基因DNA序列BLAST及系统发育树构建操作步骤

基因DNA序列BLAST及系统发育树构建操作步骤
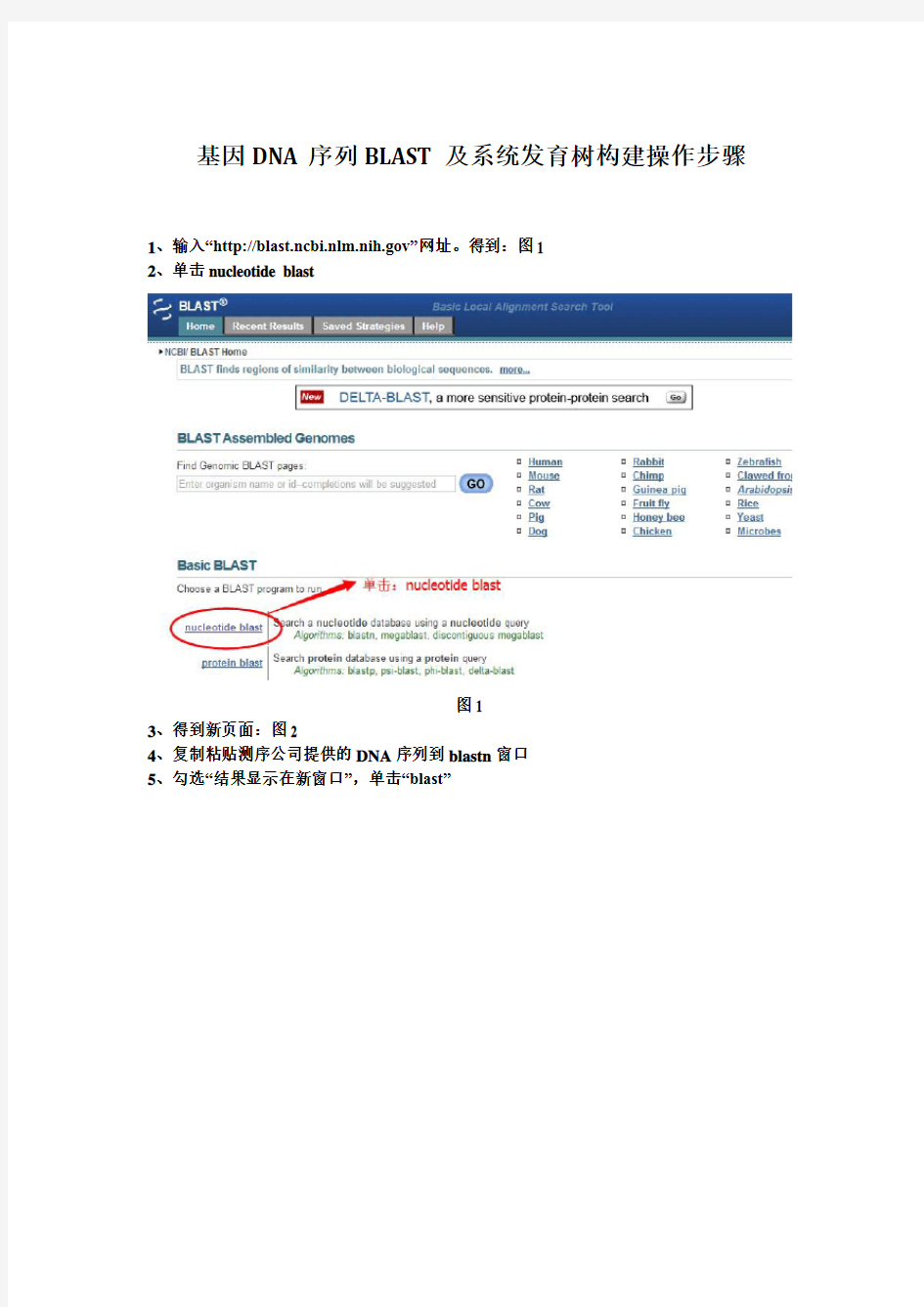
1、输入“https://www.360docs.net/doc/5415535863.html,”网址。得到:图1
2、单击nucleotide blast
图1
3、得到新页面:图2
4、复制粘贴测序公司提供的DNA序列到blastn窗口
5、勾选“结果显示在新窗口”,单击“blast”
图
2
6、得到新窗口:图3
图3
7、下拉页面,如:图4
8、选择Accession下的基因编码。入选第一个“HQ711983.1”
图4
9、得到新窗口如:图5
10、单击FASTA
图5
11、得到如:图6
12、复制菌株种名跟16SrDNA序列,并粘贴到“.txt”文件中
图6
13、新建“.txt”文件,如:图7
14、把测序菌株16SrDNA序列和选取blast所得的匹配度高的菌株16SrDNA序列粘贴在“.txt”文件下,在菌株种名前加“>”,每个序列后空一行
图7
15、复制“.txt”文件,改“.txt”文件为“.fasta”文件,如:图8
图8
16、用MEGA 4.0.2软件打开“.fasta”文件,如:图9
图9
17、单击Alignment里的Alinment Explorel/CLUSAC,如:图10
图10
18、单击Alignment里的Align by clustalW,如:图11
图11
19、在弹出窗口中单击OK,如:图12
图12 19、在弹出窗口中单击OK,如:图13
图13 20、等待数据运算,如:图14
图14
21、运算结束,关闭当前窗口。如:图15
22、在弹出的第一个窗口单击NO
图15 23、在弹出的第一个窗口单击YES,如:图16
图16
24、将文件以“.meg”格式保存,如:图17
图17
25、关闭其余窗口,打开刚刚保存的“.meg”文件,如:图18
图18
26、选择phyloeny下的Bootsteap Test of phylogeng下的Neighbor-Joining...,如:图19
图19
27、在弹出窗口单击Compulte,如:图20
图20
28、单击树状图,得到系统发育树,建议截图保存,如:图21
图21
系统发育树构建步骤
如何建树 step 1. 将16S rDNA序列在NCBI上进行BLAST比对(https://www.360docs.net/doc/5415535863.html,/BLAST/) BLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990 [62];1997[63])。国际著名生物信息中心都提供基于Web的BLAST服务器。BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为内核向两端延伸,以找出尽可能长的相似序列片段。首先登录到提供BLAST服务的常用网站,比如国内的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。这些网站提供的BLAST服务在界面上差不多,但所用的程序有所差异。它们都有一个大的文本框,用于粘贴需要搜索的序列。把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。如果是DNA序列,一般选择BLASTN搜索DNA数据库。这里以NCBI为例。登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。BLASTN结果如何分析(参数意义): 例如: >gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete sequence Score = 2020 bits (1019), Expect = 0.0 Identities = 1382/1497 (92%), Gaps = 8/1497 (0%) Strand = Plus / Plus Query: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggaaaggccctttcgggggt 60 |||||||||||||||||||||||||||||||||||||||||| ||||||||| ||||| Sbjct: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggtaaggcccttc--ggggt 58 Query: 61 actcgagcggcgaacgggtgagtaacacgtgggtaacctgccttcagctctgggataagc 120 || ||||||||||||||||||||||||||||||| | |||||| ||||||||||||| Sbjct: 59 acacgagcggcgaacgggtgagtaacacgtgggtgatctgcctcgtactctgggataagc 118 其中,Score指的是提交的序列和搜索出的序列之间的分值,越高说明越相似。