第一性原理

第一章引言
在21世纪的今天,全球都面对着资源的短缺和环境的污染这两大问题。氢能源的出现,不仅仅解决了能源短缺的问题(氢能源是二次能源),同时氢能源的使用对环境几乎是没有任何的污染(氢气和氧气的反应产物只有水)。因此,从上个世纪70年代就开始关注氢能源的研发。到21世纪的今天氢能源也逐渐开始走上舞台,但是对于氢能源在应用过程中会出现的问题也亟待解决。
本章内容之一将主要介绍氢能源应用中面临的一个严峻的问题——氢气的
。其二,存储,我们将会详细的论述最新出现的一种储氢材料:储氢合金——AlH
3
简述历年来在实验和理论上对于该材料在常压下的研究成果。同时,提出对于在高压条件下进行研究的必要性以及在现阶段的成果,指出我们理论研究的AlH
3
必要性。最后,将简单的介绍高压物理学在当今学科发展中的重要性以及高压物理的发展历史,当然我们将会简述由于现实实验条件上的限制,高压物理的理论研究对于材料性能的分析和高压物理未来发展方向上的重要性和必然性。
§1.1储氢材料简介
伴随着人类社会的发展和进步,人类赖以生存的环境却让全世界都开始担忧。环境的破坏的危机以及能源的短缺的意识,迫使人们一方面去寻求新的能源,另一方面又要考虑新能源对环境所造成的破坏问题。于是氢能源作为存储量丰富,无公害,无污染的新型能源而得到了全球的关注。在以氢作为能源媒介的氢能体系中,氢的存储和氢的运输成为氢能源的实际应用中的关键环节。近年来,人们注意到储氢合金由于其材料结构上的优势而成为一种新型的储氢功能材料。由于某些合金具备特殊的晶体结构,能够使氢原子很容易的进入晶格间隙中并且形成金属氢化物,由于这种氢与金属的结合力很弱,在加热的时候,氢就能从金属中释放出来。但是这些储氢合金的储氢量很大,可以存储比其自身体积要大上1000-1300倍的氢。目前,对于储氢合金的研究也进行的如火如荼。
1.1.1氢能
随着全球人口急增,人类的能源消耗大幅度的增长;而作为主要能源的煤炭和石油,它们又都是不可再生的能源,其储量极为有限。另外,大量矿物能源的燃烧,造成大气污染、"酸雨"和"温室效应"等环境问题。因此,从20世纪60年代以来,人类为了解决未来能源的供应和生存环境问题,高呼"能源革命"。"
能源革命"是以绿色能源利用为目的,包括新能源和可再生能源逐步代替资源有限、对环境产生污染的化石能源。
一直以来,氢能(即氧和氢反应放出的能量)由于其放热效率高:每千克氢
燃烧后的热量,约为汽油的3倍,酒精的3.9倍,焦炭的4.5倍。燃烧的产物是水,对环境无污染。资源丰富,氢气可以由水制取,而水是地球上最为丰富的资源。适用范围广,氢燃料电池既可用于汽车、飞机、宇宙飞船、可发电,又可用
于分布式电源等其他场合,如可以代替煤气、暖气、电力管线而走进家庭生活而愈来愈受到世人关注。越来越多的科学家也认为:在21世纪的今天,氢能是能够解决化石能源危机和缓解环境污染问题的绿色能源。它势必将在未来的能源领域中扮演重要的角色。
如今,氢能的发展在全球都在如火如荼的进行。美国从2003年布什政府投
资17亿美元启动氢燃料开发计划到2004年建立第一座氢气站,其次美国已成功研制出世界上第一辆以氢为原料的汽车;欧盟从投资2500万——3000万欧元研究氢能和燃料电池到成立“北欧能源研究机构”,再力争在2020年建立一个燃料电池和氢能源的庞大市场;日本从发展燃料电池到在全国各地建造不少“加氢站”能为近百辆燃料电池车服务,再计划2030年发展到1500万辆的计划;等等。在中国,2002年1月18号,中科院正式启动"863"项目——科技创新战略行动计划重大项目,该项目是为研究和开发具有自主知识产权的燃料电池发动机以及氢能源的成套技术。
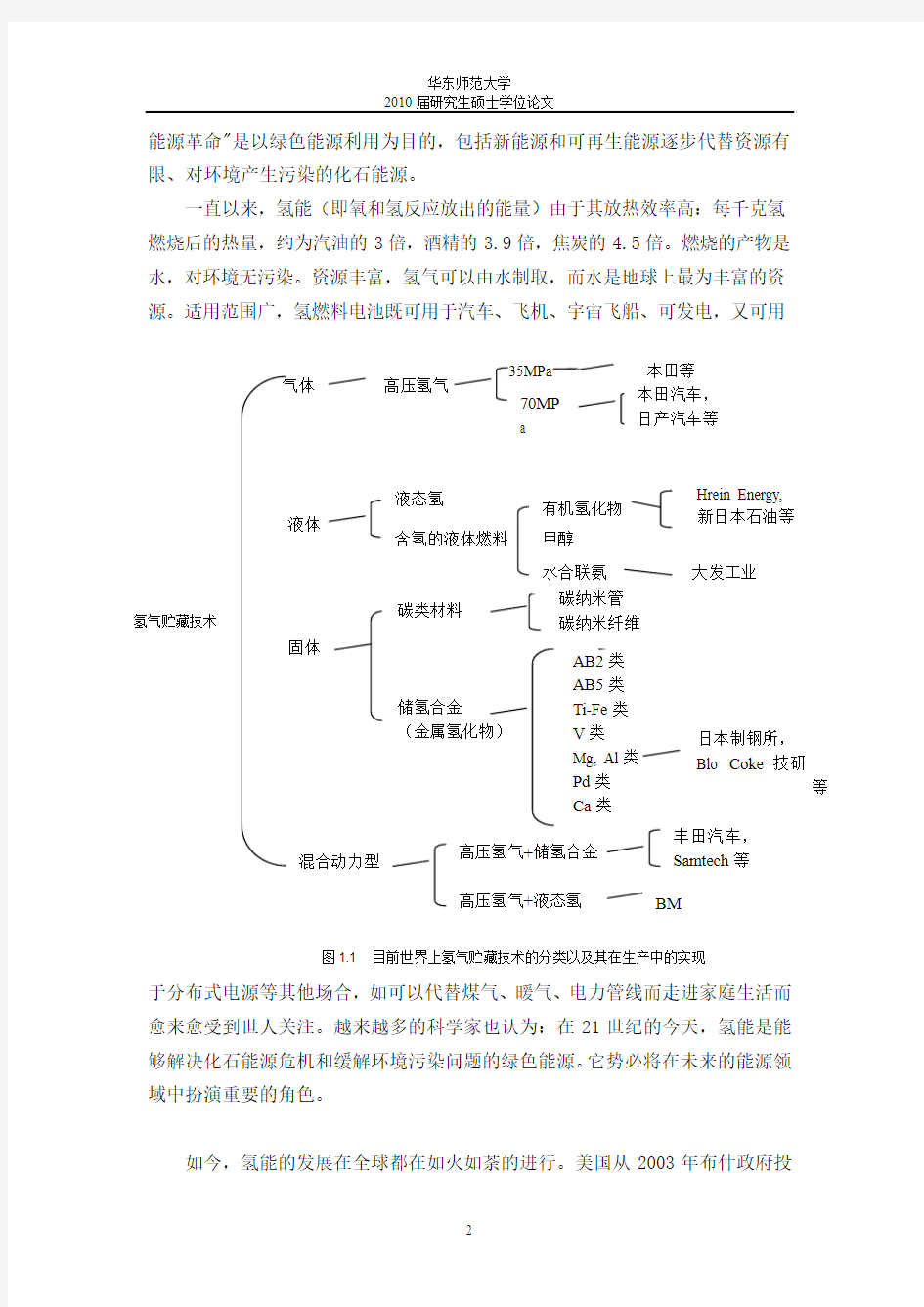
2003年11月,由包括中国在内的美国,澳大利亚,巴西,加拿大,意大利,英国,冰岛,挪威,法国,俄罗斯,日本,韩国,印度,欧盟一起组成的委员会共同制定了“氢经济国际伙伴计划(International Partnership For The Hydrogen Economy)”[1],这也标志着国际社会在发展氢经济上已经初步达到共识。在氢能的发展包括了氢气的生成,氢气的储藏以及氢气的运输。其中,氢气的储藏方式包括了高压气态存储、低温液氢存储和以及储氢材料存储。在图1.1中,我们可以看到目前氢气贮藏技术的各种方式以及他们在生产中的实现。
目前高压氢气罐由于其存储简单以及耐高压的储氢压力容器和材料而使用比较普遍。但是这种方式却存在着以下几个主要问题:(1)氢容量小;在储氢压力为15Mpa时,氢的重量仅占总重量的1%,体积容量约0.008kgH2/L。(2)安全性差;高压容器本身就需要特殊的照顾与维护,况且容器中装的是易燃易爆又易渗漏的氢气。车祸时可能有严重的后果。(3)实施上的困难;容器压力愈高,充氢站的建设、压缩运行所化的代价愈高。而且充装1立方米氢气要耗电0.5度左右、而1立方米氢气经燃料电池发电仅得2度电。其次对于低温液氢存储方式,虽然从理论上看,在各种储氢方式中,无论是从体积密度还是从重量密度的角度看,只有氢气以液态储存才能达到最高的储存密度。目前,液氢存储的重量比约5%-7.5%,体积容量约0.04kgH2/L。不过,由于低温容器的热漏损,液氢的生产、储存、运输对注,以及氢液化消耗大量的能量等问题,使携带液氢规模实施是不可行的。因此,当20世纪60年代出现了应用储氢材料的方式来储藏氢气以来,人们就开始把焦点放在寻找储氢量高,成本低而且质量轻的储氢材料。同时,美国能源部(DOE)提出要实现氢能源实用化的目标是:至2010年达到室温和安全压力下重量储氢率6.5%(质量分数),体积储氢率45g/L;2015年重量储氢率9.0%(质量分数),体积储氢率81g/L[2]。我国也十分重视储氢材料的研
究,在“863”高新技术发展规划和“973”计划中,都将储氢材料作为重点研究项目。其中,Mg类及Al类储氢合金由于其材料和性能上的优势而得到积极地开发。根据图1.2中列出的各种氢气存储技术的氢元素密度含量的对比,可以清楚地看到AlH3这类储氢合金材料无论在单位面积还是单位重量的氢元素密度均是处于很高的位置。
1.1.2储氢合金AlH3 介绍
由于其特殊的材料性质而得到了广泛的应用,从20世纪60年代开始,AlH
3
其中包括作为复合固体推进剂,电池或燃料电池,有机合成化学,高分子合成化学和原子层积技术许多领域[3-5]。近十几年以来,氢能源作为一种理想的能源
作为其中一种储氢合金,且它的氢气存储量在得到了很多的关注。同时, AlH
3
理论上可以达到10.0wt.%。因此,国内外的许多科研工作者在这种材料上面不断地进行研究,不论是理论上[6-17]还是实验上[18-29]都取得的很大的成功。
常温常压下的结构性质的研究已经取得了比较全面的成果,截止目前,对AlH
3
发现该材料可以存在至少十个相,分别是α[18]、'α[19]、β[20]、γ[21,22]、δ、ε,ζ,P4/n,Pnma和Pbcm[6]相。其中前面四种结构在理论和实验上得到了相互的正式。但是AlH
这种材料具有独特的热力学性质(受热易分解)[30],
3
在以上的所有相中α相是最稳定的结构,而β和γ都是亚稳态,在受热的情况下
很容易发生相变,变到比较稳定的 相。以上的研究都是出于常压的条件下进行的,对于高压下该材料结构和性质的研究起步于70年代。由于所有材料在足够高的压强下都会变到金属相,那么氢在高压条件下会不会出现高温超导现象?根据BCS理论[31],对于那些氢含量比较高的材料,由于它的质子质量会很小,从而导致很高的徳拜温度,那么就会出现超导现象。我们知道,单纯的氢气只有到250,000个大气压(约25GPa)下,才会呈现出金属的性质[32]。但是对于氢化
物,它的压强要求就有可能没有这么高。这种现象在材料SiH
4[33-36],GeH
4
[37]
和SnH
4[38]都得到了验证。同时,能够得到一些高压下AlH
3
的材料特点,那么
就可以得到有关储氢材料在高压下的稳定性结构。目前,对于AlH
3
高压下的研
究,在国外进行的比较多不管是理论[39,40]或是实验上[41]。其中在理论和实验上也都得到了该材料在高压下具有了金属性的结构:Pm-3n。但是由于受到一些条件的限制,目前在理论上的研究达到的最大压强是175GPa[40],实验上也只有164GPa[41].本文就是在此基础上,利用第一性原理的模拟计算,使理论压
强达到210GPa,通过物态方程的处理,得到了AlH
3
在更高压强条件下的相变以及其稳定的结构。
§1.2高压物理学简介
高压物理学主要研究的是在实验上如何获得高压技术,在理论上分析固体的物态方程(温度,压强和体积的关系),高压相变过程和相变机制,还涉及高压下的材料的物理性能和应用等等。高压物理在合成新型材料,材料的高压加工,材料在高压下的特殊性能等等方面都有着广阔的发展前景。同时,高压物理是在力学,化学,材料科学以及各种实验加工和精密测量技术的支持下发展起来的。尤其是近代高压物理学,它涉及到激光技术,无线电技术,和物理技术,核磁共振以及超声测量等等,因此,高压物理学是一门多学科相互交叉的科学。高压物理学的发展为其它学科的研究和发展开辟了新的方法。例如:实验高压条件下产生的几百万大气压已经接近地心的压力,这为研究地球化学和地质力学提供了有力的依据。高压物理状态方程的研究,是人们看到了高压物理和天体物理之间的联系等等。可以说,高压物理学的不断自身发展的同时也逐渐的渗透到其它学科的发展中。
1.2.1高压在物理学研究中的重要性
在图1.3中,可以看到高压如何改变着物体的微观世界的。
图1.3 高压作用下物体结构的变化
结构相变:在高压下,可使得物质的各态间出现转化,即气态转化为固态。
