概述 生产过程中所用的生产设备均可能有残留物遗留,为了最大程度的避免由于上一批次生产产品的残留影响下一批次或其他品种,故必须对生产所用的设
备进行清洁。清洁后要对该清洗方法进行取样检测残留量。一般通常的取样方法为棉签擦拭法和淋洗法。 目的 本验证方案的目的是考察清洗验证涉及取样过程和所用检测方法的过程,是对人员取样操作、残留物转移、测试过程的考察,考察项目最低定量限、线性、综合回收率等。 3. 验证小组成员与职责 3.1验证小组成员 组长:xxx 组员:xxx、xxx 3.2验证小组职责 组长:质量副总经理xxx,负责批准验证方案和验证报告。 组员:xxx,参与验证方案的制定,对验证操作过程监督检查,收集验证资料和数据,参与起草验证方案和验证报告。 组员:xxx,负责参与验证方案的制定,对所取样品进行化验,收集数据并报告结果,审核验证报告。 组员:xxx,负责审核验证方案,审核清洁验证方案和报告,协助验证方案的实施,并审核验证报告。 4. 验证正文 4.1验证前确认 棉花/棉签材质:棉签 紫外分光光度计编号:校验有效期□接受□不接受
天平编号:校验有效期□接受□不接受 批号: 4.2 检测方法描述 检测过程 结构中含有很强的紫外吸收官能团,如双键的苯环等,故可采用紫外分光光度法。 比色皿,在合适的最大的吸收波长处,以甲醇为空白,测定样品溶液的吸收度。根据测得的吸收度来计算样品残留的量。 贮备液的配制 取对照品约10mg,精密称定,置于200ml容量瓶中,用甲醇溶解并稀释至刻度,混匀,得贮备液(50μg/ml)。 计算 测定时同时进行对照品的测定,根据朗伯比尔定律,按下列公式计算: A样 C样=×C对 A对 A:分别为样品和贮备液的吸收度 C:分别为样品和贮备液的溶液浓度 4.3 验证内容 检测方法验证部分
分析方法验证报告三篇
分析方法验证报告三篇 篇一:分析方法验证报告三篇 1.工作曲线的测定 1.1工作曲线的测定条件 分析日期:年月日 温度21.3℃湿度64% 测定波长650.00nm 1.2工作曲线的测定 表1工作曲线测定值 标准溶液加入体积 质量(ug)吸光度(A)序号(n) (mL) 1 0.00 0.000 0.000 2 0.05 0.016 3 0.200 0.041 4 0.8 0.068 5 3.2 0.126 6 7.2 0.195 7 12.8 0.26 8 20 0.325 回归方程y=0.1609x+0.002
相关系数r 0.9992 1.3标准曲线的绘制 回归方程y=0.1609x+0.0021 相关系数r=0.9992 2.空白值测定结果及方法检出限的计算第页共页 依据《环境监测分析方法标准制修订技术导则》HJ 168-20XX附录A方法特性指标确定方法。 方法检出限的一般确定方法: 按照样品分析的全部步骤,重复n(≥7)次空白试验,将各测定结果换算为样品中的浓度或含量,计算n次平行测定的标准偏差,按公式(A.1)计算方法检出限。 ×S(A.1) MDL=t (n-1,0.99) 式中:MDL——方法检出限; n——样品的平行测定次数;
t——自由度为n-1,置信度为99%时的t分布(单侧); S——n次平行测定的标准偏差。 其中,当自由度为n-1,置信度为99%时的t值可参考表A.1取值。表A.1 t值表 平行测定次数(n)自由度(n-1)T (n-1,0.99) 7 6 3.143 8 7 2.998 9 8 2.896 10 9 2.821 表2空白值测定结果及方法检测限的计算结果 分析日期:年月日 空白测定次数(n)吸光度(A)浓度(mg/L) 1 0 -0.013 2 0 -0.013 3 -0.002 -0.025 4 0 -0.013 5 0 -0.013 6 0.003 0.006 7 -0.004 -0.038 8 0 -0.013 9 -0.002 -0.025
糖类新抗原CA199方法学验证报告
糖类抗原CA199检测系统/方法的分析性能验证评价 验证内容:正确度、精密度、线性范围、临床可报告范围的验证、干扰物质及参考区间的确认 验证人员:王爱林马力张卫李琳 一、检测系统信息 项目:CA199 仪器名称全自动电化学发光免疫分析仪 仪器型号Cobas e601 试剂及厂商:罗氏诊断有限公司 检测方法:双抗体夹心法 二、厂商提供的相关参数 三、验证过程 1、正确度 目的:评价仪器测量结果与真值的一致程度。通过实验室检测数据的偏倚从而评价和验证实验室检测结果的正确度 评价方法:参加卫生部临检中心的室间质评,本组参加室间质评的项目一律用回报结果作为评价标
正确度验证试验数据记录表
2精密度(Precision) 重复性精密度 目的:考察仪器检测方法的随机误差 原理:在检测系统处于优良的条件下,连续测定20个结果,判断这20个独立结果间的一致程度 方法:选择新鲜混合血清标本(病人高值、低值)20份,测量前先定标,再做质控,质控结果在控制范围内,连续重复测定20次,质控结果在控制范围内,计算SD,CV,得到重复性精密度 标本来源:高低值标本均为混合血 结果:本室CA199重复性精密度低值CV为:1.7% 、高值CV为:2.2% 结果判断方式:重复性精密度CV值<1/4CV总误差 重复性精密度验证试验数据记录表