Expect指的是比对的期望值。比对越好,expect越小,一般在核酸层次的比对,expect小于1e-10,就比对很好了,多数情况下为0。Identities指的是提交的序列和参比序列的相似性,如上所指为1497个核苷酸中二者有1382个相同。Gaps指的是一般翻译成空位,指的是对不上的碱基数目。Strand指的是链的方向,Plus / Minus意味着提交的序列和参比序列是反向互补的,如果是Plus / Plus则二者皆为正向。 挑选与目的菌株具有较近亲源关系的模式种(type strain)序列将这些序列用记事本保存成dna.seq文件。 >M.mulatta AAGCTTTTCT GGCGCAACCA TCCTCATGAT >M.fascicularis AAGCTTCTCC GGCGCAACCA CCCTTA TAA T step 2. 用CLUSTALX对已知DNA序列做多序列比对 1 双击clustalx.exe运行程序。 2 点File→Load Sequence,打开dna.seq。
全基因组关联分析的原理和方法
全基因组关联分析(Genome-wide association study;GWAS)是应用基因组中 数以百万计的单核苷酸多态性(single nucleotide ploymorphism ,SNP)为分子 遗传标记,进行全基因组水平上的对照分析或相关性分析,通过比较发现影响复杂性状的基因变异的一种新策略。 随着基因组学研究以及基因芯片技术的发展,人们已通过GWAS方法发现并鉴定了大量与复杂性状相关联的遗传变异。近年来,这种方法在农业动物重要经济性状主效基因的筛查和鉴定中得到了应用。 全基因组关联方法首先在人类医学领域的研究中得到了极大的重视和应用,尤其是其在复杂疾病研究领域中的应用,使许多重要的复杂疾病的研究取得了突破性进展,因而,全基因组关联分析研究方法的设计原理得到重视。 人类的疾病分为单基因疾病和复杂性疾病。单基因疾病是指由于单个基因的突变导致的疾病,通过家系连锁分析的定位克隆方法,人们已发现了囊性纤维化、亨廷顿病等大量单基因疾病的致病基因,这些单基因的突变改变了相应的编码蛋白氨基酸序列或者产量,从而产生了符合孟德尔遗传方式的疾病表型。复杂性疾病是指由于遗传和环境因素的共同作用引起的疾病。目前已经鉴定出的与人类复杂性疾病相关联的SNP位点有439 个。全基因组关联分析技术的重大革新及其应用,极大地推动了基因组医学的发展。(2005年, Science 杂志首次报道了年龄相关性视网膜黄斑变性GWAS结果,在医学界和遗传学界引起了极大的轰动, 此后一系列GWAS陆续展开。2006 年, 波士顿大学医学院联合哈佛大学等多个研究机构报道了基于佛明翰心脏研究样本关于肥胖的GWAS结果(Herbert 等. 2006);2007 年, Saxena 等多个研究组联合报道了与2 型糖尿病( T2D ) 关联的多个位点, Samani 等则发表了冠心病GWAS结果( Samani 等. 2007); 2008 年, Barrett 等通过GWAS发现了30 个与克罗恩病( Crohns ' disrease) 相关的易感位点; 2009 年, W e is s 等通过GWAS发现了与具有高度遗传性的神经发育疾病——自闭症关联的染色体区域。我国学者则通过对12 000 多名汉族系统性红斑狼疮患者以及健康对照者的GWAS发现了5 个红斑狼疮易感基因, 并确定了4 个新的易感位点( Han 等. 2009) 。截至2009 年10 月, 已经陆续报道了关于人类身高、体重、 血压等主要性状, 以及视网膜黄斑、乳腺癌、前列腺癌、白血病、冠心病、肥胖症、糖尿病、精神分 裂症、风湿性关节炎等几十种威胁人类健康的常见疾病的GWAS结果, 累计发表了近万篇 论文, 确定了一系列疾病发病的致病基因、相关基因、易感区域和SNP变异。) 标记基因的选择: 1)Hap Map是展示人类常见遗传变异的一个图谱, 第1 阶段完成后提供了 4 个人类种族[ Yoruban ,Northern and Western European , and Asian ( Chinese and Japanese) ] 共269 个个体基因组, 超过100 万个SNP( 约1
构建系统发育树需要注意的几个问题
构建系统发育树需要注意的几个问题 1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。 2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。 3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。 4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。 5 枝长可以用来表示类间的真实进化距离。 6 重要的是理解系统发育分析中的计算能力的限制。任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。 7 没有一种方法能够保证一颗系统发育树一定代表了真实进化途径。然而,有些方法可以检测系统发育树检测的可靠性。第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样(bootstrap),来检测他们统计上的重要性。 分子进化研究的基本方法 对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。 表型的(phenetic)和遗传的(cladistic)数据有着明显差异。Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(species tree)、基因树等等一些相同或含义略有差异的名称。 系统进化树分有根(rooted)和无根(unrooted)树。有根树反映了树上物种或基