电子相变:电子具有空间和自旋特征。高压可引起电子在空间分部,这表现为绝缘体到金属转变,亦可引起自旋态转变(如高低自旋态转变)。
因此,同高温效应一样,高压效应也是人们认识物质本质的重要途径。此外,高压研究也使人类得到许多新的,甚至自然界未发现的材料。例如:在高温高压下石墨转变成金刚石;高温高压合成的立方氮化硼具有类似金刚石的晶体结构,它的硬度仅次于金刚石,但耐热性却优于金刚石;高压下加热非晶物质能制得平常难以得到的超导亚稳合金等等。目前,高压科学有涉及以下几个前沿问题:(1)高压下的结晶和非晶相变;(2)压制金属化;(3)压力对超导性和电子——声子相互作用的影响;(4)高压下新型功能材料的设计和制备。
1.2.2高压物理的发展历史
高压物理是今年来发展迅速的一门专业学科,它研究的是物质在高压条件下的物理行为。由于它研究的对象多数是凝聚态物质。因此,高压物理也可以说是在高压极端条件下的凝聚态物理。
高压物理学的发展从1762年坎顿对水的压缩性实验开始;到19世纪末,阿马伽创建了活塞式压力计,从此为高压实验打开了除了研究液体压缩性以外的大门;后来,塔曼研究了固体体积随压力变化的时候出现的不连续现象,得到了熔点和相变点,从此开创了高压相变。但是在高压物理刚开始发展将近150年的时间里,由于受到设备条件的设置,高压物理学的研究范围一直处于5000大气压以内。直到1906年,P.W.布里奇曼(美国人)把高压物理实验带入了一个崭新的时代。他通过技术上的改进和对高压设备材料上的革新,在1952年,他发明了一种完善的高压装置——可使高压稳定在100000大气压。P.W.布里奇曼也因发明产生超高压的装置及利用这些装置在超高压领域的做出的众多贡献,获得了1946年的诺贝尔物理奖。同时,雅各布、劳逊发展了高压下物质 X射线结构分析技术;劳逊与纳赫特里布研究了固体中原子扩散的高压效应。这样,就初步形成了以原子行为为基础的高压物理的研究内容。所以我们说高压物理学是包括了高压的产生和高压下对物体各种物理量的检测,两者都需要专门的,特殊技巧的实验技术和方法。
在高压的产生技术方面,目前主要有静态高压技术和动态高压技术。静态高压是指可以相对长期维持的高压强。所谓相对长期是指有足够的时间,把压缩功所产生的热量通过热传导的方式与环境温度平衡。因此静态高压是等温压缩过程。但是静高压研究中允许使用的试件用量极少,样品很薄,这使得样品测量的信息常常混有加载装置的信息,而对于样品的分析却非常困难。动态高压技术是一种用脉冲加载方法产生高压的技术。它是基于在瞬态脉冲加载下,利用材料惯性响应特性而达到高压的原理,从而避免了静态高压装置中由于制成高压容器材料的强度极限对提高压力带来的困难。采用动态高压技术可以获得远大于静态高压技术所能达到的最高压力水平。但在这么高的压力条件下,试样和装置会彻底损坏,允许进行物理测量的时间又极短,都使得提供物理信息的实验手段受到很大的限制。所以对高压物理实验新方案的探索,也是进一步发展高压物理研究所必须考虑的一个重要问题。
目前,实验上普遍流行的是在静态高压实验装置中采用金刚石对顶砧(DAC)技术,可以获得高达数百GPa(1GPa约等于1万大气压)的压强。在实验中经常将金刚石对顶钻(DAC)技术与中子衍射、拉曼谱和红外谱等实验手段相结合使用。高压物理的发展的同时也为其他科学的发展开辟了新的途径。百万大气压已接近地心压力,高压物理研究为地球化学,地质力学的发展提供了重要的依据。高压
物态方程的研究,使人们看到了高压物理与天体物理的联系。目前,把高压物理和其他学科有机结合,形成了许多交叉学科。如:高压地球物理、高压材料物理、高压化学、高压生物、高压传感等。
虽然高压实验技术在不断地更新与提高,使得产生的压强可以更加的高,但当今的实验技术依旧还是存在很多的局限性。于是高压物理的理论计算的研究作为高压物理研究的重要的组成部分应运而生。于是高压理论和实验相互补充,相互验证又相互促进。从基于第一性原理的理论上探索高压下的稳定结构已有近三十年的历史。
总之,在高压物理学发展的今天,高压研究手段除了用同步辐射结合DAC 技术+X射线衍射、非弹性中子散射、光电子谱等以外,高压物理的理论计算也已经逐渐走向成熟,目前主要理论手段表现在: 第一性原理计算+分子动力学(考虑温度效应),研究热动力学特征、声子谱及力学特征等,程序有:VASP,Abinit,Wien2k等。
第二章理论原理和计算方法
本章将详细的介绍在上一章中提出的对于储氢材料——AlH
在高压条件下
3
相变的研究方法的理论依据。在固体材料分析中所说的第一性原理(the
first-principle theory )是以电子和原子核相互作用为基础的量子力学原理。同时我们将重新来介绍一下会应用到我的文章里面的关于多体问题的概念,以及多体问题的处理方法。在这一章我们主要分两部分对其理论与计算方法进行论述。
第一部分,由于原子核的质量是电子质量的一千倍,引入绝热近似或称玻恩-奥本海默近似(Born-Oppenheimer approximation)[42]。在这个条件下,原子核就看成是一个固定的参数,只需要考虑多电子问题。于是,Hohenberg-Kohn 定理[43]指出只需要电子密度函数就可以来描述基态系统的所有物理性质。后来的密度泛函理论就是建立在HK定理基础之上的。接着,由Kohn和Sham提出了单电子方程[44]:用无相互作用粒子模型来代替有相互作用的粒子系统。我们所用的软件也是在理在以Kohn-Sham方程为基础理论上的。同时,在交换关联势的选择和基矢以及赝势的选择上做简单的分析。由于在我们的工作中考虑的温度这个物理量,因此我们会对从头计算分子动力学(即第一性原理分子动力学方法)做了大概的称述。在第一部分的最后我们简单的介绍了我们所用的软件VASP发展和它所包括的一些重要的文件。
在第二部分由于论文涉及到得是高压下的材料相变情况,因此我们对在高压下的相变进行分类介绍。同时对其高压下的固体物态方程选择性的介绍论文中应用到的三阶Birch-Murnaghan物态方程进行简单的论述。
§2.1 第一性原理概述
第一性原理(First Principle)是与经验参数相对应的一个概念。第一性原理通常情况下是与计算相互联系起来。它与经验参数相对立,在进行计算的时候,只需要告诉程序你所使用的原子类型和它们所处的位置,就可以对材料的各种物理特性进行分析。它并不需要其它的实验的,经验的或者是半经验的参量,它是完全建立在量子力学原理基础上的计算。但是,由于组成物质的粒子系统非常的
庞大,这样势必会导致计算上出现一些问题。因此,我们在实际应用中,通常会加入一些有说服力的假设(例:玻恩-奥本海默近似),那么建立在这些假设之上的第一性原理就称为“半经验的”。
2.1.1 玻恩-奥本海默近似(Born-Oppenheimer approximation ))
对于由N i 个原子核和N e 个电子组成的多粒子体系的薛定谔方程为: ()()()
,,,H r R r R E r R Φ=Φ (1.1) 哈密顿量可以写成: ()
,()()()()(,)i ii e ee ie H r R T R U R T r U r U r R =++++ (1.2) 其中第一项为核的动能,第二项为核与核相互作用能,第三项为电子动能,第四项为电子与电子相互作用能,最后一项则表示电子与原子核的相互作用能。
{}{}R R i i r r 用表示所有电子坐标的集合,用表示所有电原子核坐标的集合。
哈密顿量中各个部分具体形式为:(用原子单位即1e m e
--==) 12T ()2R i R M k k
k =-?∑ (1.3) Z Z k k U ii R R k k k k -'=+∑-'<' (1.4)
21()2i e r i
T r =-?∑ (1.5) 1()ee i j i j
U r r r <=+-∑ (1.6)
,(,)k ie i k i k
Z U r R r R =--∑ (1.7)
由于原子核的质量比电子大很多,因此在电子处于高速运动的中,原子核只是在它们周围的平衡位置附近振动。于是我们可以把()i T R
这一项从哈密顿算附中分离出来作为干扰。于是(1.2)式可写成: ()
,()(,)i e i H r R T R H r R +=+ (1.8) (),()()()(,)e i ii e ee ie H r R U R T r U r U r R +=+++ (1.9) 这时候我们可以把整个问题分成两部分考虑:考虑电子运动时原子核实处在
它们的瞬时位置上,而考虑核的运动的时候则不考虑电子在空间的分布。这就是著名的Born-Oppenheimer approximation(玻恩-奥本海默近似)。在该条件下,
在固体材料方面的分析主要关注电子部分的哈密顿量,于是把原子核的运动当作系统的一个微扰。用微扰方法得出来的波函数误差为()2o k 数量级,能级误差为 ()5o k 数量级,这对大多数半导体和金属来说是足够好的近似[45]。
2.1.2 Hartree-Fock 近似
Hartree 在绝热近似的条件下,得到了多电子的薛定谔方程,并且采用了独
立坐标的表述方式,将系统的波函数用互不相关的单电子波函数的连乘积的形式来表示:即价电子总数为N 的体系的波函数为
12()()()N r r r ψ???=??? (1.10)
()i i r ?表示每一个电子波函数。但是这里没有考虑电子(费米子)交换的反
对称性。于是,Slater 提出了Slater 行列式来表示多电子系统的波函数,这样即采用了Hatree 的单电子近似,同时又考虑了交换反对称性。
112131N 1121N N N ()()()()()()()
r r r r r r r ???ψ????????????????=???