日间精密度: 目的:考察目前实验室检测方法批间精密度 原理:在检测系统处于优良的条件下,连续测定20天,取得20个结果,判断这20个独立结果间的一致程度。 方法:新鲜混合血清(高值、低值)分装20份冷冻,每天取两份混合血清随标本连续测定10天,测定前定标做质控且结果在控制范围内,计算CV,SD,得到日间精密度 结果:本室CA199日间精密度平均低值CV:3.9% 平均高值CV:3.4% 结果判断方式:日间精密度CV值<1/3CV总误差
检测方法确认报告(邻苯二甲酸酯)
检测方法验证报告 部门: 检测方法气相色谱-质谱法检测项目PVC中六种邻苯二甲酸酯的测定 方法类别■标准方法□非标准方法方法编号GB/T 22048-2015 一、目的:(描述方法验证的目的和用途) 确认该方法是否能满足标准及客户要求。 二、职责:(描述参与部门、人员及其相应职责) 1.检测室负责检测方法的选择和验证; 2.技术负责人负责方法验证的审批。 三、参考标准概述: ①标准来源 ChemService(美国) ②适用范围(测定值范围及材质等) 适用于含聚氯乙烯材料的玩具和儿童用品。 ③检测项目 PVC中六种邻苯二甲酸酯:DBP、BBP、DEHP、DNOP、DINP、DIDP。 ④检测过程要点 用二氯甲烷在索氏(Soxhlet)抽提器或者溶剂萃取器(参见附录B)中对试样中的邻苯二甲酸酯进行提取,对提取液定容后,用气相色谱/质谱联用仪(GC/MS)测定,采用总离子流色谱图(TIC)进行定性,选择离子检测(SIM)进行定量。 ⑤检测结果符合性指标 方法检出限:DBP、BBP、DEHP、DNOP:10mg/kg;DINP、DIDP:50mg/kg。 加标回收率:85%—115% 重复性测试:精密度≤20% 四、验证方式:■方法检出限;■加标回收率;■重复性测试;□再现性测试;□能力验证;□实验室间比对;□标准物质检测;□加标实验;□标准方法对比;□其他。
五、验证过程与结果(针对不同的验证方式分别描述验证过程和结果): 1.过程描述: 方法检出限:配制“DBP 、DEHP 、BBP 、DNOP 各1ppm ;DINP 、DIDP5ppm ”的样品;用GCMS 连 续检测7次该样品;对各组分进行检出限的计算(用以下公式):MDL=t ×7次检测数据的标准偏差×稀释比例。(其中t=3.14) 加标回收率:向两份平行样品中,一份准确加入一定量的标准溶液,一份不加。两个样品经 过相同的步骤前处理及GC-MS 测试,根据测试结果求出加标回收率。 重复性测试:对同一样品进行2次平行测试,按标准规定,求出结果的精密度。 2.数据处理结果: ①方法检出限: 项目名称 检测数据(mg/L ) 指标分析 ① ② ③ ④ ⑤ ⑥ ⑦ 平均值(mg/L ) 标准偏差(mg/L ) 方法检出限(mg/kg ) DBP 0.96 0.93 0.79 0.78 0.81 0.76 0.77 0.83 0.08 0.26 BBP 1.01 1.15 1.09 1.21 1.05 0.99 1.06 1.08 0.08 0.24 DEHP 0.98 1.09 1.12 0.93 1 0.85 1.13 1.01 0.10 0.33 DNOP 1.04 1.02 0.92 0.91 0.98 0.81 0.83 0.93 0.09 0.28 DINP 4.56 5.56 5.25 5.81 5.16 4.8 4.82 5.14 0.45 1.40 DIDP 5.52 5.21 4.62 4.65 5.09 4.44 4.94 4.92 0.38 1.19
方法验证报告模板最终
分析方法验证报告 ZBYZ-2015-000 项目名称:磷酸盐的测定 分析方法:磷钼蓝分光光度法 方法编号:GB5750.5-2006 验证人员:乔柱史红艳 验证日期:2015.10.20 众邦环保检测技术 Shaanxi ZhongBang Environmental Protection Testing Technology Co.Ltd.
1.工作曲线的测定第页共页1.1工作曲线的测定条件 分析日期:年月日 1.2工作曲线的测定 表1 工作曲线测定值 序号( n) 标准溶液加入体积 (mL) 质量(ug)吸光度(A) 1 0.00 0.0000.000 2 0.050.016 3 0.2000.041 4 0.80.068 5 3.20.126 6 7.20.195 7 12.80.26 8 200.325 回归方程y = 0.1609x+0.002 相关系数r 0.9992 1.3标准曲线的绘制 回归方程 y = 0.1609x+0.0021 相关系数 r=0.9992 温度21.3℃湿度64% 测定波长650.00nm
2.空白值测定结果及方法检出限的计算第页共页 依据《环境监测分析方法标准制修订技术导则》 HJ 168-2010 附录A 方法特性指标确定方法。 方法检出限的一般确定方法: 按照样品分析的全部步骤,重复n (≥7)次空白试验,将各测定结果换算为样品中的浓度或含量,计算n次平行测定的标准偏差,按公式(A.1)计算方法检出限。 MDL = t (n-1,0.99)× S (A.1) 式中:MDL ——方法检出限; n ——样品的平行测定次数; t ——自由度为n -1,置信度为99%时的t分布(单侧); S——n次平行测定的标准偏差。 其中,当自由度为n -1,置信度为99% 时的t 值可参考表A.1 取值。 表A.1 t值表 表2 空白值测定结果及方法检测限的计算结果 分析日期:年月日