如何查找基因的序列(全)
如何查找基因序列?(转载) (2010-08-01 11:47:41) 如何查找基因序列? ——在Genbank中寻找目的基因的实例 ——献给受类似问题困扰的广大酷友,以及给我动力和信心发表原创帖的基因酷的朋友们。 酷友感言:网络的世界很精彩,网络的查询很无奈。为了我们的科学研究事业,为了我们能够顺利毕业,我们的广大酷友们在网络的海洋里遨游…遨游…咋就找不到彼岸呢?今天要设计这个基因的PCR引物,明天又要查那个基因的信息,那么大一张网,唉想起来就郁闷……鉴此,我们推出了利用Genbank查找基因序列的帖子,希望对大家有所帮助,并请大家多多指教!当然,如果您已经是此中高手,那就权当我是班门弄斧了,呵呵。 1. 根据文献 搞reasearch肯定要读文献的,如果你曾经在文献中看到过你感兴趣的基因,而且文中还提到了该基因在Genbank中的ID号,那就好办了,直接打开https://www.360docs.net/doc/5415535863.html,,在Search后的下拉框中选择Nucleotide,把Genbank ID号输入GO前面的文本框中,点“GO”,就可以找到他了。 举例说明,例如:在2003年JBC的文章(Conditional Knock-out of
Integrin-linked Kinase Demonstrates an Essential Role in Protein Kinase B/Akt Activation)中出现了“calreticulin (GenBank accession number gi 16151096)”,那么把“16151096”输入GO前面的文本框中,点“GO”,就可以找到该基因了(当然包括基因序列等相关信息)。 在出现了检索结果界面(下图)后,直接点击红箭头所指的 AY047586就可以看到基因的相关信息了...(呵呵,是不是有点太......easy 了) 这里需要指出一下,在显示基因的页面右侧有一个Link,点击后出现一个小菜单,里面是与该基因相关的链接,很有用的,值得一个一个地去看看,这里我就不多说了。点击 AY047586后出现的界面如下:如果你只想获得序列(例如去设计PCR引物的时候),那就可以选择FASTA,这样就得到了FASTA格式的序列文件,没有其他数字和格式的干扰。 (缩略图,点击图片链接看原图)这就是FASTA格式的序列: (缩略图,点击图片链接看原图)2. 根据已经获得的基因的相关信息进行查找(待续......) 鼓励一下吧,累坏了正如路漫漫所说,如果只是知道基因的名字,怎么查序列呢?还是举例说明,比如我想做的基因名称是人的VEGF基因,那么怎么在Genbank中找到它呢?还是一步一步来...打开https://www.360docs.net/doc/5415535863.html,/ 在search后面的下拉框中选择Gene,然后在中间的文本框中输入基
分子进化与系统进化树的构建
分子进化与系统进化树的构建 分子进化与系统进化树的构建 分子进化与系统进化树的构建 主要内容: 1、分子进化的研究方法 2、系统进化树的构建方法 3、系统进化树构建常用软件汇集 4、系统进化树构建方法及软件的选择 5、Phylip分子进化分析软件包简介及使用 6、如何利用MEGA3.1构建进化树 声明: 1、本篇涉及的资源主要源于网络及相关书籍,由酷友搜集、分析、整理、审改,供大家学习参考用,如有转载、传播请注明源于基因酷及本篇的工作人员;若本篇侵犯了您的版权或有任何不妥,请Email genecool@https://www.360docs.net/doc/5415535863.html,告知。 2、由于我们的学识、经验有限,本篇难免会存在一些错误及缺陷,敬请不吝赐教:请到基因酷论坛(https://www.360docs.net/doc/5415535863.html,/bbs)本篇对应的专题跟贴指出或Email genecool@https://www.360docs.net/doc/5415535863.html,。 致谢: 整编者:flashhyh 主要参考资料:《生物信息学札记》樊龙江;《分子进化分析与相关软件的应用》作者不详;《进化树构建》ZHAO Yangguo;《如何用MEGA 3.1构建进化树》作者不详;《MEGA3指南》作者不详; 分子进化的研究方法 分子进化的研究方法 分子进化的研究方法 分子进化研究的意义 自20世纪中叶,随着分子生物学的不断发展,进化研究也进入了分子进化(molecularevolution)研究水平,并建立了一套依赖于核酸、蛋白质序列信息的理论和方法。随着基因组测序计划的实施,基因组的巨量信息对若干生物领域重大问题的研究提