?????????
(1.11) 最后通过变分法处理重新得到单电子近似方程,即Hartree-Fock 方程: 2V ()()(,)()()()eff H F i i i i r r r r V r d r r E r r r ρρ????
??''-??'-?+-=??'-??
????
? 有效势 (1.12) 由于式中与ρ有关的相互作用相里面含有解?,所以可以用自洽迭代求解的方法来求解方程。这就是哈特利-福克自洽场近似方法。
2.1.3 密度泛函理论(The density functional theory )
类似于哈特利-福克方法,密度泛函理论(DFT )也把多点字问题简化为电子
方程。同时,DFT 的表达形式是准确的,没有任何近似过程而且它的有效势是局域的。
与前面所述的寻找多粒子系统的波函数方法相比,DFT 的基本思想是可以用粒子密度函数来描述原子,分子和固体的基本物理量。因此,可以把求解问题从3e N ?空间转换到了3维空间。在接下来的理论部分,我们首先解释怎样把多体
问题转化为只与电子的密度有关。然后给出从无相互作用的多粒子系统推到出这样的密度函数。
1 Hohenberg-Kohn 定理
密度泛函理论的基础是建立在P.Hohenberg 和W.Kohn 在1964年关于非均匀电子气理论基础之上的[46]。HK 定理归结如下:
定理一:不计自旋的全同费米子的基态能量是粒子数密度()r ρ
的唯一泛函。 或:对于一个共同的外部势()ext V r
,相互作用的多粒子系统的所有基态性质都由非简并基态的电子密度分布()r ρ 唯一的决定。
在定理一的基础上可以得出对于任何粒子系和任何外部势,整个系统的基态能量泛函可写为:
[()]()()ext E r V r r dr T U ρρψψ=++?
(1.13)
其中定义:[]F T U ρψψ
=+,是一个与外场没有关系的泛函。
定理二:能量泛函[]E ρ在粒子数不变的条件下对正确的粒子数密度函数()r ρ 取极值,并等于基态能量。
系统的基态能量: []()()[]ext E V r r dr F ρρρ=+? (1.14) 其中: ()()1
[][][]2x c r r F d r d r T E r r ρρρρρ''=++'-??
(1.15) 第一项和第二项分别是非相互作用电子体系的经典Coulomb 排斥项和动能项,第三项为相互作用电子体系的交换关联能。HK 定理已经建立了密度泛函理论的框架,但是在实际应用上遇到了严重的困难。其中包括:1.对于粒子数密度函数()r ρ
的确定问题;2.动能泛函项[]T ρ的确定问题;3.对于交换关联泛函[]x c E ρ的确定问题。对于第一和第二的两个问题,已经由W. Kohn 和J. L. Sham 提出的Kohn-Sham 方程解决。对于第三个问题,为了使DFT 理论能够付诸实施,Kohn-Sham 提出了局域密度近似(Local Density Approximation, LDA )。 2 Kohn-Sham 方程
Kohn-Sham 方程是由W. Kohn 和L. J. Sham 提出的单电子形式的DFT 方程。
根据HK 定理,基态能量和基态粒子数密度函数可以由能量泛函(1.14)式对密度函数()r ρ
的变分得到:即 [()][()](')()()'0()
'()xc E r T r r d r r v r d r r r r r δρδρρδρδρδρ??+++=??-????
(1.16) 由于粒子数不变条件()0d r r δρ=? ,所以得
[()][()]
(')()'()'()xc E r T r r v r d r r r r r δρδρρμδρδρ+++=-? (1.17)
其中μ是系统的化学势,且令有效势形式为:
[()](')()()''()x c e ff E r r V r v r d r r r r δρρδρ=++-? (1.18)
对于未知的[]T ρ动能泛函,W. Kohn 和L. J. Sham 提出:假设动能泛函[]T ρ可以用一个已知的无相互作用粒子的动能泛函[]s T ρ来代替,它也具有与有相互
作用系统同样的密度函数,这是只要把[]s T ρ与[]T ρ的差别中无法转换的复杂部
分并入[()]xc E r ρ 中。此时,交换关联项[()]xc
E r ρ 依旧是未知的。这样就解决了上面的第二个问题。对于问题一中的密度泛函,还是用N 个单粒子波函数()i r ψ
来构造,可以得到:
2
1()()N i i r r ρψ==∑
(1.19) 这样对于无相互作用的粒子的动能函数[]s T ρ可以用密度函数表示为:
*21[]()()()N
s i i i T d r r r ρψψ==-?∑?
(1.20)
此时可以将能量泛函(1.14)式对密度函数()r ρ 的变分转化到对()i r ψ
的变
分,且此时的拉格朗日乘子用i E 来代替可以得到: 2{[()]}()()K S i i i V r r E r ρψψ-?+= (1.21) 其中:
[()]()[()][()]K S C oul xc V r v r V r V r ρρρ=++ (1.22) 且
(')[()]''C o u l r V r d r r r ρρ=
-? (1.23) [()]
[()]()xc xc E r V r r δρρδρ= (1.24)
我们把(1.19),(1.21)和(1.22)三个式子一起成为Kohn-Sham 方程。该方程的形式类似与薛定谔方程,但是,在这里已经把系统从3N 维直接降到了3维。同时,从该方程中,我们看到关于粒子数密度函数()r ρ
和动能泛函项[]T ρ的确定问题已经得到了解决,这里只是把一些复杂的部分放入到交换关联泛函[]x c E ρ中。此时,用DFT 方法来求解系统就可以实现了。 在图2.1中我们给出了具体的求解过程的流程图。在图中虽然给出了DFT 方法求解的具体步骤,但是对于交换关联泛函[]x c E ρ的确定问题依旧存在。在
接下来的部分我们介绍了几种常见的交换关联泛函的处理过程以及如何针对性的对不用的泛函进行选择处理不同的材料问题。
图2.1用 DFT 方法求解的流程图
2.1.4 交换关联泛函
那现在接下来的主要目的就是解决交换关联泛函[]x c E ρ这一项。到目前
为止,由于各个学科发展的需要,提出了集中不用的交换关联函数的选择。这里我们介绍一些常见的交换关联函数形式。
1局域密度近似( Local Density Approximation 简称LDA )
对相关能的最简单的处理方法就是Kohn -Sham 提出的局域密度近似(LDA ),LDA 近似认为,一个电子在电子气r 上时的交换关联能xc E 和在r 处相等的均匀电子气
的交换关联能xc ε相同,即认为总的交换相关能可以表示为: [()][()]()xc xc E r r r dr ρερρ=? (1.25) ()r ρ
表示为无相互作用的均匀电子气密度函数。 根据(1.24)式可得交换关联式为 ()[]()[()]()E
xc V r r xc xc r δδ
ρρερδρδρ== (1.26)
这样就可以把(1.25)式代入Kohn-Sham 方程进行自洽计算。下面列出集中常见的交换关联势的形式:
Kohn-Sham-Gaspar 交换势近似[47]
1/31/32(3/)[()]xc V r πρ=-
Wigner 关联能近似[48] 2/37.79
()0.882(7.79)R V R c R +=-+
因此建立在DFT 上的局域密度近似(LDA ),可以从求解一组单粒子在有效势场中的运动方程而得到次点在密度分布,从而求得固体的基态性质。它比哈特利-福克自洽场近似更为严格,更加精确。因此,LDA 是研究固体能带,界面,表面,低维材料和超晶格材料强而有力的工具。对于大多数的半导体和金属能给出与实验符合良好的价带,同时对于很多的半导体和一些金属的基态物理性质研究,例如:晶格常数,结合能,晶体力学性质等等都能给出与实验值符合很好的结果。但是同时也遇到了一些问题,比如LDA 对于金属d 带宽度以及对于半导体禁带宽度的计算与实验室相差有35%-50%。因此,对于依然存在着 一些缺陷,LDA 方法有待进一步的修正和法展。
以上对于材料的研究都没有涉及到磁性材料的研究,对于磁性晶体以及含有原子序数较大的元素晶体,还应该考虑到交换相互作用中的自旋轨道耦合。于
是要将电子密度分布重新进行定义,按照自旋朝上和朝下两部分表示为:
()()()r r r ρρρ↑↓=+
这种近似方法被称为局域自旋密度近似(Local Spin Density Approximation,简称LSDA )。
2 广义梯度近似(Generalized Gradient Approximation 简称GGA )
交换相关能的局域密度近似(LDA )是交换关联泛函的零级表示,它仅仅采用空间点r 处的电子密度()n r 来决定那点交换-相关能密度的形式。对于交换-相关能密度由密度相同的均匀电子气完全确定,那么泛函的交换部分就准确的用均匀电子气的微分表达。各种不同的局域密度近似(LDA )仅仅是相关部分表示方法不同,所有现代应用的局域密度泛函都基于Ceperly 和Alder’s 在80年代对均匀电子气总能量的Monte Carlo 模拟。GGA 是Jacob 阶梯[49]的第二个台阶,属于一级校正,它不仅与电子密度有关,还与电子密度的空间变化有关。GGA泛函包含了两个主要的方向:一个称为“无参数”,泛函中新的参数通过已知形式中参数或在其它准确理论帮助下得到。另外一个就是经验方法,未知参数来自于对实验数据的拟和或通过对原子和分子性质准确的计算。Perdew,Burke and Emzerhof(PBE)[50]以及Perdew-Wang 1991( PW91)[51,52]是无参数的。在量子化学中广泛采用的GGA ,比如Becke,Lee,Parr and Yang(BLYP)是经验性。LYP 校正采用了密度的二阶Laplace 算符,因此严格上讲属于Jacob 阶梯的第三阶,但通常仍然归类为GGA 。
在实际应用中,是选择LDA 还是GGA 主要是依赖于模拟结果与实验结果哪个符合的比较好。一般来说,LDA 比较适合密度系数变化比较缓慢的体系(例金属),高密度的体系(例过渡金属)以及大多数的结构优化过程。对于GGA 比较适合模拟结合能或粘附能方面。所以,对于选择哪一种方法来进行拟合,主要是依靠材料的本身的一些性质来决定的[53]。