供了有力的帮助,分子进化研究再次成为生命科学中最引人注目的领域之一。这些重大问题包括:遗传密码的起源、基因组结构的形成与演化、进化的动力、生物进化等等。分子进化研究目前更多地是集中在分子序列上,但随着越来越多生物基因组的测序完成,从基因组水平上探索进化奥秘,将开创进化研究的新天地。 分子进化研究最根本的目的就是从物种的一些分子特性出发,从而了解物种之间的生物系统发生的关系。通过核酸、蛋白质序列同源性的比较进而了解基因的进化以及生物系统发生的内在规律。 分子进化研究的基础 假设假设::核苷酸和氨基酸序列中含有生物进化历史的全部信息核苷酸和氨基酸序列中含有生物进化历史的全部信息。。 分子钟理论:在各种不同的发育谱系及足够大的进化时间尺度中,许多序列的进化速率几乎是恒定不变的。如下图: 直系同源与旁系同源 直系同源(orthologs):同源的基因是由于共同的祖先基因进化而产生的; 旁系同源(paralogs):同源的基因是由于基因复制产生的。 两者之间的关系如下图所示: 注:用于分子进化分析中的序列必须是直系同源的用于分子进化分析中的序列必须是直系同源的 用于分子进化分析中的序列必须是直系同源的,才能真实反映进化过程。 分子进化研究的基本方法 对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。 表型的(phenetic)和遗传的(cladistic)数据有着明显差异。Sneath 和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(speciestree)、基因树等等一些相同或含义略有差异的名称. 系统进化树分有根(rooted)和无根(unrooted)树。有根树反映了树上物种或基因的时间顺序,而无根树只反映分类单元之间的距离而不涉及谁是谁的祖先问题。下图表示了
如何构建系统发育树
如何构建系统发育树 Bioinformatics2009-11-03 10:45 阅读159 评论0 字号:大中小小 (2009-06-11 22:44:13) 标签:系统发育树构建系统发育树分子生物学发育分析it 转自丁香园 构建系统发育树需要注意的几个问题 1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。 2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。 3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。 4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。 5 枝长可以用来表示类间的真实进化距离。 6 重要的是理解系统发育分析中的计算能力的限制。任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。 7 没有一种方法能够保证一棵系统发育树一定代表了真实进化途径。然而,有些方法可以检测系统发育树检测的可靠性。第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样,来检测他们统计上的重要性。 分子进化研究的基本方法 对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。 表型的(phenetic)和遗传的(cladistic)数据有着明显差异。Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(speciestree)、基因树等等一些相同或含义略有差异的名称. 系统进化树分有根(rooted)和无根(unrooted)树。有根树反映了树上物种或基因的时间顺序,而无根树只反映分类单元之间的距离而不涉及谁是谁的祖先问题。 用于构建系统进化树的数据有二种类型:一种是特征数据(characterdata),它提供了基因、个体、群体或物种的信息;二是距离数据(distancedata)或相似性数据(similaritydata),它涉及的则是成对基因、个体、群体或物种的信息。距离数据可由特征数据计算获得,但反过来则不行。这些数据可以矩阵的形式表达。距离矩阵(distancematrix)是在计算得到的距离数据基础上获得的,距离的计算总体上是要依据一定的遗传模型,并能够表示出两个分类单位间的变化量。系统进化树的构建质量依赖于距离估算的准确性。 一1) 打开clustal X,载入上述序列,“load sequences”→“output format options”: “CLASTAL FORMA T”;CLASTAL SEQUENCES NUMBERS:ON; ALIGNMENT PARAMETERS: “RESET NEW GAPS BEFOR ALIGNMENT” “MULTIPLE ALIGNMENT PARAMETERS”→设置相关参数 2) “DO COMPLETE ALIGNMENT”→FILE→SA VE AS,掐头去尾。 3) 打开MEGA4,FILE→CONVERT TO MEGA FORMA TE→SA VE→FILE→OPEN DA TA→CONTAINING PROTAIN SEQUENCES? NO →PHYLOGENY→BOOTSTRAP TEST OF PHYLOGENY→N J → 设置相关参数。最后看到系统发育树 二这里要介绍的是Bioedit-Mega建树法,简单实用,极易上手。 1 将所测得的序列在NCBI上进行比对,这个就不多讲了。 2 选取序列保存为text格式。 3 运行Bioedit,使用其中的CLUSTAL W进行比对。 4 运用MEGA 4 建树,首先将前面的文件转化格式为mega格式,然后进行激活,最后进行N-J建树。
系统发育树构建方法优劣
1.邻接法邻接法(neighbor-joiningmethod,NJ)由Saitou和Nei(1987)提出,NJ法是基于最小进化原理经常被使用的一种算法,它不检验所有可能的拓扑结构,能同时给出拓扑结构和分支长度。在重建系统发生树时,它取消了UPGMA法所做的假定,认为在进化分支上,发生趋异的次数可以不同。最近的计算机模拟已表明它是最有效的基于距离数据重建系统树的方法之一。该方法通过确定距离最近(或相邻)的成对分类单位来使系统树的总距离达到最小。它的特点是重建的树相对准确,假设少,计算速度快,只得一棵树。其缺点主要表现在将序列上的所有位点等同对待,且所分析序列的进化距离不能太大。故NJ法适用于进化距离不大,信息位点少的短序列。邻接法在距离建树中经常会用到,而不用理会使用什么样的优化标准。完全解析出的进化树是通过对完全没有解析出的“星型”进化树进行“分解”得到的,分解的步骤是连续不断地在最接近(实际上是最孤立的)的序列对中插入树枝,而保留进化树的终端。于是,最接近的序列对被巩固了,而“星型”进化树被改善了,这个过程将不断重复。这个方法相对而言很快,也就是说,对于一个50个序列的进化树,只需要若干秒甚至更少。 2.最大简约法最大简约法(maximum parsimony method,MP)最早是基于形态特征分类的需要发展起来的,具体的算法有许多不同版本,其中有些已被广泛地应用于分子进化研究中。利用MP方法重建系统发生树,实际上是一个对给定OTUs其所有可能的树进行比较的过程。对某一个可能的树,首先对每个位点祖先序列的核苷酸组成做出推断,然后统计每个位点用来阐明差异的核苷酸最小替换数目。在整个树中,所有信息简约位点最小核苷酸替换数的总和称为树的长度(常青和周开亚,1998)。MP法是一种优化标准,这种标准遵循“奥卡姆剃刀原则(Occam’S Razor principle)”:对数据最好的解释也是最简单的,而最简单的所需要的特别假定也最少。MP法基于进化过程中所需核苷酸(或氨基酸)替代数目最少的假说,对所有可能正确的拓扑结构进行计算并挑选出所需替代数最小的拓扑结构作为最优系统树,也就是通过比较所有可能树,选择其中长度最小的树作为最终的系统发生树,即最大简约树(maximum parsimony tree)。与其他建树方法相比,MP法无需引入处理核苷酸或者氨基酸替代时所必需的假设(替代模型)。同时,MP法对于分析某些特殊的分子数据(如插入序列和插入/缺失)有用。在分析的序列位点上没有回复突变或平行突变,且被检验的序列位点数很大的时候,MP法能够获得正确的(真实)系统树。但MP法推导的树不是唯一的,在分析序列上存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,最大简约法可能会出现建树错误。故MP法适用于序列残基差别小,具有近似变异率,包含信息位点比较多的长序列。 3.最大似然法最大似然法(maximum likelihood method,MI。)是20世纪60年代末期由于对地生物信息学分析实践震波和水声信号等处理的需要而发展起来的一种非线性谱估计方法。最早由凯佩用这种方法对空间阵列接收信号进行频率波数谱估值,后来推广到对时问信号序列的功率谱估值。 最大似然法最早应用于系统发育分析是在对基因频率数据的分析上。其原理是考虑到每个位点出现残基的似然值,将每个位置所有可能出现的残基替换概率进行累加,产生特定位点的似然值。MI。法对所有可能的系统发育树都计算似然函数,似然函数值最大的那棵树即为最可能的系统发育树。利用最大似然法来推断一组序列的系统发生树,需首先确定序列进化的模型,如Jukes—Cantor模型、Kimura二参数模型及一般二参数模型等。在进化模型选择合理的情况下,MI。法是与进化事实吻合最好的建树算法。其缺点是计算强度非常大,极为耗时。
全基因组从头测序(de novo测序)