2.1.5基组的近似和选择 即使在经过上面的各种近似之后,虽然理论上已经可以实现DFT 的求解。在理论原则上对于粒子数密度函数(基矢的选择),所用的函数可以是任一组完备函数集合。但这种完备集合是无穷大的。因此,我们不得不对其进行近似处理。随着时间推移人们倾向的模拟晶体的计算方法是不断变化的。下面简单介绍几种常见的基函数方法。
首先,对于基函数的展开,可以使用一组完备基组展开为:
(,,)()(,)nl lm r R r Y ψθφθφ=
其中:()n l R r 是径向部分,(,)lm Y θφ表示角度部分(s,p,d,f...),对于角度部分
由轨道符号直接确定, 不需要给定, 而且把原子的轨道取为球形的, 包括所有的分量, 比如对p 必然包括了,,x y z p p p 三个轨道, 其取向就是原子在笛卡儿坐
标中的取向。在计算程序中,对于不同的基函数方法主要体现在径向部分。
下面列举三种基函数选取方法以及给出它们对应的径向部分。
原子轨道(STO):
1(0)n r N r e ζζ--?=-∞
Gaussian 函数(GTF): 2exp()(0)N r αα?-=-∞
平面波函数(PW): (主要用于固体的计算)
exp()(0)ip r p -?=-∞
由于在实际应用上不能取到无穷大,所以可以使用的基组主要有原子轨道函数的变形Slater 型轨道,Gaussian 型轨道以及对于周期性体系的计算经常用到的平面波基矢。
2.1.6赝势方法(Pseudo-Potential Method )
在实际的固体中原子核附近库仑吸引作用是周期势场偏离平均值很远。在离子实内部势场对电子波函数的影响是很大的,电子波函数变化非常的厉害。这样,势场就不能被看成是很小的微扰势场。但是在近自由电子模型中假定周期势场的起伏是很小的。这样的矛盾必须用引进赝势这个概念来解决。
所谓赝势,即表示在离子实的内部用假想的势能来代替真实的势能,在求解薛定谔方程的时候,不改变能量本征值和离子实之间区域的波函数。这个假想的势能就称为赝势。其基本思想是,所研究体系的哈密顿算符仅显含价电子部分,而将原子内层的全部电子连同原子核构成的核实对外层价电子的作用用适当的赝势表示,同时引入表示投影算符的势以便将价电子波函数与内层电子波函数分离开来,然后对价电子进行变分并用自洽迭代处理,计算出价电子轨道波函数和
能级值等。所引入的两个势均通过全电子的原子从头计算确定,随后用于分子计算。此时,由赝势求出的波函数称为赝波函数。如图2.2所示:
图2.2 真实势()V r ,电子波函数()r ψ,赝势()W r ,赝势波函数()r ?
从图中可以看出来在离子实之间的区域,真实的势和赝势给出同样的波函数。并且可以看出赝势波函数的与真实电子波函数相比更加的光滑 。
在实际程序设计和应用中,经常会把赝势方法和基函数相结合,可以得到一些常见的模拟计算方法。例如:在固体材料分析中最常见的把赝势和平面波基函数相结合得到平面波赝势方法。赝势方法也渐渐地得到不同类型的表示。例如:模型赝势,模守恒赝势(NCPP )[54]和建立在第一性原理上的超软赝势等等。其中,我们所用的软件VASP[55]主要运用的是超软赝势。
2.1.7 从头计算分子动力学(ab-initio molecular dynamical )
由于本文涉及到用VASP 软件来模拟在一定温度条件下的结构分析,因此我采用了从头计算分子动力学方法来模拟。该方法其实于1985年,由Car 和Parrinello[60]提出。一次也称该方法为CP 方法。在此之前,人们是利用完全的经典牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系综中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。CP 方法是在传统的分子动力学方法中引入了电子的虚拟动力学。
CP 方法提出了一个全新的求解牛顿运动方程的方法,它要求在求解牛顿运动方程的又要求解出波函数的方程,同时要求解针对原子坐标的运动方程。从头计算分子动力学把电子和核的自由度作了统一的考虑,开创性的把密度泛函理论
和分子动力学有机的结合起来。该理论的实现可以通过图 2.2来体现其基本轮廓。
图2.2 从头计算分子动力学流程图
2.1.8 VASP简介
VASP是Vienna Ab-initio Simulation Package的缩写[55,56,57],它是使用赝势和平面波基组,进行从头量子力学分子动力学计算的软件包,它基于CASTEP 1989版开发。VAMP/VASP中的方法基于有限温度下的局域密度近似(用自由能作为变量)以及对每一MD步骤用有效矩阵对角方案和有效Pulay混合求解瞬时电子基态。这些技术可以避免原始的Car-Parrinello 方法存在的一切问题,而后者是基于电子、离子运动方程同时积分的方法。离子和电子的相互作用超缓Vanderbilt赝势(US-PP)或投影扩充波(PAW)方法描述。两种技术都可以相当程度地减少过渡金属或第一行元素的每个原子所必需的平面波数量。力与张量可以用VAMP/VASP很容易地计算,用于把原子衰减到其瞬时基态中。
VASP 包括四个主要的输入文件:计算控制参数文件(INCAR),描述体系结构文件(POSCAR),K点取样设置文件(KPOINTS)和赝势文件(POTCAR)。用时也包括几个主要的输出文件:最主要的输出文件(OUTCAR),电子态密度文件(DOSCAR),波函数文件(WAVECAR),原子迟豫或MD后的体系结构文件(CONTCAR)等等。
本文在采用VASP进行模拟计算时,所采取的一些计算方法设计如下:交换关联能——广义梯度近似GGA-PW91;用PAW来代替离子和电子之间的相互作用;布里渊区的积分用Monkhost-Pack方法【58】。
§2.2 高压物理理论基础
由于物体(这里主要之固体)是由大量的原子或者分子组成的凝聚体,在高压的作用下,凝聚体的体积必将会缩小,原子或分子之间的距离会发生缩短的现象。从而使物体的内部结构发生重组,得到新的相或者新的材料。那么在宏观上则表现为温度,压强和体积的变化,物体的状态方程则是高压物理学所关心的基本问题之一。一般情况下,我们通常会给出等温状态方程:即表示在一定温度条件下物质体积和压强之间的关系式。因为等温状态方程即能表征物质的重要的热力学性质,同时又能体现组成的原子或分子在相互接近的过程中的变化信息。目前,在不同的温度范围内,物质的状态方程有不用的固体状态方程(P-V关系式)。
2.2.1相变理论
相变的根源是多粒子相互作用的结果,是热运动与相互作用竞争的一个结
第一性原理计算原理和方法
第二章 计算方法及其基本原理介绍 化学反应的本质就是旧键的断裂与新建的形成,参与成键原子的电子壳层重新组合就是导致生成稳定多原子化学键的明显特征。因此阐述化学键的理论应当描写电子壳层的相互作用与重排,借助求解满足适当的Schrodinger 方程的波函数描写分子中电子分布的量子力学,为解决这一问题提供了一般的方法,然而,对于一些实际的体系,不引入一些近似,就不可能求解其Schrodinger 方程。这些近似使一般量子力学方程简化为现代电子计算机可以求解的方程。这些近似与关于分子波函数的方程形成计算量子化学的数学基础。 2、1 SCF-MO 方法的基本原理 分子轨道的自洽场计算方法 (SCF-MO)就是各种计算方法的理论基础与核心部分,因此在介绍本文计算工作所用方法之前,有必要对其关键的部分作一简要阐述。 2、1、1 Schrodinger 方程及一些基本近似 为了后面介绍各种具体在自洽场分子轨道(SCF MO)方法方便,这里将主要阐明用于本文量子化学计算的一些重要的基本近似,给出SCF MO 方法的一些基本方程,并对这些方程作简略说明,因为在大量的文献与教材中对这些方程已有系统的推导与阐述[1-5]。 确定任何一个分子的可能稳定状态的电子结构与性质,在非相对论近似下,须求解 R AB =R 图2-1分子体系的坐标
定态Schrodinger 方程 ''12121212122ψψT p B A q p A p pA A pq AB B A p A A A E R Z r R Z Z M =??????? ?-++?-?-∑∑∑∑∑∑≠≠ (2、1) 其中分子波函数依赖于电子与原子核的坐标,Hamilton 算符包含了电子p 的动能与电子p 与q 的静电排斥算符, ∑∑≠+?-=p q p pq p e r H 12121?2 (2、2) 以及原子核的动能 ∑?-=A A A N M H 2121? (2、3) 与电子与核的相互作用及核排斥能 ∑∑≠+-=p A B A AB B A pA A eN R Z Z r Z H ,21? (2、4) 式中Z A 与M A 就是原子核A 的电荷与质量,r pq =|r p -r q |,r pA =|r p -R A |与R AB =|R A -R B |分别就是电子p 与q 、核A 与电子p 及核A 与B 间的距离(均以原子单位表示之)。上述分子坐标系如图2、1所示。可以用V(R,r)代表(2、2)-(2、4)式中所有位能项之与 ∑∑∑-+=≠≠p A pA A B A q p pq AB B A r Z r R Z Z r R V ,1 2121),( (2、5) 原子单位 上述的Schrodinger 方程与Hamilton 算符就是以原子单位表示的,这样表示的优点在于简化书写型式与避免不必要的常数重复计算。在原子单位的表示中,长度的原子单位就是Bohr 半径
第一性原理简介
第一性原理是什么 第一性原理怎么用 1什么是第一性原理 根据原子核和电子互相作用的原理及其基本运动规律,运用,从具体要求出发,经过一些近似处理后直接求解的算法,称为第一性原理。广义 的第一原理包括两大类,以Hartree-Fock自洽场计算为基础的从头算和 (DFT计算。 从定义可以看出第一性原理涉及到量子力学、、Hartree-Fock自洽场、等许多对我来说很陌生的物理化学定义。因此我通过向师兄请教和上网查资料一点点的了解并学习这些知识。 2第一性原理的作用 以密度泛函理论(DFT)为基础以及在此基础上发展起来的简单而具有一定精度的局域密度近似(LDA)和广义梯度近似(GGA)的第一性原理电子结构计算方法,与传统的解析方法一样,不但能够给出描述体系微观电子特性的物理量如波函数、态密度、费米面、电子间互作用势等,以及在此基础上所得到的体现体系宏观物理特性的参量如结合能、电离能、比热、电导、光电子谱、穆斯堡尔谱等等,而且它还可以帮助人们预言许多新的