全基因组从头测序(de novo测序) https://www.360docs.net/doc/5415535863.html,/view/351686f19e3143323968936a.html 从头测序即de novo 测序,不需要任何参考序列资料即可对某个物种进行测序,用生物信息学分析方法进行拼接、组装,从而获得该物种的基因组序列图谱。利用全基因组从头测序技术,可以获得动物、植物、细菌、真菌的全基因组序列,从而推进该物种的研究。一个物种基因组序列图谱的完成,意味着这个物种学科和产业的新开端!这也将带动这个物种下游一系列研究的开展。全基因组序列图谱完成后,可以构建该物种的基因组数据库,为该物种的后基因组学研究搭建一个高效的平台;为后续的基因挖掘、功能验证提供DNA序列信息。华大科技利用新一代高通量测序技术,可以高效、低成本地完成所有物种的基因组序列图谱。包括研究内容、案例、技术流程、技术参数等,摘自深圳华大科技网站 https://www.360docs.net/doc/5415535863.html,/service-solutions/ngs/genomics/de-novo-sequencing/ 技术优势: 高通量测序:效率高,成本低;高深度测序:准确率高;全球领先的基因组组装软件:采用华大基因研究院自主研发的SOAPdenovo软件;经验丰富:华大科技已经成功完成上百个物种的全基因组从头测序。 研究内容: 基因组组装■K-mer分析以及基因组大小估计;■基因组杂合模拟(出现杂合时使用); ■初步组装;■GC-Depth分布分析;■测序深 度分析。基因组注释■Repeat注释; ■基因预测;■基因功能注释;■ ncRNA 注释。动植物进化分析■基因家族鉴定(动物TreeFam;植物OrthoMCL);■物种系统发育树构建; ■物种分歧时间估算(需要标定时间信息);■基因组共线性分析; ■全基因组复制分析(动物WGAC;植物WGD)。微生物高级分析 ■基因组圈图;■共线性分析;■基因家族分析; ■CRISPR预测;■基因岛预测(毒力岛); ■前噬菌体预测;■分泌蛋白预测。 熊猫基因组图谱Nature. 2010.463:311-317. 案例描述 大熊猫有21对染色体,基因组大小2.4 Gb,重复序列含量36%,基因2万多个。熊猫基因组图谱是世界上第一个完全采用新一代测序技术完成的基因组图谱,样品取自北京奥运会吉祥物大熊猫“晶晶”。部分研究成果测序分析结果表明,大熊猫不喜欢吃肉主要是因为T1R1基因失活,无法感觉到肉的鲜味。大熊猫基因组仍然具备很高的杂合率,从而推断具有较高的遗传多态性,不会濒于灭绝。研究人员全面掌握了大熊猫的基因资源,对其在分子水平上的保护具有重要意义。 黄瓜基因组图谱黄三文, 李瑞强, 王俊等. Nature Genetics. 2009. 案例描述国际黄瓜基因组计划是由中国农业科学院蔬菜花卉研究所于2007年初发起并组织,并由深圳华大基因研究院承担基因组测序和组装等技术工作。部分研究成果黄瓜基因组是世界上第一个蔬菜作物的基因组图谱。该项目首次将传
全基因组重测序大数据分析报告
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。 3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:
基因组序列的差异分析
基因组序列的差异分析 ----mVISTA的在线使用说明 当然,除了在线版的,我们还可以在网站上填写信息申请离线的软件。但我试用了一下,需要先自己比对,然后要按照一定的格式来制作文件,当然你还必须得安装java才能运行软件;总之,我感觉没有在线版的方便。 1 将数据放入服务器中 在首页,你将被要求确定你想要分析的基因组序列的数量。输入这个数字之后,点击“提交”,将带你到主提交页面。 mVISTA服务器最多可以同时处理100条序列。 1.1主提交页面必填的内容 E-mail 地址 通过E-mail,我们可以提示你的在线处理已经得到结果。
序列 你可以用2种方式来上传你的序列: 1.使用“Browse”按钮从你的电脑上,上传纯文本的Fasta格式文件。如果是一个作为参 考的生物体的DNA序列必须作为一个contig提交(可以进行一定的定向排列将多个片段合并为一个contig),而其他非参考序列可以在一个或多个contig中提交(draft)。 Fasta格式的示例序列(您可以在NCBI站点上找到关于该格式的更多细节): >mouse ATCACGCTCTTTGTACACTCCGCCATCTCTCTCT … !!!注意:序列里面我们只接受字母CAGTN和X。请确保提交序列是作为一种纯文本格式,而不是Word或HTML文件格式。 如果您以FASTA格式提交序列,我们建议您为它取一个有意义的名称(比如直接是你的物种名之类的),因为这些名称将出现在我们生成的图形中。如果您使用的是一个draft草图序列,那么结果中每个contigs的命名都将按照您在“>”符号后指示的命名进行。 2.您可以给出它的GenBank登录号,系统将自动从GenBank数据库里进行检索序列。 在这两种情况下,序列的总大小都不应超过10M,而且任何一条序列都不应超过2M。 1.2主提交页面选填的内容 这些选项允许您自定义您的VISTA分析。您可以使用独立获得的基因注释,选择合适的Repeat Masker选项,给分析的序列指定名称,并改变序列保存分析的参数。如果您没有填写这些选填选项,我们将使用它们的默认值。 比对程序 根据您分析的具体内容(参见“about”-链接中的详细信息),您可以选择以下比对程序之一:1、AVID----全局两两比对。如果您选择使用这个程序,其中一个序列应该被完成比对,其他 所有序列可以完成或以草图draft格式完成。对于集合中所有已完成的序列,AVID生成所有相对所有成对的比对结果,可以使用任何序列作为基础(参考)来显示。如果某些序列是草图格式,AVID将生成它们与最终序列的比对,这将被用作基础(参考)。这是该服务器上唯一可以处理草图序列的比对程序。 (小知识:草图序列与完整序列DNA sequence, draft: Sequence of a DNA with less accuracy than a finished sequence. In a draft sequence, some segments are missing or are in the wrong order or are oriented incorrectly. A draft sequence is as opposed to a finished DNA sequence.)2、LAGAN----完成完整序列的全局两两比对和多重比对。如果某些序列是草图格式,您的查 询将被重定向到AVID以获得两两比对。多重比对将由VISTA可视化,它将计算并显示序列的保守区,以您指示的任何序列作为参考。这是该服务器上唯一能够产生真正的多重
人类全基因组测序
1 技术优势 全基因组测序(Whole Genome Sequencing,WGS)是利用高通量测序平台对人类不同个体或群体进行全基因组测序,并在个体或群体水平上进行生物信息分析。可全面挖掘DNA 水平的遗传变异,为筛选疾病的致病及易感基因,研究发病及遗传机制提供重要信息。 全基因组测序 平台优势 HiSeq X 测序平台 读长:PE150 通量:1.8T/run 测序周期:3 天 专为人全基因组测序准备、测序周期短、通量高
生物信息分析 技术路线 技术参数 样品要求 样本类型:DNA 样品 样本总量:≥1.0 μg DNA (提取自新鲜及冻存样本) ≥1.5 μg DNA (提取自FFPE 样本)样品浓度:≥ 20 ng/μl 测序平台及策略HiSeq X PE150 测序深度 肿瘤:癌组织(50X),癌旁组织/血液样本(30X)遗传病:30~50 X 项目周期37天
3 案例解析 该研究选取3个家系中6个患者和1个正常个体,首先使用基因芯片寻找纯合突变位点,然后对其中无亲缘关系的2例患者采用全基因组测序研究,在2例患者非编码区域均发现相同的变异,10号染色体PTF1A 末端发生一个点突变(chr10:23508437 A>G),且变异在患病人群和细胞试验中均得到了验证。研究解释了生长发育启动子隐性变异是罕见孟德尔遗传病的常见致病原因,同时说明许多疾病的致病突变也可能位于非编码区。 图1 检出的变异信息 智力障碍是影响新生儿心智发育的一类疾病。这项研究选取50个经过基因芯片和全外显子测序未确诊致病因子的trio 家系,全基因组测序检出84个de novo SNVs 和8个de novo CNVs,及一些结构变异(如VPS13B、STAG1、IQSEC2-TENM3),检出率为42%。揭示编码区的de novo SNVs 和de novo CNVs 是导致智力障碍的主要因素,全基因组测序可以作为可靠的遗传性检测应用工具。 案例一 单基因病研究——全基因组测序鉴定PTF1A末端增强子常染色体隐性突变导致胰腺 发育不全[1] 案例二 复杂疾病研究——全基因组测序解析智力障碍的主要致病因素[2] 图2 PTF1A 的家系图谱
构建系统进化树的方法步骤
构建系统进化树的方法步骤 1. 建树前的准备工作 1.1 相似序列的获得——BLAST BLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。国际著名生物信息中心都提供基于Web的BLAST服务器。BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为内核向两端延伸,以找出尽可能长的相似序列片段。 首先登录到提供BLAST服务的常用网站,比如国内的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。这些网站提供的BLAST服务在界面上差不多,但所用的程序有所差异。它们都有一个大的文本框,用于粘贴需要搜索的序列。把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。如果是DNA序列,一般选择BLASTN搜索DNA数据库。 这里以NCBI为例。登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。 