物理现象和物理规律。密度泛函计算的一些结果能够与实验直接进行比较一些应用程序的发展乃至商业软件的发布,导致了基于密度泛函理论的第 一原理计算方法的广泛应用。 密度泛函理论(DFT)为第一性原理中的一类,在物理系、化学、材料科学以及其他工程领域中,密度泛函理论(DFT及其计算已经快速发展成 为材料建模模拟的一种“标准工具”。 密度泛函理论可以计算预测固体的晶体结构、晶格参数、能带结构、态密度(DOS、光学性能、磁性能以及原子集合的总能等等。 3第一性原理怎么用 目前我所学到的利用第一性原理的软件为Material Studio 、VASP软件。其中Materials Studio (简称MS是专门为材料科学领域研究者幵发的一款可运行在PC上的模拟软件。使化学及材料科学的研究者们能更方便地建立三维结构模型,并对各种晶体、无定型以及高分子材料的性质及相关过程进行深入的研究。模拟的内容包括了催化剂、聚合物、固体及表面、晶体与衍射、化学反应等材料和化学研究领域的主要课题。 模块简介 Materials Studio 采用了大家非常熟悉的Microsoft标准用户界面, 允许用户通过各种控制面板直接对计算参数和计算结果进行设置和分析。 目前,Materials Studio 软件包括如下功能模块: Materials Visualizer: 提供了搭建分子、晶体及高分子材料结构模型所需要的所有工具,可以操作、观察及分析结构模型,处理图表、表格或文本等形式的数据,并提供软件的基本环境和分析工具以及支持Materials Studio 的其他产品。是Materials Studio 产品系列的核心模块。 Discover: Materials Studio 的分子力学计算引擎。使用多种分子力学和动力学 方法,以仔细推导的力场作为基础,可准确地计算出最低能量构型、分子体系的结构和动力学轨迹等。
第一性原理计算原理和方法精编
第一性原理计算原理和 方法精编 Document number:WTT-LKK-GBB-08921-EIGG-22986
第二章 计算方法及其基本原理介绍 化学反应的本质是旧键的断裂和新建的形成,参与成键原子的电子壳层重新组合是导致生成稳定多原子化学键的明显特征。因此阐述化学键的理论应当描写电子壳层的相互作用与重排,借助求解满足适当的Schrodinger 方程的波函数描写分子中电子分布的量子力学,为解决这一问题提供了一般的方法,然而,对于一些实际的体系,不引入一些近似,就不可能求解其Schrodinger 方程。这些近似使一般量子力学方程简化为现代电子计算机可以求解的方程。这些近似和关于分子波函数的方程形成计算量子化学的数学基础。 SCF-MO 方法的基本原理 分子轨道的自洽场计算方 法(SCF-MO)是各种计算方法的理论基础和核心部分,因此在介绍本文计算工作所用方法之 前,有必要对其关键的部分作 一简要阐述。 Schrodinger 方程及一些基本近似 为了后面介绍各种具体在自洽场分子轨道(SCF MO)方法方便,这里将主要阐明用于本文量子化学计算的一些重要的基本 R AB =R 图2-1分子体系的坐标
近似,给出SCF MO 方法的一些基本方程,并对这些方程作简略说明,因为在大量的文献和教材中对这些方程已有系统的推导和阐述[1-5]。 确定任何一个分子的可能稳定状态的电子结构和性质,在非相对论近似下,须求解定态Schrodinger 方程 ''12121212122ψψT p B A q p A p pA A pq AB B A p A A A E R Z r R Z Z M =??????? ?-++?-?-∑∑∑∑∑∑≠≠ () 其中分子波函数依赖于电子和原子核的坐标,Hamilton 算符包含了电子p 的动能和电子p 与q 的静电排斥算符, ∑∑≠+?-=p q p pq p e r H 12121?2 以及原子核的动能 ∑?-=A A A N M H 2121? 和电子与核的相互作用及核排斥能 ∑∑≠+-=p A B A AB B A pA A eN R Z Z r Z H ,21? 式中Z A 和M A 是原子核A 的电荷和质量,r pq =|r p -r q |,r pA =|r p -R A |和R AB =|R A -R B |分别是电子p 和q 、核A 和电子p 及核A 和B 间的距离(均以原子单位表示之)。上述分子坐标系如图所示。可以用V(R,r)代表-式中所有位能项之和 ∑∑∑-+=≠≠p A pA A B A q p pq AB B A r Z r R Z Z r R V ,12121),( 原子单位
如何分析能带图及第一性原理的计算
分析能带图 能带结构是目前采用第一性原理(从头abinitio)计算所得到的常用信息,可用来结合解释金属、半导体和绝缘体的区别。能带可分为价带、禁带和导带三部分,倒带和价带之间的空隙称为能隙,基本概念如图所示: 如何能隙很小或为0 ,则固体为金属材料,在室温下电子很容易获得能量而跳跃至传倒带而导电;而绝缘材料则因为能隙很大(通常大于9电子伏特),电子很难跳跃至传导带,所以无法导电。一般半导体材料的能隙约为1至3电子伏特,介于导体和绝缘体之间。因此只要给予适当条件的能量激发,或是改变其能隙之间距,此材料距能导电。 能带用来定性地阐明了晶体中电子运动的普遍特点。价带(valence band),或称价电带,通常指绝对零度时,固体材料里电子的最高能量。在导带(conduction band)中,电子的能量范围高于价带,而所有在传导带中的电子均可经由外在的电
场加速而形成电流。对与半导体以及绝缘体而言,价带的上方有一个能隙(band gap),能隙上方的能带则是传导带,电子进入传导带后才能在固体材料内自由移动,形成电流。对金属而言,则没有能隙介于价带与传导带之间,因此价带是特指半导体与绝缘体的状况。 费米能级(fermi level)是绝对零度下的最高能级。根据泡利不相容原理,一个量 子态不能容纳两个或两个以上的费米子(电子),所以在绝度零度下,电子将从低到高依次填充各能级,除最高能级外均被填满,形成电子态的“费米海”。“费米海” 中每个电子的平均能量为(绝对零度下)为费米能级的3/5。海平面即是费米能级。一般来说,费米能级对应态密度为0的地方,但对于绝缘体而言,费米能级就位于价带顶。成为优良电子导体的先决条件是费米能级与一个或更多的能带相交。 能量色散(dispersion of energy)。同一个能带内之所以会有不同能量的量子态, 原因是能带的电子具有不同波向量(wave vector),或是k-向量。在量子力学中, k-向量即为粒子的动量,不同的材料会有不同的能量-动量关系(E-K relationship)。能量色散决定了半导体材料的能隙是直接能隙还是间接能隙。如导带最低点与价带最高点的K值相同,则为直接能隙,否则为间接能隙。 能带的宽度。能带的宽度或三度,即能带最高和最低能级之间的能量差,是一个非常重要的特征,它是由相互作用的轨道之间的重叠来决定的,因而反应出轨道之间的重叠情况,相邻的轨道之间重叠越大,带宽就越大。
第一性原理
第二章 第一性原理计算方法与软件介绍 19世纪末,科学家们发现经典力学和经典电动力学在描述物质的微观系统时存在明显不足,对实验中的许多现象不能做出真正合理的解释。鉴于此,20世纪初物理学家们在旧量子论的基础上建立了量子力学,主要研究原子、分子、凝聚态物质等内部微观粒子的结构、运动规律等性质,目前已广泛应用于物理、化学、材料等学科领域。随着量子力学理论的不断完善,并结合日趋成熟的计算机技术,量子计算模拟成为了现代科学中必不可少的研究手段之一。第一性原理计算(First-principles calculation),亦称为从头算(Ab-initio calculation)。该计算方法可根据量子力学基本原理,基于密度泛函理论对材料微观体系的状态和性质进行理论上的预测,且计算过程中不需要使用任何经验参数,只需要一些基本物理量(电子电荷质量e 、电子静止质量m 0、光速c 、普朗克常数h 、波尔兹曼常数k B )。本工作所选用的计算程序为Materials Studio 软件中的CASTEP 量子力学模块,该模块是基于密度泛函理论的从头算量子力学程序。本章节将简要的介绍密度泛函理论和CASTEP 计算模块。 2.1密度泛函理论概述 第一性原理主要的研究对象是多原子体系。它依据量子力学原理,且在无任何实验参数引入的情况下,将多原子体系当作由自由电子和原子核组成的多粒子体系进行处理。然而,关于量子力学中多粒子体系处理的出发点则为著名的薛定谔方程(Schr?dinger Equation)。Schr?dinger 方程是量子力学的一个基本方程,也是第一性原理计算方法的核心,它是由奥地利物理学家薛定谔(Schr?dinger)于1926年提出的。该方程可用于描述微观粒子的运动规律,故亦被称为薛定谔波动方程(Schr?dinger Wave Equation),其定态方程描述如下: 2 2[()]()(,)2V r r,t i r t t ψψμ?-?+=? (2-1) 式中?为约化普朗克(Plank)常数;μ和V(r)分别表示粒子质量和势场;r 和t 则为体系中所有电子与原子核的位置坐标;Ψ(r,t)是系统波函数,即运动的微观粒子
第一性原理简介
第一性原理是什么? 第一性原理有什么用? 第一性原理怎么用? 怎样将第一性原理与实 践结合起来? 什么是第一性原理?1原理,量子力学根据原子核和电子互相作用的原理及 其基本运动规律,运用第一性称为经过一些近似处理后直接求解薛定谔方程的算法,从具体要求出发,计算为基础的从头算。广义的第一原理包括两大类,以