BLASTN结果如何分析(参数意义): >gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete sequence Score = 2020 bits (1019), Expect = 0.0 Identities = 1382/1497 (92%), Gaps = 8/1497 (0%) Strand = Plus / Plus Query: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggaaaggccctttcgggggt 60 |||||||||||||||||||||||||||||||||||||||||| ||||||||| ||||| Sbjct: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggtaaggcccttc--ggggt 58 Query: 61 actcgagcggcgaacgggtgagtaacacgtgggtaacctgccttcagctctgggataagc 120 || ||||||||||||||||||||||||||||||| | |||||| ||||||||||||| Sbjct: 59 acacgagcggcgaacgggtgagtaacacgtgggtgatctgcctcgtactctgggataagc 118 Score :指的是提交的序列和搜索出的序列之间的分值,越高说明越相似;
三、全基因组序列分析--基因组学的新内容
三、全基因组序列分析--基因组学的新内容1.数据存放。 2.碱基百分含量分析。无论是GC富含区还是AT富含区,都可能是一些特殊功能的区域。 肺炎支原体GC百分含量高和GC百分含量低的区域对应于重组值较低的区域,包括着丝粒和端粒,而尿殖道支原体GC百分含量最低的区域对应于rRNA和tRNA。流感嗜血杆菌GC百分含量高的区域也对应于6个rRNA基因。 3.ORF分析。首先要用多个不同的软件来要找到并估测基因组中的每一个ORF。 通过比较确知其功能的; 在数据库中有相匹配的蛋白质序列,但不知其能的; 在数据库中找不到任何相匹配蛋白质序列的新基因。 1995年,J.C. Venter所领导的TIGR(The Institute of Genomic Reseach)完成了第一个单细胞自由生物基因组,流感嗜血杆菌(Haemopophilus influenzae Rd)全序列测定。 1996年他们又完成了拥有最小基因组的单细胞生物尿殖道支原体(Mycoplasma genitalium)和一种不同于原核、真核生物的单细胞生物--产甲烷古细菌(Methanococcus jannaschi) 的全序列测定。德国人则测定了肺炎支原体(Mycoplasma pneumoniae)基因组全序列。
与此同时,历时七年(1989-1996年)的第一个真核生物酿酒酵母 (Saccharomyces cevevisiae)基因组计划在欧共体及美、日、加、 英等各国实验室共同努力下得以完成。 1997年大肠杆菌(Escherichia. Coli S)的基因组计划完成,美丽隐 杆线虫(caenothabditis elegans)的基因组计划也于1998年完成。 最受瞩目的人类基因组计划(HGP, Human GenomeProject)也将 于2000年底前完成。 (1)通过流感嗜血杆菌能量代谢类群的ORF分析,了解到在这种生物中缺乏三羧酸循环(TCA)中必需的三个酶,即柠檬酸合成酶基因、异柠檬酸脱氢酶基因和顺乌头酸酶基因。由此推断流感嗜血杆菌TCA缺失,不能合成谷氨酸,因为谷氨酸的供体是TCA的中间产生物α-酮戊二酸。 (2)在尿殖道支原体基因组中有一个称为MgPa的ORF。考察全基因组,共发现有9个与MgPa同源的重复序列,这些重复序列之间发生重组可能诱导尿殖道支原体群体中抗原性改变,帮助细菌逃避宿主免疫攻击。
构建系统进化树的详细步骤
构建系统进化树的详细步骤 1. 建树前的准备工作 1.1 相似序列的获得——BLAST BLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool 的缩写,意 为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。国际著名生物信息中心 都提供基于Web的BLAST服务器。BLAST算法的基本思路是首先找出检测序列和目标序 列之间相似性程度最高的片段,并作为核向两端延伸,以找出尽可能长的相似序列片段。 首先登录到提供BLAST服务的常用,比如国的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。这些提供的BLAST服务在界面上差不多,但所用的程序有所差异。它 们都有一个大的文本框,用于粘贴需要搜索的序列。把序列以FASTA格式(即第一行为说明 行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是 任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就 可以开始搜索了。如果是DNA序列,一般选择BLASTN搜索DNA数据库。 这里以NCBI为例。登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。 BLASTN结果如何分析(参数意义): >gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete sequence Score = 2020 bits (1019), Expect = 0.0 Identities = 1382/1497 (92%), Gaps = 8/1497 (0%) Strand = Plus / Plus