Hartree-Fock自洽场原理DFT)计算。密度泛函理论和(自从定义可以看出第一性原理涉及到量子力学、薛定谔方程、Hartree-Fock因此我通过向师兄密度泛函理论等许多对我来说很陌生的物理化学定义。洽场、请教和上网查资料一点点 的了解并学习这些知识。 2第一性原理的作用为基础以及在此基础上发展起 来的简单而具有一定精(DFT)以密度泛函理论,的第一性原理电子结构计算方法 和广义梯度近似(GGA)度的局域密度近似(LDA)不但能够给出描述体系微观电子特性的物理量如波函与传统的解析方法一样,以及在此基础上所得到的体现体系宏,数、态密度、费米面、电子间互作用势等,穆斯堡尔谱等等比热、电导、观物理特性的参量如结合能、电离能、光电子谱、密度泛函计算的一些而且它还可以帮助人们预言许多新的物理现象和物理规律。. 导致了,结果能够与实验直接进行比较,一些应用程序的发展乃至商业软件的发布基于密度泛函理论的第一原理计算方法的广泛应用。为第一性原理中的一类,在物理系、化学、材料科学以(DFT)密度泛函理论)及其计算已经快速发展成为材料建模DFT及其他工程领域中,密度泛函理论(模拟的一种“标准工具”。密度泛函理论可以计算预测固体的晶体结构、晶格参数、能带结构、态密度(DOS)、 光学性能、磁性能以及原子集合的总能等等。 3第一性原理怎么用?其中ASP、软件。V目前我所学到的利用第一性原理的软件为Material Studio)是专门为材料科学领域研究者开发的一款可运行在MSMaterials Studio(简称使化学及材料科学的研究者们能更方便地建立三维结构模型,上的模拟软件。PC模拟无定型以及高分子材料的性质及相关过程进行深入的研究。并对各种晶体、的内容包括了催化剂、聚合物、固体及表面、晶体与衍射、化学反应等材料和化学研究领域的主要课题。模块简介Materials Studio采用了大家非常熟悉的Microsoft标准用户界面,允许用户通过各种控制面板直接对计算参数和计算结果进行设置和分析。目前,Materials Studio软件包括如下功能模块: Materials Visualizer: 提供了搭建分子、晶体及高分子材料结构模型所需要的所有工具,可以操作、观察及分析结构模型,处理图表、表格或文本等形式的数据,并提供软件的基本环境和分析工具以及支持Materials Studio的其他产品。是Materials Studio产品系列的核心模块。 Discover: Materials Studio的分子力学计算引擎。使用多种分子力学和动力学方法,以仔细推导的力场作为基础,可准确地计算出最低能量构型、分子体系的结构和动力学轨迹等。. COMPASS: 支持对凝聚态材料进行原子水平模拟的功能强大的力场。是第一个由凝聚态性质以及孤立分子的各种从头算和经验数据等参数化并经验证的从头算力场。可以在很大的温度、压力范围内精确地预测孤立体系或凝聚态体系中各种分子的结构、构象、振动以及热物理性质。 Amorphous Cell: 允许对复杂的无定型系统建立有代表性的模型,并对主要性质进行预测。通过观察系统结构和性质之间的关系,可以对分子的一些重要性质有更深入的了解,从
第一性原理计算
实验一、第一性原理计算 1. 实验目的 (1) 掌握第一性原理和密度泛涵的计算方法; (2) 学会使用Visualizer 的各种建模和可视化工具; (3) 熟悉CASTEP 模块的功能。 2. 实验原理 CASTEP 是基于密度泛涵理论平面波赝势基础上的量子力学计算。 密度泛涵理论的基本思想是原子、分子和固体的基本物理性质可以用粒子密度函数进行描述。可以归纳为两个基本定理: 定理1:粒子数密度函数是一个决定系统基态物理性质的基本参量。 定理2:在粒子数不变的条件下能量对密度函数变分得到系统基态的能量。不计自旋的全同费米子的哈密顿量为:H T U V =++ 其中动能项为:()()T dr r r ψψ+=??? 库仑作用项为:11'()(')()(')2 ' U drdr r r r r r r ψψψψ++=-? V 为对所有粒子均相同的局域势u(r)表示的外场影响:()()()V dru r r r ψψ+=?粒子数密度函数为: ()()()r r r ρψψ+=ΦΦ 对于给定的()r υ,能量泛函[]E ρ定义为: []()()E dr r r T U ρυρ=+Φ+Φ ?;[]F T U ρ=Φ+Φ系统基态的能量: ' ''''[]''''[][]()()[][]()()[] E T U V G E F dr r r E G G F dr r r E G ρρυρφρυρρΦ=Φ+Φ+ΦΦ==+>?=+=? 3. 实验内容 材料的电子结构计算; 4. 实验设备和仪器 (1) 硬件:多台PC 机和一台高性能计算服务器。 软件:主要利用Materials studio 软件包里的Materials Visualizer 和CASTEP 模块 5. 实验步骤
第一性原理计算原理和方法
第二章 计算方法及其基本原理介绍 化学反应的本质是旧键的断裂和新建的形成,参与成键原子的电子壳层重新组合是导致生成稳定多原子化学键的明显特征。因此阐述化学键的理论应当描写电子壳层的相互作用与重排,借助求解满足适当的Schrodinger 方程的波函数描写分子中电子分布的量子力学,为解决这一问题提供了一般的方法,然而,对于一些实际的体系,不引入一些近似, 确定任何一个分子的可能稳定状态的电子结构和性质,在非相对论近似下,须求解定态Schrodinger 方程 ''12121212122 ψψT p B A q p A p pA A pq AB B A p A A A E R Z r R Z Z M =??? ?????-++?-?-∑∑∑∑∑∑≠≠ (2.1) 其中分子波函数依赖于电子和原子核的坐标,Hamilton 算符包含了电子p 的动能和电子p
与q 的静电排斥算符, ∑∑≠+?-=p q p pq p e r H 12121?2 (2.2) 以及原子核的动能 ∑?-=A A A M H 2? (2.3) 和电子与核的相互作用及核排斥能 ∑∑≠+-=p A B A AB B A pA A eN R Z Z r Z H ,21? (2.4) 式中Z A 和M A 是原子核A 的电荷和质量,r pq =|r p -r q |,r pA =|r p -R A |和R AB =|R A -R B |分别是电子p 和q 、核A 和电子p 及核A 和B 间的距离(均以原子单位表示之)。上述分子坐标系如图2.1所示。可以用V(R,r)代表(2.2)-(2.4)式中所有位能项之和 ∑∑∑-+= ≠≠p A pA A B A q p pq AB B A r Z r R Z Z r R V ,1 2121),( (2.5) 原子单位 上述的Schrodinger 方程和Hamilton 算符是以原子单位表示的,这样表示的优点在于简化书写型式和避免不必要的常数重复计算。在原子单位的表示中,长度的原子单位是Bohr 半径 能量是以Hartree 为单位,它定义为相距1Bohr 的两个电子间的库仑排斥作用能 质量则以电子制单位表示之,即定义m e =1 。
第一性原理
第一章引言 在21世纪的今天,全球都面对着资源的短缺和环境的污染这两大问题。氢能源的出现,不仅仅解决了能源短缺的问题(氢能源是二次能源),同时氢能源的使用对环境几乎是没有任何的污染(氢气和氧气的反应产物只有水)。因此,从上个世纪70年代就开始关注氢能源的研发。到21世纪的今天氢能源也逐渐开始走上舞台,但是对于氢能源在应用过程中会出现的问题也亟待解决。 本章内容之一将主要介绍氢能源应用中面临的一个严峻的问题——氢气的 。其二,存储,我们将会详细的论述最新出现的一种储氢材料:储氢合金——AlH 3 简述历年来在实验和理论上对于该材料在常压下的研究成果。同时,提出对于在高压条件下进行研究的必要性以及在现阶段的成果,指出我们理论研究的AlH 3 必要性。最后,将简单的介绍高压物理学在当今学科发展中的重要性以及高压物理的发展历史,当然我们将会简述由于现实实验条件上的限制,高压物理的理论研究对于材料性能的分析和高压物理未来发展方向上的重要性和必然性。 §1.1储氢材料简介 伴随着人类社会的发展和进步,人类赖以生存的环境却让全世界都开始担忧。环境的破坏的危机以及能源的短缺的意识,迫使人们一方面去寻求新的能源,另一方面又要考虑新能源对环境所造成的破坏问题。于是氢能源作为存储量丰富,无公害,无污染的新型能源而得到了全球的关注。在以氢作为能源媒介的氢能体系中,氢的存储和氢的运输成为氢能源的实际应用中的关键环节。近年来,人们注意到储氢合金由于其材料结构上的优势而成为一种新型的储氢功能材料。由于某些合金具备特殊的晶体结构,能够使氢原子很容易的进入晶格间隙中并且形成金属氢化物,由于这种氢与金属的结合力很弱,在加热的时候,氢就能从金属中释放出来。但是这些储氢合金的储氢量很大,可以存储比其自身体积要大上1000-1300倍的氢。目前,对于储氢合金的研究也进行的如火如荼。 1.1.1氢能 随着全球人口急增,人类的能源消耗大幅度的增长;而作为主要能源的煤炭和石油,它们又都是不可再生的能源,其储量极为有限。另外,大量矿物能源的燃烧,造成大气污染、"酸雨"和"温室效应"等环境问题。因此,从20世纪60年代以来,人类为了解决未来能源的供应和生存环境问题,高呼"能源革命"。"
第一性原理计算方法讲义
第一性原理计算方法 引言 前面讲述的有限元和有限差分等数值计算方法中,求解的过程中需要知道一些物理参量,如温度场方程中的热传导系数和浓度场方程中的扩散系数等,这些参量随着材料的不同而改变,需要通过实验或经验来确定,所以这些方法也叫做经验或者半经验方法。而第一性原理计算方法只需要知道几个基本的物理参量如电子质量、电子的电量、原子的质量、原子的核电荷数、布朗克常数、波尔半径等,而不需要知道那些经验或半经验的参数。第一性原理计算方法的理论基础是量子力学,即对体系薛定额方程的求解。 量子力学是反映微观粒子运动规律的理论。量子力学的出现,使得人们对于物质微观结构的认识日益深入。原则上,量子力学完全可以解释原子之间是如何相互作用从而构成固体的。量子力学在物理、化学、材料、生物以及许多现代技术中得到了广泛的应用。以量子力学为基础而发展起来的固体物理学,使人们搞清了“为什么物质有半导体、导体、绝缘体的区别”等一系列基本问题,引发了通讯技术和计算机技术的重大变革。目前,结合高速发展的计算机技术建立起来的计算材料科学已经在材料设计、物性研究方面发挥着越来越重要的作用。 但是固体是具有?1023数量级粒子的多粒子系统,具体应用量子理论时会导致物理方程过于复杂以至于无法求解,所以将量子理论应用于固体系统必须采用一些近似和简化。绝热近似(Born-Oppenheimei 近似)将电子的运动和原子核的运动分开,从而将多粒子系统简化为多电子系统。Hartree-Fock 近似将多电子问题简化为仅与以单电子波函数(分子轨道)为基本变量的单粒子问题。但是其中波函数的行列式表示使得求解需要非常大的计算量;对于研究分子体系,他可以作为一个很好的出发点,但是不适于研究固态体系。1964年,Hohenberg和Kohn提出了严格的 密度泛函理论(Density Functional Theory, DFT )。它建立在非均匀电子气理论基础之上,以粒子数密度(『)作为基本变量。1965年,Kohn和Sham提出Kohn-Sham方程将复杂的多电子问题及其对应的薛定谔方程转化为相对简单的单电子问题及单电子Kohn-Sham方程。将精确的密度泛函理 论应用到实际,需要对电子间的交换关联作用进行近似。局域密度近似(LDA、广义梯度近似(GGA 等的提出,以及以密度泛函理论为基础的计算方法(赝势方法、全电子线形缀加平面波方法(FLAPW)等、的提出,使得密度泛函理论在化学和固体物理中的电子结构计算取得了广泛的应用,从而使得固体材料的研究取得长足的进步。 第一性原理计算方法的应用 1、体系的能量
第一节第一性原理计算方法综述
第一性原理计算的理论方法 随着科技的发展,计算机性能也得到了飞速的提高,人们对物理理论的认识也更加的深入,利用计算机模拟对材料进行设计已经成为现代科学研究不可缺少的研究手段。这主要是因为在许多情况下计算机模拟比实验更快、更省,还得意于计算机模拟可以预测一些当前实验水平难以达到的情况。然而在众多的模拟方法中,第一性原理计算凭借其独特的精度和无需经验参数而得到众多研究人员的青睐,成为计算材料学的重要基础和核心计算。本章将介绍第一性原理计算的理论基础,研究方法和ABINIT 软件包。 1.1第一性原理 第一性原理计算(简称从头计算,the abinitio calculation),指从所要研究的材料的原子组分出发,运用量子力学及其它物理规律,通过自洽计算来确定指定材料的几何结构、电子结构、热力学性质和光学性质等材料物性的方法。基本思想是将多原子构成的实际体系理解成为只有电子和原子核组成的多粒子系统,运用量子力学等最基本的物理原理最大限度的对问题进行”非经验”处理。【1】第一性原理计算就只需要用到五个最基本的物理常量即(b o k c h e m ....)和元素周期表中各组分元素的电子结构,就可以合理地预测材料的许多物理性质。用第一性原理计算的晶胞大小和实验值相比误差只有几个百分点,其他性质也和实验结果比较吻合,体现了该理论的正确性。
第一性原理计算按照如下三个基本假设把问题简化: 1.利用Born-Oppenheimer 绝热近似把包含原子核和电子的多粒子问题转化为多电子问题。 2.利用密度泛函理论的单电子近似把多电子薛定谔方程简化为比较容易求解的单电子方程。 3.利用自洽迭代法求解单电子方程得到系统基态和其他性质。 以下我将简单介绍这些第一性原理计算的理论基础和实现方法:绝热近似、密度泛函理论、局域密度近似(LDA)和广义梯度近似(GGA)、平面波及赝势方法、密度泛函的微扰理论、热力学计算方法和第一性原理计算程序包ABINIT 。 1.2量子力学与Born-Oppenheimer 近似 固体是由原子核和核外的电子组成的,在原子核与电子之间,电子与电子之间,原子核与原子核之间都存在着相互作用。从物理学的角度来看,固体是一个多体的量子力学体系【2】,相应的体系哈密顿量可以写成如下形式: ),(),(R r E R r H H ψψ= (1-1) 其中r,R 分别代表所有电子坐标的集合、所有原子核坐标的集合。在不计外场作用下,体系的哈密顿量日包括体系所有粒子(原子核和电子)的动能和粒子之间的相互作用能,即 N e N e H H H H -++= (1-2) 其中,以是电子部分的哈密顿量,形式为:
第一性原理计算
钙钛矿型PbZrO3电子能带结构的第一性原理计算 班级:s1467 姓名:学号:201421801014 锆酸铅(PbZrO3)是最早发现的反铁电体之一,在工业上的一个重要应用是其固溶物Pb(Zr,Ti)O3。由于反铁电材料在相开关、电荷存储、电流源、电容、微电子及微型机电设备等方面有重要应用,其电子结构和物理特性一直为人们所关注。PbZrO3的有三个不同的相,在233℃以上为立方顺电相,具有钙钛矿结构,所属的空间群为Pm3m;当晶体处于233℃以下,将发生氧八面体的扭曲畸变和阳离子相对于O的移动,形成结构相变;230~233℃为正交铁电相,而230℃以下的基态为正交晶系,空间群为Pbam。基态正交相中离子移动主要由Pb、O之间的相对位移提供,由于相邻晶格之间Pb-O的位移相反,因此其为反铁电体。 1、原理及计算 采用第一性原理局域密度近似下的投影缀加平面波方法精确计算并比较了钙钛矿材料PbZrO3低温正交相(反铁电相)、高温立方相(顺电相)的电子能带结构,计算了PbZrO3材料正交相、立方相的电子结构。PbZrO3立方相的空间群为Pm3m,计算采用实验得到的晶格常量为a=4.11nm,Wyckoff坐标为Pb:(0,0,0),Zr:(0.5,0.5,0.5),O:(0.5,0.5,0)。正交相的空间群为Pmam,采用的晶格常数a=5.9411nm,b=11.8024nm,c=8.2564nm,各原子坐标见表1。正交相和立方相的多面体结构模型如图1所示。平面波截断能取为500eV,布里渊区积分分别采用5×5×5及7×3×5的K点网格,高斯展宽因子为0.1eV。 表1 正交相PbZrO3原胞内的原子位置
第一性原理简介
1什么是第一性原理? 根据原子核和电子互相作用的原理及其基本运动规律,运用量子力学原理,从具体要求出发,经过一些近似处理后直接求解薛定谔方程的算法,称为第一性原理。广义的第一原理包括两大类,以Hartree-Fock自洽场计算为基础的从头算和密度泛函理论(DFT)计算。 从定义可以看出第一性原理涉及到量子力学、薛定谔方程、Hartree-Fock自洽场、密度泛函理论等许多对我来说很陌生的物理化学定义。因此我通过向师兄请教和上网查资料一点点的了解并学习这些知识。 2第一性原理的作用 以密度泛函理论(DFT)为基础以及在此基础上发展起来的简单而具有一定精度的局域密度近似(LDA)和广义梯度近似(GGA)的第一性原理电子结构计算方法,与传统的解析方法一样,不但能够给出描述体系微观电子特性的物理量如波函数、态密度、费米面、电子间互作用势等,以及在此基础上所得到的体现体系宏观物理特性的参量如结合能、电离能、比热、电导、光电子谱、穆斯堡尔谱等等,而且它还可以帮助人们预言许多新的物理现象和物理规律。密度泛函计算的一些
结果能够与实验直接进行比较,一些应用程序的发展乃至商业软件的发布,导致了基于密度泛函理论的第一原理计算方法的广泛应用。 密度泛函理论(DFT)为第一性原理中的一类,在物理系、化学、材料科学以及其他工程领域中,密度泛函理论(DFT)及其计算已经快速发展成为材料建模模拟的一种“标准工具”。 密度泛函理论可以计算预测固体的晶体结构、晶格参数、能带结构、态密度(DOS)、光学性能、磁性能以及原子集合的总能等等。 3第一性原理怎么用? 目前我所学到的利用第一性原理的软件为Material Studio、V ASP软件。其中Materials Studio(简称MS)是专门为材料科学领域研究者开发的一款可运行在PC上的模拟软件。使化学及材料科学的研究者们能更方便地建立三维结构模型,并对各种晶体、无定型以及高分子材料的性质及相关过程进行深入的研究。模拟的内容包括了催化剂、聚合物、固体及表面、晶体与衍射、化学反应等材料和化学研究领域的主要课题。 模块简介 Materials Studio采用了大家非常熟悉的Microsoft标准用户界面,允许用户通过各种控制面板直接对计算参数和计算结果进行设置和分析。目前,Materials Studio软件包括如下功能模块: Materials Visualizer: 提供了搭建分子、晶体及高分子材料结构模型所需要的所有工具,可以操作、观察及分析结构模型,处理图表、表格或文本等形式的数据,并提供软件的基本环境和分析工具以及支持Materials Studio的其他产品。是Materials Studio产品系列的核心模块。 Discover: Materials Studio的分子力学计算引擎。使用多种分子力学和动力学方法,以仔细推导的力场作为基础,可准确地计算出最低能量构型、分子体系的结构和动力学轨迹等。
第一节第一性原理计算方法综述
第一性原理计算的理论方法 随着科技的发展,计算机性能也得到了飞速的提高,人们对物理理论的认识也更加的深入,利用计算机模拟对材料进行设计已经成为现代科学研究不可缺少的研究手段。这主要是因为在许多情况下计算机模拟比实验更快、更省,还得意于计算机模拟可以预测一些当前实验水平难以达到的情况。然而在众多的模拟方法中,第一性原理计算凭借其独特的精度和无需经验参数而得到众多研究人员的青睐,成为计算材料学的重要基础和核心计算。本章将介绍第一性原理计算的理论基础,研究方法和ABINIT软件包。 1.1 第一性原理 第一性原理计算( 简称从头计算,the abinitio calculation) ,指 从所要研究的材料的原子组分出发,运用量子力学及其它物理规律,通过自洽计算来确定指定材料的几何结构、电子结构、热力学性质和光学性质等材料物性的方法。基本思想是将多原子构成的实际体系理解成为只有电子和原子核组成的多粒子系统,运用量子力学等最基本的物理原理最大限度的对问题进行”非经验”处理。【1】第一性原理计算就只需要用到五个最基本的物理常量即( m o.e.h.c.k b ) 和元素周期表中各组分元素的电子结构,就可以合理地预测材料的许多物理性质。用第一性原理计算的晶胞大小和实验值相比误差只有几个百分点,其他性质也和实验结果比较吻合,体现了该理论的正确性。
第一性原理计算按照如下三个基本假设把问题简化: 1.利用Born-Oppenheimer 绝热近似把包含原子核和电子的多粒子问题转化为多电子问题。 2.利用密度泛函理论的单电子近似把多电子薛定谔方程简化为比较容易求解的单电子方程。 3.利用自洽迭代法求解单电子方程得到系统基态和其他性质。以下我将简单介绍这些第一性原理计算的理论基础和实现方法:绝热近似、密度泛函理论、局域密度近似(LDA)和广义梯度近似(GGA)、平面波及赝势方法、密度泛函的微扰理论、热力学计算方法和第一性原理计算程序包ABINIT。 1.2量子力学与Born-Oppenheimer 近似固体是由原子核和核外的电子组成的,在原子核与电子之间,电子与电子之间,原子核与原子核之间都存在着相互作用。从物理学的角度来看,固体是一个多体的量子力学体系【2】,相应的体系哈密顿量可以写成如下形式: H (r,R) E H(r ,R) (1-1) 其中r,R 分别代表所有电子坐标的集合、所有原子核坐标的集合。在不计外场作用下,体系的哈密顿量日包括体系所有粒子( 原子核和电子) 的动能和粒子之间的相互作用能,即 H H e H N H e N (1-2) 其中,以是电子部分的哈密顿量,形式为: 22 1 e2 H e(r) r2i 1 e(1-3)
第一性计算原理
Vasp 我所用第一原理是基于密度泛函(DFT)的从头计算,是以电子密度作为基本变量(HK定理),通过求解kohn-sham方程,迭代自洽得到体系的基态电子密度,然后求体系的基态性质。还有一种是基于hartree-fock自洽计算,通过自洽求解HF方程,获得体系的波函数,求基态性质。KS方程的计算水平达到了HF水平,同时还考虑了电子间的交换关联作用。关于DFT中密度泛函的Function其实是交换关联泛函,包括LDA,GGA,杂化泛函等等。一般LDA为局域密度近似,在空间某点用均匀电子气密度作为交换关联泛函的唯一变量,多数为参数化的CA-PZ方案;GGA为广义梯度近似,不仅将电子密度作为交换关联泛函的变量,也考虑了密度的梯度为变量,包括PBE,PE.RPBE等方案。 在处理计算体系中原子的电子态时有两种方法,一种是考虑所有电子叫做全电子法,比如WIEN2K中的FLAPW方法(线性缀加平面波);另一种是只考虑价电子而把芯电子和原子核构成离子实放在一起考虑即赝势法,一般贋势法是选取一个截断半径,截断半径以内波函数变化较平滑,和真实的不同,截断半径以外则和真实情况相同,而且贋势法得到的本征值和全电子法应该相同。贋势的测试标准应是贋势与全电子法计算结果的匹配度,而不是贋势与实验结果的匹配度,因为和实验结果的匹配可能是偶然的。 关于Ecut的收敛测试。一般情况下,总能相对于不同Ecut做计算,当截断能增大时总能变化不明显即可。但是在需要考虑体系应力时,还需要对应力进行收敛测试,而且应力相对于截断能要比总能更为苛刻。也就是某个截断能下总能已经收敛了,但应力未必收敛。(力的计算是在能量的基础上进行的,能量对坐标的一阶导数得到力。计算量的增大和误差的传递导致力收敛慢。) K点也是需要经过测试的。 何时需要考虑自旋?例如BaTiO3中,三个元素分别为=+2,+4,-2价,离子全部为各个轨道满壳层的结构,此时就不必考虑自旋了。对于BaMnO3中,由于Mn+4价时d轨道还有电子但未满,因此需要考虑Mn(4s23d5)的自旋,Ba和O就不必考虑。其实设定自旋就是给定一个原子磁矩的初始值,只在刚开始计算时作为初始值使用。 几何优化包括晶格常数和原子位置的优化,一般情况下也有不优化几何结构直接计算电子结构的,但是对于缺陷形成的计算则往往要优化。 软件大致分为基于平面波的软件,如CASTEP,PWSCF.ABINIT等,计算量大概和体系原子数目的三次方相关;还有基于原子轨道线性组合的软件,比如openmx等,计算量和体系原子数目相关,一般可模拟较多原子数目的体系。 V ASP是使用贋势和平面波基组,进行从头量子力学分子动力学计算的软件包。V ASP中的方法基于有限温度下的局域密度近似(用自由能作为变量)以及对每一MD步骤用有效矩阵对角方案和有效Pulay混合求解瞬时电子基态。这些技术可以避免元氏的Car-Parrinello 方法存在的一切问题,而后者是基于电子、离子运动方程同时积分的方法。离子和电子的相互作用超缓Vanderbilt贋势(US-PP)或投影扩充波(PAW)方法描述。两种技术都可以相当程度地减少过度金属或第一行元素的每个原子所必须的平面波数量。V ASP可以很容易地计算力与张力,用于把原子衰减到其瞬时基态中。!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! V ASP程序亮点: 1、使用PAW方法或超软贋势,因此基组尺寸非常小,描述材料一般需要原子不超过100 个平面波,大多数情况下甚至每原子50个平面波就能得到可靠结果。 2、2. 在平面波程序中,某些部分代码的执行是三次标度。在VASP中,三次标度部分的前 因子足可忽略,导致关于体系尺寸的高效标度。因此可以在实空间求解势的非局域贡献,
第一性原理
第一性原理的理解及其应用 第一性原理,英文First Principle,是一个计算物理或计算化学专业名词,广义的第一性原理计算指的是一切基于量子力学原理的计算。 我们知道物质由分子组成,分子由原子组成,原子由原子核和电子组成。量子力学计算就是根据原子核和电子的相互作用原理去计算分子结构和分子能量(或离子),然后就能计算物质的各种性质。 从头算(ab initio)是狭义的第一性原理计算,它是指不使用经验参数,只用电子质量,光速,质子中子质量等少数实验数据去做量子计算。但是这个计算很慢,所以就加入一些经验参数,可以大大加快计算速度,当然也会不可避免的牺牲计算结果精度。 根据原子核和电子互相作用的原理及其基本运动规律,运用量子力学原理,从具体要求出发,经过一些近似处理后直接求解薛定谔方程的算法,习惯上称为第一性原理[1]。 广义的第一原理包括两大类,以Hartree-Fork自洽场计算为基础的ab initio从头算,和密度泛函理论(DFT)计算。也有人主张,ab initio专指从头算,而第一性原理和所谓量子化学计算特指密度泛函理论计算。 第一性原理通常是跟计算联系在一起的,是指在进行计算的时候除了告诉程序你所使用的原子和他们的位置外,没有其他的实验的,经验的或者半经验的参量,且具有很好的移植性。作为评价事物的依据,第一性原理和经验参数是两个极端。第一性原理是某些硬性规定或推演得出的结论,而经验参数则是通过大量实例得出的规律性的数据,这些数据可以来自第一性原理(称为理论统计数据),也可以来自实验(称为实验统计数据)。 但是就某个特定的问题,第一性原理和经验参数没有明显的界限,必须特别界定。如果某些原理或数据来源于第一性原理,但推演过程中加入了一些假设(这些假设当然是很有说服力的),那么这些原理或数据就称为“半经验的”。 那为什么使用“第一性原理”这个字眼呢?据说这是来源于“第一推动力”这个宗教词汇。第一推动力是牛顿创立的,因为牛顿第一定律说明了物质在不受外力的作用下保持静止或匀速直线运动。如果宇宙诞生之初万事万物应该是静止的,后来却都在运动,是怎么动起来的呢?牛顿相信这是由于上帝推了一把,并且牛顿晚年致力于神学研究。现代科学认为宇宙起源于大爆炸,那么大爆炸也是有原因的吧。所有这些说不清的东西,都归结为宇宙“第一推动力”问题。 科学不相信上帝,我们不清楚“第一推动力”问题只是因为我们科学知识不完善。第一推动一定由某种原理决定。这个可以成为“第一原理”。爱因斯坦晚年致力与“大统一场理论”研究,也是希望找到统概一切物理定律的“第一原理”,可惜,这是当时科学水平所不能及的。现在也远没有答案。 但是为什么称量子力学计算为第一性原理计算?大概是因为这种计算能够从根本上计算出来分子结构和物质的性质,这样的理论很接近于反映宇宙本质的原理,就称为第一原理了。 第一性原理计算方法的应用
第三节第一性原理计算简介
第一性原理计算简介 在物理学中,第一性原理计算或称从头计算是指,基于构建物理学的基础定理,不作任何假设,例如:经验模型和拟合参数,所进行的计算研究。特别地,在凝聚态物理中,指的是运用薛定愕方程在一定的近似情况下,但不包括拟合实验数据所得到的参数和模型,对物质的电子结构进行计算r 从而得到所研究物质的性质的一种研究方法。近些年,随着计算机技术的飞速发展,其运算能力越来越强大,使得人们可以处理更庞大更繁杂的物质结构体系,同时也使得计算物理成为了现代物理学,尤其是在凝聚态物理领域的一个重要分支。众所周知,固体是由相对重且带正电的粒子——原子核,以及相对轻且带负电的粒子——电子聚集在一起构成的。如果有个原子,需要处理的问题是包含有N+ZN(Z 为原子核所含的质子的个数)个粒子的电磁相互作用,是一个多体问题。另一方面,由于处理的是微观粒子的运动,所以需要运用量子力学来描述其基本的运动规律和相互作用。对于该系统,精确的多粒子哈密顿量可以写作: i 2i i i 1122R H M ?=--∑∑ Fuuuuuuuuj 其中位于為处的原子核的质量为M,.,位于巧处的电子的质量为m 一第一项是原子核的动能算符,第二项是电子的动能算符。后三项分别是描述电子与原子核,单个电子与其它电子以及单个原子核与其它原子核之间的库伦相互作用。很显然,直接精确求解(1.64)式几乎是不可能的。为了在合理的近似条件下得到体系的本征值,需要作不同层次的近似。 1.3.1波恩-奥本海默(Bom-Oppenheimer)近似 由于原子核的质量远大于电子质量,所以,原子核的运动速度远小于电子。因此,可以将原子“冻结”在固定的位置,并假设电子在瞬时与原子核是平衡的。或者说,只有电子在这个多体问题中是考察对象,原子核仅仅被当作一个带正电的外源场,相对于电子云是外在独立的。该近似被称为波恩-奥本海默(Bom-Oppenheimer)近似。原来的多体问题被简化成在原子的静电势下,瓜个带负电的粒子的相互作用。波恩-奥本海默认为,原子核不再运动,其动能为零,因此,(1.64)式的第一项被消除,最后一项退化为常数。(1.64)式简化为只含有电子气的动能,电 子与电子之间的相互作用所产生的势能,以及电子在可看作外源的原子核的势中的势能。(1.64)式可重写为: H = f + V + V^, 值得注意的是,(1.65)式中的动能以及电子与电子间的相互作用只取决于所处理的是系统是多电子系统,而不是多质子系统中强的原子内部作用力,并不依赖于特定的多电子系统本身,例如,Br2或者水分子,Cu 还是Fe, bcc-Fe 还是fcc-Fe,等等。因此,前两项是普适的,包含特定系统信息的部分均在第三项中。 1.3.2 密度泛函理论(Density functional theory) 在波恩-奥本海默近似后,该量子多体问题得到了极大的简化,但是,依然很难直接求解。存在许多方法将方程(1.65)进一步近似变为易于处理的形式,历史上非常重要的是Hartree-Fock 方法。该方法在处理原子以及分子时效果很好,因此在量子化学中被广泛使用。但对于处理固体问题,其精度不够高。本文中使用的是更为现代且可能更强大的方法:密度泛函理论。 密度泛函理论的建立可以追溯到1964年Hohenberg 和Kohn[7]提出的两条定理。 1.3. 2.1 Hohenberg-Kohn 定理 两条定理的原始表述如下: 第一定理:多电子体系(原子,分子,固体)基态时的电荷密度pOO 与外源的势之间存在着一一